

Abstract
Neural stem/progenitor cell (NSPC) proliferation and self-renewal, as well as insult-induced differentiation, decrease markedly with age. The molecular mechanisms responsible for these declines remain unclear. Here, we show that levels of NAD+ and nicotinamide phosphoribosyltransferase (Nampt), the rate-limiting enzyme in mammalian NAD+ biosynthesis, decrease with age in the hippocampus. Ablation of Nampt in adult NSPCs reduced their pool and proliferation in vivo. The decrease in the NSPC pool during aging can be rescued by enhancing hippocampal NAD+ levels. Nampt is the main source of NSPC NAD+ levels and required for G1/S progression of the NSPC cell cycle. Nampt is also critical in oligodendrocytic lineage fate decisions through a mechanism mediated redundantly by Sirt1 and Sirt2. Ablation of Nampt in the adult NSPCs in vivo reduced NSPC-mediated oligodendrogenesis upon insult. These phenotypes recapitulate defects in NSPCs during aging, giving rise to the possibility that Nampt-mediated NAD+ biosynthesis is a mediator of age-associated functional declines in NSPCs.
Keywords: adult neural stem cells, aging, differentiation, Nampt, proliferation
Introduction
Aging manifests as a physiological breakdown of robustness, leading to a significant functional decline, increased vulnerability to environmental insults, and many different diseases in a variety of tissues and organs. Cognitive decline is one such example, and cognitive impairment, even without dementia, occurs in 22% of people over age 71 in the United States (Plassman et al, 2008). With this prevalence, efforts to understand the mechanisms underlying age-associated cognitive decline and to develop therapeutic strategies to prevent it have become particularly poignant. With these goals in mind, the regulation of adult neurogenesis has been an intense focus of research. Throughout the lives of all mammals, including humans, two distinct populations of neural stem cells (NSCs) are maintained in the brain: one in the subgranular zone (SGZ) of the dentate gyrus (DG) and the other in the subventricular zone (SVZ) bordering the lateral ventricles. NSCs have the ability to self-renew and to differentiate into transient amplifying progenitors (collectively referred to as neural stem/progenitor cells or NSPCs), which in turn undergo finite, lineage-restricted cell divisions to produce neurons, oligodendrocytes, or astrocytes (Deng et al, 2010; Artegiani & Calegari, 2012; Jadasz et al, 2012).
Aging is one of the strongest negative regulators of adult NSPC proliferation (Artegiani & Calegari, 2012). In mice, the number of proliferating NSPCs in the SGZ declines exponentially in the first 9 months of life (Ben Abdallah et al, 2010), and they are almost completely absent by 24 months of age (Jin et al, 2003; Lugert et al, 2010; Encinas et al, 2011). Aging is also associated with impaired oligodendrocyte differentiation and remyelination in response to insult (Sim et al, 2002; Franklin & Ffrench-Constant, 2008). Given the potential of NSPCs to contribute to cognitive function and their ability to proliferate and differentiate into the major cell types of the brain (Deng et al, 2010; Artegiani & Calegari, 2012; Jadasz et al, 2012), it is of great importance to understand key signaling pathways regulating the NSPC decisions of proliferation versus quiescence and self-renewal versus terminal differentiation. NSPCs can be reactivated in the aged brain (Decker et al, 2002; Jin et al, 2003; Lugert et al, 2010). Therefore, elucidation of the key signaling pathways that are involved in age-associated decline in NSPC functionality will provide critical insight into preventing age-associated cognitive decline.
We have previously found that aging significantly reduces levels of the essential cofactor nicotinamide adenine dinucleotide (NAD+) in multiple peripheral tissues (Yoshino et al, 2011). This age-associated decrease in NAD+ levels is due to a decline in protein levels of nicotinamide phosphoribosyltransferase (Nampt), the rate-limiting enzyme in the biosynthetic pathway of NAD+ from nicotinamide (Fig1A) (Revollo et al, 2004; Yoshino et al, 2011). Nampt converts nicotinamide, a major precursor in mammalian NAD+ biosynthesis, and 5′-phosphoribosyl-1-pyrophosphate to nicotinamide mononucleotide (NMN), a key NAD+ intermediate. NMN is then converted to NAD+ by nicotinamide/nicotinic acid mononucleotide adenylyltransferase (Nampt) (Revollo et al, 2004, 2007). We and others have previously reported that the expression of Nampt in the brain is extremely low compared to peripheral tissues (Revollo et al, 2007; Friebe et al, 2011). However, Nampt has uniquely strong expression in the hippocampus (Zhang et al, 2010; Wang et al, 2011a). Because recent studies show that the energetic demands of stem cell proliferation and lineage specification require distinct metabolic programs (Folmes et al, 2012), we hypothesized that NSPCs would be particularly sensitive to changes in NAD+ levels and accordingly alter their proliferation, self-renewal, and differentiation.
Figure 1. Hippocampal NAD+ levels and Nampt expression decline with age.

A NAD+ biosynthesis from nicotinamide. Nicotinamide phosphoribosyltransferase (Nampt) converts nicotinamide and 5′-phosphoribosyl-1-pyrophosphate (PRPP) to nicotinamide mononucleotide (NMN). Nicotinamide/nicotinic acid mononucleotide adenylyltransferase (Nampt) converts NMN and adenosine-5′-triphosphate (ATP) to NAD+. While NAD+ is commonly used in redox reactions, cells primarily require NAD+ as a co-substrate for several families of enzymes, one of which is the sirtuin family of protein deacetylases. The sirtuin family includes Sirt1 and Sirt2, which cleave NAD+ at its glycosidic bond, releasing nicotinamide (Stein & Imai, 2012). Red text indicates inhibitors used in subsequent experiments.
B HPLC analysis of NAD+ levels in hippocampal extracts (1 month, n = 5; 3–4 months, n = 16; 6 months, n = 10; 10–12 months, n = 28).
C, D Quantification of immunofluorescence for Nampt in the subgranular zone (SGZ). Measurement of thresholded levels of Nampt immunoreactivity (C) and the number of highly immunoreactive Nampt+ cells (D) along the SGZ (n = 5).
E Representative images of immunofluorescence for Dapi (blue) and Nampt (red) in the SGZ in young (6-month-old) and old (18-month-old) mice. Dotted lines denote the SGZ. Scale bars denote 20 μm.
Data information: Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Here, we show that hippocampal NAD+ and Nampt levels decrease with age. Nampt mediates NSPC NAD+ biosynthesis. Pharmacological inhibition and genetic ablation of Nampt-mediated NAD+ biosynthesis in NSPCs impair NSPC proliferation, self-renewal, and formation of oligodendrocytes in vivo and in vitro. Furthermore, augmentation of NAD+ levels during aging with NMN administration maintains the NSPC pool. Thus, enhancing NAD+ levels in NSPCs may be an effective intervention to preserve the endogenous NSPC population to repair the aged, diseased, or damaged brain.
Results
Hippocampal NAD+ levels and Nampt expression decline with age
Since aging significantly reduces levels of Nampt and NAD+ in multiple peripheral tissues (Yoshino et al, 2011), we hypothesized that aging also reduces Nampt-mediated NAD+ biosynthesis in the brain, particularly in the hippocampus, affecting the function of NSPCs. To address this hypothesis, we first measured NAD+ levels in hippocampi isolated from 1-, 3- to 4-, 6-, and 10- to 12-month-old C57Bl6 mice. NAD+ levels gradually decreased with age, reaching 63% in 10- to 12-month-old mice compared to that of 1-month-old mice (Fig1B). Consistent with this finding, quantifying Nampt immunoreactivity in the SGZ of the DG by both a thresholded level of Nampt intensity and a count of the number of thresholded Nampt+ cells demonstrated that 18-month-old mice exhibit 52–66% of the Nampt immunoreactivity present in 6-month-old mice (Fig1C–E). These results suggest that Nampt-mediated NAD+ biosynthesis in the hippocampus declines with age at a time course similar to that of NSPC proliferation (Ben Abdallah et al, 2010).
Nampt is expressed in a subpopulation of SGZ NSPCs
Previously, it was reported that Nampt is predominantly expressed in hippocampal neurons, but not in stellate astrocytes (Zhang et al, 2010; Wang et al, 2011a). Consistent with this finding, immunohistochemistry for Nampt and cell type-specific markers revealed that almost all NeuN+ neurons in the granule layer of the DG expressed Nampt, while almost no S100β+ glial cells did (Supplementary Fig S1A–E). However, we also noticed that many intensely Nampt immunoreactive cells along the SGZ of the DG did not express NeuN (Supplementary Fig S1B and E). Since NSPCs engage in the energetically costly tasks of proliferation and differentiation (Folmes et al, 2012), we performed co-immunohistochemistry for NSPC markers (Sox2+, radial Gfap+) and found that a majority of NSPCs expressed Nampt (Fig2A–D). To assess in vivo colocalization between Nampt and Nestin, we crossed mice expressing Cre recombinase under the Nestin promoter (Nestin-CreERT2) (Lagace et al, 2007) to a GFP reporter mouse strain that expresses a loxP-flanked STOP cassette that prevents transcription of the downstream enhanced GFP (see the Materials and Methods section), generating iNSPC-GFP mice. Nampt also colocalized with GFP driven by the Nestin promoter (NestinGFP, Fig2C and D) (Lagace et al, 2007). Quantification of these observations revealed that along the SGZ, 32% of Sox2+ cells, 55% of radial Gfap+ cells, and 78% of NestinGFP+ cells expressed Nampt (Fig2D, Supplementary Fig S1F). Additionally, Ki67+ and Olig2+ cells along the SGZ also expressed Nampt (Supplementary Fig S1G–I). To confirm that Nampt is highly expressed in NSPCs, we cultured NSPCs from the hippocampi of postnatal pups as neurospheres. Neurospheres showed 22 or 32% higher expression levels of Nampt than did whole hippocampal extracts taken from postnatal (P12) or adult mice (2.5–4.5 months), respectively (Fig2E), indicating that NSPCs have higher expression levels of Nampt compared to other hippocampal cell types. Together, these results suggest that Nampt is expressed in a large subpopulation of NSPCs.
Figure 2. Nampt is expressed in a subpopulation of SGZ NSPCs.
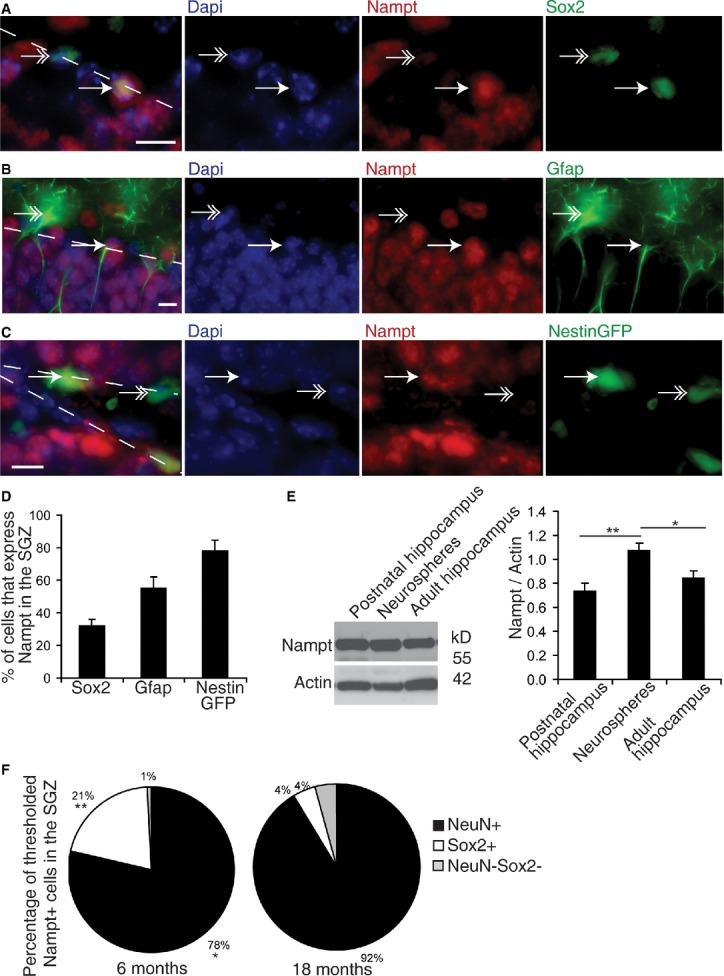
A–C Representative images of immunofluorescence for Dapi (blue), Nampt (red), and NSPC markers (Sox2, Gfap, and NestinGFP 3 days post-tamoxifen injection; green) in the SGZ. Dotted lines denote the SGZ. Single arrows indicate examples of colocalization. Double arrows indicate examples of non-colocalization. Scale bars denote 10 μm.
D Quantification of the percentages of NSPC marker-positive cells in the SGZ that also express Nampt in 3- to 6-month-old mice. At least 350 cells from 7 to 14 mice were assessed per group.
E A representative immunoblot and quantification of immunoblots for Nampt normalized by actin in neurospheres cultured from postnatal mice (n = 6 independent samples, 16 replicates), as well as hippocampal tissue extracts (HC) isolated from either postnatal (n = 12) or adult mice (n = 12).
F Nampt immunoreactivity was thresholded and the number of highly immunoreactive Nampt + cells along the SGZ was assessed for colocalization with the neuronal marker NeuN or the NSPC marker Sox2 in the subgranular zone (SGZ, n = 5).
Data information: Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Having found that Nampt expression is present in both neurons and NSPCs, we next asked which cell populations lose Nampt expression with age. To address this question, we thresholded Nampt immunoreactivity and assessed the thresholded Nampt+ cells for colocalization with the neuronal marker NeuN and the NSPC marker Sox2. With age, the percentage of intensely Nampt immunoreactive cells that colocalized with NeuN increased slightly, whereas the percentage of intensely Nampt immunoreactive cells that colocalized with Sox2 decreased from 21 to 4% (Fig2F). Similarly, in the SGZ, the percentage of NeuN+ that expressed Nampt increased with age, while the percentage of Sox2+ cells that expressed Nampt decreased (Supplementary Fig S1E and F). Thus, at least part of the decrease in Nampt expression in the SGZ with age is due to the loss of expression from Sox2+ NSPCs.
Adult NSPC-specific deletion of Nampt impairs NSPC self-renewal in vivo
Having seen enrichment of Nampt in NSPCs, we next asked whether inactivating Nampt specifically in adult NSPCs could recapitulate age-associated phenotypic changes in NSPC functionality in vivo. To address this question, we generated adult NSPC-specific inducible Nampt-knockout mice by crossing Namptflox/flox mice (Rongvaux et al, 2008) with Nestin-CreERT2 mice (iNSPC-Nampt-KO mice). To trace the progeny of adult NSPCs in which Nampt was inactivated and to confirm the specificity and magnitude of the deletion induced by tamoxifen, we also crossed iNSPC-Nampt-KO mice to the aforementioned iNSPC-GFP mice. After tamoxifen injection, these mice expressed NestinGFP in the SGZ and SVZ but not in non-neurogenic regions of the brain such as the corpus callosum or cortex (Supplementary Fig S2A and B). Immunohistochemistry and recombination PCR for NestinGFP confirmed that there was undetectable recombination present in vehicle-injected mice (Supplementary Fig S2A and C). To verify that the NestinGFP+ population consisted of NSPCs, we costained for the NSPC markers Sox2 and Gfap. 61% of Sox2+ cells and 34% of radial Gfap+ cells coexpressed NestinGFP 7 days post-tamoxifen (Supplementary Fig S2D). We also verified Nampt deletion efficiency by quantifying the percentage of NestinGFP+ cells that expressed Nampt 3 and 7 days post-tamoxifen injection. At 3 days post-tamoxifen injection, the percentage of NestinGFP+ cells that expressed Nampt in iNSPC-Nampt-KO mice was 40% less than littermate controls, and at 7 days post-tamoxifen injection, the percentage of NestinGFP+ cells that expressed Nampt was reduced by 62% (Supplementary Fig S2E).
To assess the cumulative effect of loss of Nampt on NSPC proliferation, we deleted Nampt in iNSPC-Nampt-KO mice at 6 weeks of age with three rounds of five consecutive days of tamoxifen injections, separated by 6 weeks (Fig3A). We then assessed control and iNSPC-Nampt-KO mice for the expression of lineage-specific markers by immunohistochemistry (Fig3B). In iNSPC-Nampt-KO mice, we found that the Nestin+ NSPC pool was significantly decreased by 49% in the DG (Fig3C). Indeed, incorporation of BrdU and the population of proliferating cells [Ki67+ (von Bohlen und Halbach, 2011)] were also decreased by 22% (P = 0.019) and 35% (P = 0.064), respectively (Fig3D and E). Consistent with this defect in the NSPC pool and proliferation, the pool of newborn neurons [doublecortin, Dcx+ (von Bohlen und Halbach, 2011)] was also significantly decreased by 26% (Fig3F and G). In contrast, we did not observe any significant difference in the maturation of newborn neurons, as assessed by categorization of Dcx+ cells with no or horizontal projections as immature neurons and Dcx+ cells with vertical projections spanning the granule cell layer as mature neurons (Supplementary Fig S2F). We next assessed NSPC/daughter cell survival by immunostaining for activated caspase-3. Only rare activated caspase-3+ cells were observed in both neurogenic and non-neurogenic regions of the brain (Supplementary Fig S2A and B), and these rare activated caspase-3+ cells were never observed in GFP+ cells in iNSPC-Nampt-KO DG, providing evidence against a potential contribution of cell death to the observed effects.
Figure 3. Adult NSPC-specific deletion of Nampt impairs NSPC proliferation and self-renewal in vivo.
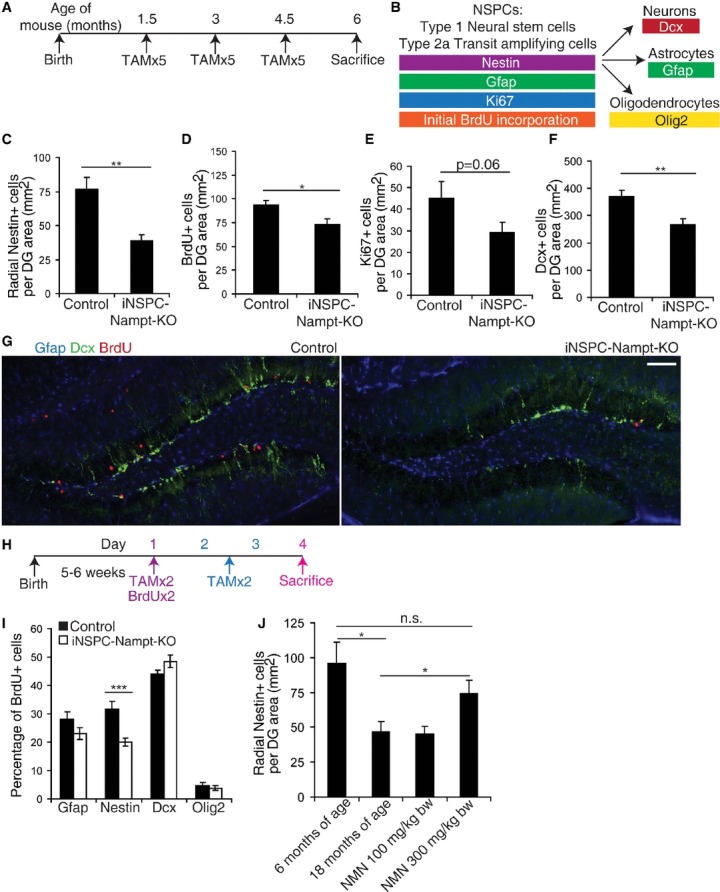
A To assess proliferation, iNSPC-Nampt-KO mice and littermate controls were subjected to three rounds of five tamoxifen (TAM) injections (one injection per day, 6 weeks apart). Sacrifice was performed at 6 months of age.
B A scheme for the specificity of the markers assessed.
C–F Quantification of radial Nestin+ NSPCs (n = 15–16 mice) (C), BrdU+ proliferating cells (n = 14–16 mice) (D), Ki67+ proliferating cells (n = 7 mice) (E), and newborn neurons (Dcx+, n = 15–20 mice) (F), per unit area of the dentate gyrus (DG) in control and iNSPC-Nampt-KO mice. For BrdU labeling, four injections of BrdU at 100 mg/kg body weight were given intraperitoneally over 48 h.
G Representative images of immunofluorescence for Gfap (blue), Dcx (green), and BrdU (red) in the subgranular zone (SGZ). Scale bar denotes 200 μm.
H To assess differentiation, control littermates and iNSPC-Nampt-KO mice were subjected to four total TAM injections (two injections on the first day coupled with BrdU at 100 mg/kg body weight as well as two total injections on the subsequent 2 days).
I Quantification of the percentage of BrdU+ cells in the DG that also express markers of NSPCs (Gfap+, Nestin+), newborn neurons (Dcx+), and OPCs/oligodendrocytes (Olig2+) (n = 6–13 mice).
J Quantification of radial Nestin+ NSPCs in 6- and 18-month-old C57Bl6 mice and 18-month-old C57Bl6 mice treated with 100 or 300 mg/kg body weight NMN in their drinking water for 12 months (n = 5 mice).
Data information: Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001; n.s., not significant.
To assess the acute effect of loss of Nampt on NSPC fate decisions, we induced deletion of Nampt at 6 weeks of age with four total tamoxifen injections followed by sacrifice 72 h after the first injection (Fig3H). To facilitate assessment of differentiation, we labeled dividing cells by injecting the mice with BrdU concurrently with the first day of tamoxifen treatment. iNSPC-Nampt-KO mice displayed significantly reduced levels of colocalization of BrdU with radial Nestin+ cells (Fig3I), suggesting decreased self-renewal decisions. However, iNSPC-Nampt-KO mice exhibited normal levels of BrdU colocalization with neuronal (Dcx+), astrocytic (Gfap+, also marks NSPCs), and oligodendrocytic (Olig2+) markers, indicating that alterations in differentiated cell lineage decisions were undetectable under basal conditions. The lack of increase in colocalization of BrdU with cell type-specific markers may imply that a larger percentage of BrdU+ cells have failed to differentiate in iNSPC-Nampt-KO mice. iNSPC-Nampt-KO NSPCs could have stalled during differentiation after losing Nestin expression.
While NSPC proliferation declines exponentially throughout life (Artegiani & Calegari, 2012), quiescent NSPCs can be reactivated in the aged murine hippocampus by multiple environmental stimuli (Decker et al, 2002; Jin et al, 2003; Lugert et al, 2010). If NSPCs quiesce due to reduced NAD+ levels, systemic administration of NMN may be able to correct age-associated defects in NSPC functionality. Intraperitoneal injection of NMN (500 mg/kg body weight) increased hippocampal NAD+ levels 34–39% within 15 min, strongly suggesting that NMN can cross the blood–brain barrier (Supplementary Fig S2G). To see whether NMN supplementation can maintain NSPC proliferation and self-renewal with age, we treated 6-month-old mice with NMN at the daily dose of 100 or 300 mg/kg body weight in their drinking water until 18 months of age. The number of Nestin+ cells along the SGZ was significantly lower in the 18-month-old control mice relative to 6-month-old mice, as previously reported (Encinas et al, 2011) (Fig3J). Remarkably, mice treated with 300 mg/kg body weight NMN showed improved maintenance of the type 1 (radial Nestin+) population with age. However, the population of proliferating cells (Ki67+) remained similar to controls (Supplementary Fig S2H). While not statistically significant, the population of newborn neurons (Dcx+) tended to increase (Supplementary Fig S2I). The age-related depletion of the NSPC pool is thought to be caused by an increase in terminal fate decisions relative to self-renewal fate decisions (Encinas et al, 2011). Thus, it is possible that NMN administration maintains the NSPC pool by preventing the age-associated increase in terminal fate decisions.
Inhibition of Nampt in NSPCs in vitro impairs NAD+ biosynthesis and proliferation
Having shown that NAD+ levels in the hippocampus and Nampt expression in the SGZ decreased with age, we next asked whether Nampt mediates NSPC-specific NAD+ biosynthesis by using hippocampal neurospheres as the in vitro NSPC culture model. We treated neurospheres with a highly specific Nampt inhibitor, FK866, at a dosage and duration (10 nM, 48 h) that has little to no effect on cellular viability (Hasmann & Schemainda, 2003). Strikingly, FK866 reduced NAD+ levels in neurospheres to 4% of controls, a decrease completely rescued by concurrent NMN treatment ((Fig4A, Supplementary Fig S3A), strongly suggesting that Nampt activity is the predominant source of NAD+ biosynthesis in NSPCs.
Figure 4. Inhibition of Nampt in NSPCs in vitro impairs NAD+ biosynthesis and proliferation.

- HPLC analysis of NAD+ levels (n = 6).
- Quantification of the fold increase of cell number in neurospheres (n = 6–30).
- Representative bright-field image of neurospheres. Scale bars denote 10 μm.
- Cell cycle-related pathways among the top 50 biological pathways downregulated by FK866. Parametric analysis of gene enrichment (PAGE) was conducted based on microarray analyses. See the Materials and Methods section.
- Quantitative RT–PCR results for mRNA expression of cyclin E2 (Ccne2), cyclin E1 (Ccne1), cyclin A2 (Ccna2), and E2F1 (n = 3).
- FACS analysis of FK866-treated NSPCs (n = 8).
We next examined how inhibition of Nampt affects neurosphere proliferation. Consistent with the decreases in the NSPC pool and in NSPC proliferation in iNSPC-Nampt-KO mice, FK866 reduced NSPC number by 61% after 48 h, but not 24 h, of treatment (Fig4B and C, Supplementary Fig S3B). To distinguish whether this decrease in cell number was due to an inhibition of proliferation or enhancement of death, we analyzed the protein levels of markers of proliferation, apoptosis, and autophagy. Expression of the proliferation markers Ki67 and PCNA decreased 87 and 43%, respectively (Supplementary Fig S3C–E), whereas levels of activated caspase-3 became detectable and levels of the autophagy marker, glycosylated LC3B, were unchanged. Consistent with these observations, parametric analysis of gene set enrichment (PAGE) of a microarray performed on neurospheres treated with FK866 showed that out of the top 50 downregulated pathways, 13 of them were related to the cell cycle, while none of the top 50 upregulated pathways were involved in cell death (Fig4D, Supplementary Fig S3F and G). Analysis of specific gene changes by qRT–PCR revealed that cyclins E and A, the two cyclins required for cellular progression from G1 to S, as well as their upstream transcriptional regulator E2F1 (Wong et al, 2011), were the primary cell cycle factors affected by this treatment (Fig4E). These alterations in gene expression indicated that reducing Nampt activity stalls NSPCs at G0/G1. Supporting this notion, FACS analysis of neurospheres demonstrated that FK866 treatment increased the proportion of NSPCs in G0/G1 and decreased the proportion in S phase (Fig4F).
Genetic ablation of Nampt in NSPCs in vitro impairs NAD+ biosynthesis, proliferation, and differentiation
To assess the effect of chronic Nampt ablation on NSPC functionality, we genetically ablated Nampt by infecting neurospheres from Namptflox/flox mice with Cre recombinase- or LacZ-expressing (control) adenoviruses. Neurospheres infected with Cre recombinase (Nampt Ad-Cre) at passage 1 exhibited a 94% reduction in Nampt mRNA expression 3 days post-deletion, and the corresponding decreases in Nampt protein expression and NAD+ levels appeared 6 days post-deletion (Supplementary Fig S4A–E). Eight days post-deletion, NSPCs exhibited a 73% reduction in NAD+ levels that was rescued by concurrent NMN administration, further supporting the notion that Nampt activity is the predominant source of NSPCs NAD+ levels (Fig5A).
Figure 5. Genetic ablation of Nampt in NSPCs in vitro impairs NAD+ biosynthesis, proliferation, and differentiation.

Neurospheres were isolated from Namptflox/flox mice and infected with a Cre recombinase-expressing adenovirus (Nampt AD-Cre) or a control adenovirus expressing LacZ (Nampt AD-LacZ).
A HPLC analysis of NAD+ levels with and without NMN (100 μM, 48 h) (n = 10–22).
B, C Quantification of the fold increase in cell number (B) (n = 13–50) and neurosphere diameter (C) (n = 9 independent samples, 57–96 neurospheres).
D Representative images of neurospheres 7 days after dissociation. Scale bars denote 10 μm.
E The number of neurospheres formed 7 days after plating dissociated cells at 100 cells/ml, 0.5 ml/well in 24-well plates (n = 8 independent samples, 48–84 wells).
F, G Nampt Ad-Cre- and Nampt AD-LacZ-infected neurospheres were cultured without NMN until Nampt Ad-Cre-infected neurospheres exhibited a growth defect. Cultures were then passaged and plated at equal density with or without NMN (200 μM). Fold increases in cell number (F) (n = 6) and the percentages of total Dapi+ cells that express Ki67+ cells were quantified (G) (n = 3 independent samples, 9 fields of view).
H–L The percentages of total Dapi+ cells that express the indicated cell type-specific markers (H) by immunofluorescence after 6–7 days of differentiation: O4 (I), Gfap (J), and B-III-tubulin (K) (n = 3–6 independent samples, 23–43 fields of view). The effect of NMN was also examined for O4, S100β, TUNEL, and Nestin (L) (n = 3–6 independent samples, 10–26 fields of view). *, ^, and # indicate statistical significance between Nampt AD-LacZ and Nampt AD-Cre, Nampt AD-LacZ and Nampt AD-LacZ+NMN, and Nampt AD-Cre and Nampt AD-Cre+NMN, respectively.
Data information: Data are presented as mean ± SEM. *, ^, #P < 0.05; **P < 0.01; ***, ###P < 0.001.
Like FK866-treated cultures, proliferating Nampt Ad-Cre-infected NSPCs displayed reduced cell number (Fig5B). Remarkably, Nampt Ad-Cre NSPCs were unable to increase their cell number between 24 and 144 h of culture. In contrast, Nampt AD-LacZ-infected cells were able to exponentially increase their cell number over 13-fold in this time frame. Consistent with this finding, Nampt Ad-Cre-infected NSPCs also showed a 49% reduction in diameter relative to Nampt AD-LacZ-infected NSPCs, indicative of reduced proliferation (Fig5C and D). Since NSPC self-renewal decisions can also contribute to cell number, we assessed secondary neurosphere formation, an assay that quantifies the ability of neurosphere inhabitant cells to reformulate neurospheres upon dissociation. Nampt Ad-Cre-infected cells generated 63% fewer secondary neurospheres than did Nampt AD-LacZ-infected cells (Fig5E). Nampt AD-LacZ and Nampt Ad-Cre NSPCs exhibited no difference in the percentages of TUNEL- or activated caspase-3-positive cells as well as no difference in activated caspase-3 immunoreactivity as detected by immunoblotting, indicating that the observed phenotypes upon loss of Nampt are not primarily due to cell death (Supplementary Fig S4E and F). To see whether Nampt Ad-Cre-infected neurospheres can be reactivated to proliferate, we plated equal numbers of Nampt AD-LacZ and Nampt Ad-Cre cells after the second passage and cultured them in the presence or the absence of NMN. Remarkably, NMN treatment was able to fully reactivate the proliferative potential of Nampt Ad-Cre cells (Fig5F and G). Collectively, these results suggest that Nampt-mediated NAD+ biosynthesis is critical for NSPCs to successfully progress through the cell cycle.
Whereas we did not observe a difference in NSPC fate decisions in the neurogenic environment of the SGZ in vivo, we detected a decrease in self-renewal decisions. To see whether this was true in the absence of the influences of the SGZ niche, we differentiated dissociated neurospheres and assessed the proportion of resulting cell types by immunofluorescence after 6–7 days of differentiation induced by the removal of growth factors (Fig5H, Supplementary Fig S4G). To our surprise, differentiated Nampt Ad-Cre NSPCs exhibited a 90% reduction in oligodendrocytes (O4+, Fig5I). In contrast, Nampt Ad-Cre-infected NSPCs exhibited no change in the generation of Gfap+ cells (Fig5J). Genetic knockdown of Nampt also significantly but more mildly decreased the generation of neurons (43% decrease, β-III-tubulin+, Fig5K). Thus, the decrease in oligodendrocytes was not due to an increase in neuronal fate. As Gfap can recognize both NSPCs and mature astrocytes, we employed Nestin and S100β to distinguish whether the decrease in oligodendrocytes we observed upon Nampt knockdown was due to a cell fate choice in these directions. While there was no detectable change in the generation of S100β+ mature astrocytes in Nampt Ad-Cre cultures, there was a fourfold increase in the percentage of Nestin+ cells (6% in Nampt Ad-LacZ cells; 23% in Nampt Ad-Cre cells), suggesting quiescence rather than precocious astrocytic differentiation (Fig5L). Importantly, all of these effects were completely rescued by treatment with NMN. However, we also observed a mild increase in TUNEL+ cell death under these conditions (33% increase relative to Nampt Ad-LacZ cells), implying the possibility of a minor contribution of cell death to this phenotype. Together, these data suggest that genetic knockdown of Nampt prevents the successful differentiation of oligodendrocytes from NSPCs, potentially due to quiescence as indicated by the retention of Nestin staining.
Genetic knockdown of Nampt impairs OPC formation in vitro
Since differentiation using a non-specific lineage differentiation protocol (by the removal of growth factors) revealed a specific requirement for Nampt in the successful generation of O4+ immature oligodendrocytes, we next asked which stage of NSPC differentiation into oligodendrocytes depends on Nampt by employing a differentiation protocol that promotes the oligodendrocyte lineage (Fig6A, Supplementary Fig S5A). As previously observed using a non-specific lineage differentiation protocol, the proportion of O4+ intermediate oligodendrocytes was dramatically decreased in Nampt Ad-Cre cultures at 6–7 days post-differentiation (Fig6B). Furthermore, we observed that ablation of Nampt resulted in a decreased pool of OPCs (Pdgfrα+), but an increased pool of Nestin+ NSPCs. The proportion of Gfap+ astrocytes/NSPCs also mildly increased. To investigate whether the depletion of the OPC population was preexisting to or induced upon differentiation, we assessed the OPC population present during proliferation (Supplementary Fig S5B) and after 2 days of differentiation (Fig6C), a time point that enriches for OPCs. Indeed, both of these time points also showed the loss of OPCs (Pdgfrα+, Olig2+). These results support our notion that Nampt is necessary for NSPCs to differentiate into OPCs.
Figure 6. Genetic ablation of Nampt in vitro impairs OPC formation.

A A scheme for oligodendrocyte differentiation with stage-specific markers.
B, C Neurospheres were infected with a Cre recombinase-expressing adenovirus (Nampt AD-Cre) or a control adenovirus expressing LacZ (Nampt AD-LacZ). Markers of NSPCs (Gfap, Nestin), OPCs (Pdgfrα+), and oligodendrocyte lineage cells (Olig2+, O4+) were assessed (n = 3–9 independent samples, 6–51 fields of view). (B) To assess oligodendrocyte formation, dissociated neurospheres were harvested after 6–7 days of differentiation. (C) To assess OPC formation, dissociated neurospheres were examined after 2 days of differentiation.
D Treatment of dissociated neurospheres with the selective inhibitor of Sirt1, EX527 (80 μM), or the selective inhibitor of Sirt2, AGK2 (10 μM). The formation of oligodendrocytes was evaluated after 6–7 days of differentiation (n = 6–11 independent samples, 21–32 fields of view).
E–G Knockout and control neurospheres were formed by infecting with a Cre recombinase-expressing adenovirus or a control adenovirus expressing LacZ, respectively. (E) Neurospheres were isolated from Sirt1flox/flox mice and Sirt1 flox/flox; Sirt2−/− mice. The formation of oligodendrocytes was evaluated after 6–7 days of differentiation (n = 3–11 independent samples, 12–28 fields of view). (F, G) Neurospheres were isolated from Namptflox/flox mice (F, n = 8–9) or Sirt1 flox/flox; Sirt2−/− mice (G, n = 3–7) and differentiated for 2 days. Quantitative RT–PCR results for mRNA expression of oligodendrocyte lineage genes.
Data information: Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
How does Nampt promote oligodendrogenesis? Among NAD+-consuming enzymes, the sirtuins Sirt1 and Sirt2 have been suggested to be involved in oligodendrogenesis (Li et al, 2007; Ji et al, 2011; Rafalski et al, 2013). Furthermore, we observed that Sirt2 was upregulated during oligodendrocyte differentiation in vitro and expressed in the SGZ in Nampt+ cells and NestinGFP+ NSPCs (Supplementary Fig S5C–F). To examine whether Sirt1 and Sirt2 play a role in Nampt-mediated NSPC differentiation into oligodendrocytes, we acutely treated NSPCs with the selective inhibitor of Sirt2, AGK2, or the Sirt1 inhibitor, EX527. Surprisingly, whereas both inhibitors acutely suppressed oligodendrocyte formation (O4+, Fig6D), neither chronic ablation of Sirt2 in the NSPCs isolated from Sirt2−/− mice nor Cre adenovirus-mediated knockdown of Sirt1 in Sirt1flox/flox-derived neurospheres affected oligodendrogenesis (Pdgfrα+, Olig2+, O4+), except that Sirt1 deficiency affected the production of O4+ intermediate oligodendrocytes (Fig6E). Since Sirt1 and Sirt2 have multiple redundant targets (Luo et al, 2001; Jin et al, 2008; Rothgiesser et al, 2010), we speculated that they may have compensatory roles in the regulation of oligodendrogenesis. To test this possibility, we generated Sirt1/Sirt2-double-knockout (Sirt1/2 DKO) neurospheres. Consistent with the inhibitor studies (Fig6D), dissociated Sirt1/2 DKO neurospheres were unable to form oligodendrocyte lineage cells upon differentiation (Fig6E). To assess the role of Sirt1 and Sirt2 downstream of Nampt activity, we examined the expression of genes associated with OPC formation in Nampt Ad-Cre neurospheres, Sirt1/2 DKO neurospheres, and their respective controls. Supporting our model, dissociated Nampt Ad-Cre and Sirt1/2 DKO neurospheres showed similar decreases in the mRNA expression of Pdgfrα, Sox10, Nkx2.2 after 2 days of differentiation (Fig6F and G). Moreover, dissociated Nampt Ad-Cre and Sirt1/2 DKO neurospheres, respectively, exhibited similar increases in the expression of p21 (cdkn1a). Olig1 expression showed no change or slight reduction by these genetic ablations, potentially due to its lesser expression in NSPCs relative to Olig2 (Ligon et al, 2007) and predominant roles in oligodendrocyte maturation and remyelination rather than specification (Lu et al, 2002; Arnett et al, 2004). On the other hand, as previously observed (Saharan et al, 2013), neither Cre-mediated knockdown of Sirt1 in neurospheres nor neurospheres cultured from whole-body Sirt1−/− or Sirt2−/− mice exhibited defects in proliferation (Supplementary Fig S5G–J). Taken together, we conclude that Sirt1 and Sirt2 can redundantly mediate NSPC differentiation into OPCs.
Adult NSPC-specific deletion of Nampt impairs NSPC differentiation in response to insult in vivo
While we observed a clear effect of Nampt ablation on NSPC differentiation into OPCs in vitro, we failed to detect a defect in oligodendrogenesis in the SGZ of iNSPC-Nampt-KO mice in vivo (Fig3I), a finding consistent with previous reports that adult NSPCs rarely produce oligodendrocytes under basal conditions (Steiner et al, 2004). Since the SVZ is known to be more oligodendrogenic than the SGZ (Hack et al, 2004, 2005; Jackson et al, 2006; Menn et al, 2006), we assessed the percentage of NestinGFP+ cells that expressed Olig2 in the SVZs of iNSPC-GFP and iNSPC-Nampt-KO mice (Fig7A). Indeed, iNSPC-Nampt-KO mice showed a lower percentage of oligodendrocytes generated from adult NSPCs.
Figure 7. Adult NSPC-specific deletion of Nampt impairs NSPC self-renewal and differentiation in response to insult-induced demyelination in vivo.

A Quantification of the percentage of NestinGFP-positive cells in the SVZ that also express Olig2 in iNSPC-GFP (n = 7) and iNSPC-Nampt-KO (n = 8) mice 7 days post-initial TAM injection.
B Six- to nine-week-old iNSPC-GFP control and iNSPC-Nampt-KO mice were fed a diet containing 0.2% cuprizone for 4–5 weeks. Deletion of Nampt in the adult Nestin+ population was induced by five tamoxifen (TAM) injections at 180 mg/kg body weight per day the week before starting the cuprizone diet.
C A scheme of a coronal mouse brain section. Red boxed areas indicate regions used for quantification. Red dotted line indicates the SGZ. CC, corpus callosum; DHC, dorsal hippocampal commissure; DG, dentate gyrus; HPF, hippocampal formation; SCZ, subcallosal zone; SGZ, subgranular zone; V3, third ventricle.
D Quantification of the number of NestinGFP+ cells per unit area in the CC.
E A scheme for the specificity of the markers assessed.
F–I Quantification of the percentages of NestinGFP+ cells that express NSPC markers (Nestin, Gfap) or oligodendrocyte markers (Sox10, Apc) in the CC (n = 2–11 mice). * and ^ indicate statistical significance between iNSPC-GFP control littermates and iNSPC-Nampt-KO mice and between regular chow- and cuprizone-fed iNSPC-GFP mice, respectively.
J Representative images of immunofluorescence for Dapi (blue), Nampt (red), and Olig2 (green) in the CC. Arrows indicate examples of colocalization. Scale bars denote 20 μm.
Data information: Data are presented as mean ± SEM. *P < 0.05; **, ^^P < 0.01; ***, ^^^P < 0.001.
Given that aging impairs the ability of NSPCs to respond to environmental stimuli (Decker et al, 2002; Sim et al, 2002; Doucette et al, 2010), we hypothesized that Nampt-mediated NAD+ biosynthesis is important to enable NSPCs to respond to such environmental perturbations. To test this idea, we employed the cuprizone model of demyelination and remyelination. Specifically, we fed 6- to 9-week-old iNSPC-Nampt-KO and littermate control mice (iNSPC-GFP) a diet containing 0.2% cuprizone for 4–5 weeks, inducing deletion of Nampt in the adult Nestin+ population the week before starting the cuprizone diet (Fig7B) (Skripuletz et al, 2011). To ensure that we were analyzing progeny of adult Nestin+ cells, all mice in our cohort expressed Cre recombinase under the inducible Nestin promoter (Nestin-CreERT2) (Lagace et al, 2007) and the aforementioned Cre recombinase-responsive GFP reporter transgene. Thus, we focused our analysis on lineage tracer marked (NestinGFP+) cells.
Cuprizone feeding did not alter the total number of NestinGFP+ cells present in the iNSPC-GFP DG (Supplementary Fig S6A–C), suggesting that NSPC proliferation was unaltered. On the other hand, cuprizone-fed mice exhibited an increased percentage of NestinGFP+ cells that colocalized with the NSPC markers Nestin+ (from 13 to 35%) and Gfap+ (from 19 to 41%), suggesting that cuprizone treatment prevented SGZ NSPCs from terminally differentiating and instead resulted in their retention of NSPC characteristics, which could occur through increased self-renewal decisions and/or quiescence (Supplementary Fig S6D and E). To assess whether the NestinGFP-marked NSPCs had differentiated into oligodendrocyte lineage cells in response to cuprizone, we next assessed colocalization between NestinGFP and oligodendrocyte-specific markers, Sox10 and APC. However, the SGZ did not substantially produce oligodendrocytes even in response to demyelination (Supplementary Fig S6F and G).
While cuprizone treatment affects the hippocampus, its principle target of myelin injury is the corpus callosum (CC) (Gudi et al, 2009; Norkute et al, 2009; Doucette et al, 2010; Skripuletz et al, 2011). Under basal conditions and particularly during demyelination, NSPCs can proliferate to generate oligodendrocytes which integrate into the corpus callosum (Nait-Oumesmar et al, 1999; Picard-Riera et al, 2002; Hack et al, 2005; Menn et al, 2006; Colak et al, 2008; Jablonska et al, 2010; Soundarapandian et al, 2011). Thus, we assessed the fate decisions of migratory cells derived from the adult Nestin+ population in the subcallosal zone of the corpus callosum (Supplementary Fig S6A). In the iNSPC-GFP CC, virtually no NestinGFP+ or Nestin+ cells were seen in regular chow-fed mice (Fig7C and D, Supplementary Fig S6A). There were no differences in the number of NestinGFP+ cells in the CC between control and iNSPC-Nampt-KO mice, suggesting that loss of Nampt neither affected insult-induced NSPC proliferation or migration. In the iNSPC-GFP CC, cuprizone feeding significantly increased the percentage of NestinGFP+Nestin+ cells (from 3 to 41%), but decreased the NestinGFP+Gfap+ (from 60 to 24%) double-positive cells, suggesting increased self-renewal fate decisions at the expense of astrocytic fate decisions (Fig7E–G). In control mice, cuprizone feeding also increased the number of NestinGFP+ Sox10+ (from 2 to 16%) and NestinGFP+ Apc+ (from 0 to 4%) double-positive cells, suggesting increased oligodendrocyte lineage fate decisions (Fig7H and I). In contrast, the NestinGFP+ cells in the iNSPC-Nampt-KO CC showed significantly less colocalization with Nestin, Sox10, and Apc and more colocalization with Gfap (Fig7F–I). Interestingly, Nampt was only expressed in the CC upon insult (Fig7J, Supplementary Fig S6H). Moreover, Nampt colocalized with markers of NSPCs (Sox2, Supplementary Fig S6H) and oligodendrocytes (Olig2, Fig7J). Together, these results suggest that Nampt is specifically expressed in SGZ/SVZ-derived remyelinating NSPCs and plays an important role in oligodendrogenesis in response to insult.
Discussion
Our study establishes a novel role for Nampt-mediated NAD+ biosynthesis in NSPC proliferation, self-renewal, and differentiation into oligodendrocytes. Importantly, we demonstrated that: (i) hippocampal NAD+ levels and Nampt expression decline with age; (ii) in vivo ablation of Nampt in the adult Nestin+ population impaired NSPC proliferation and self-renewal; (iii) acute ablation of Nampt in hippocampal neurospheres significantly reduced NAD+ levels in NSPCs and stalled them in G1 of the cell cycle; (iv) chronic ablation of Nampt in hippocampal neurospheres abrogated oligodendrogenesis, and in vivo ablation of Nampt in the adult Nestin+ population reduced NSPC-mediated oligodendrogenesis upon insult; (v) Sirt1 and Sirt2 are required for NSPC-mediated oligodendrogenesis, yet in a redundant manner. These results reveal that Nampt deficiency in adult NSPCs recapitulates their functional defects observed during the aging process.
The finding that both hippocampal NAD+ levels and Nampt protein expression decline with age is consistent with the recent studies showing a significant age-related decline in NAD+ and Nampt levels in multiple peripheral organs and tissues (Yoshino et al, 2011). Based on our findings in the present study, it is conceivable that loss of Nampt activity contributes to the age-related decline in NSPC proliferation and in the NSPC pool (Lugert et al, 2010; Encinas et al, 2011; Artegiani & Calegari, 2012). This idea raises the possibility that long-term NMN administration could combat age-related declines in NSPC functionality. Indeed, we showed that the NSPC pool decreased with age and that long-term NMN administration was able to maintain the NSPC pool, providing further support for the ‘depletion’ hypothesis of NSPC aging (Encinas et al, 2011; Artegiani & Calegari, 2012). However, the dosage of NMN that maintained the NSPC pool was not sufficient to drive the proliferation of aged NSPCs and only tended to increase neurogenesis itself. These results suggest that NAD+-related effects on aged NSPCs are independent of the pathways altered by environmental enrichment (Kempermann et al, 1998, 2002) and exercise (Kronenberg et al, 2006; Wu et al, 2008), as these manipulations primarily affect NSPC proliferation and newborn neuron survival rather than the NSPC pool in aged animals. As we showed Nampt to be critically important for NSPC proliferation in neurospheres, it is likely that a higher dosage of NMN is necessary to promote NSPC proliferation. Yet, our results do not exclude the possibility that another factor present in the NSPC niche is more important for the age-related decline in NSPC proliferation and/or counteracts Nampt/NMN-driven NSPC proliferation. Importantly for therapeutic purposes, this experiment and our finding that intraperitoneal injection of NMN substantially increases hippocampal NAD+ levels within 15 min (Supplementary Fig S2G) strongly suggest that NMN can cross the blood–brain barrier.
It is remarkable that loss of Nampt in adult NSPCs produces functional impairment as hippocampal NSPCs could theoretically obtain NAD+ from release by surrounding cells. Studies to date have suggested that glia may deliver NAD+ to neurons (Verderio et al, 2001). Moreover, there are multiple precursors for and pathways of NAD+ biosynthesis, all of which are expressed in the hippocampus (Guillemin et al, 2007). Our results clearly demonstrate that cell-autonomous Nampt activity is the main source of NAD+ for NSPCs. This cell-autonomous Nampt activity is critical to regulate G1/S transition in NSPCs. Given that loss of Nampt activity in many other cell types reduced cell viability (Hasmann & Schemainda, 2003; Rongvaux et al, 2008), it is surprising that loss of Nampt activity specifically caused a G1/S stall without causing detectable cell death in NSPCs. As E2F1-deficient mice have significantly reduced hippocampal NSPC death (Cooper-Kuhn et al, 2002), the decrease in E2F1 expression that we observed upon inhibition of Nampt may explain this phenomenon (Fig4E). In line with a G1/S stall, we also observed that loss of Nampt activity specifically downregulated cyclins E and A expression. E-type cyclins regulate G1 progression, and their activity is necessary and limiting for the re-entry of G0 cells into the cell cycle as well as for passing from G1 into S phase. Because we also observed downregulation of E2F1 expression, which transcriptionally regulates cyclin E, it is very likely that the downregulation of E2F1 contributes to the downregulation of cyclin E. Interestingly, we also see upregulation of p21 upon the loss of Nampt. p21 represses E2F activity and E2F activation is required to eliminate a p21-mediated block in cell cycle entry in late G1 (Polager & Ginsberg, 2009; Wong et al, 2011). Thus, the upregulation of p21 that we see upon the loss of Nampt may also contribute to the downregulation of E2F/cyclin E activity.
Another important role of Nampt in NSPCs is to regulate their differentiation into oligodendrocytes. Why does loss of Nampt-mediated NAD+ biosynthesis particularly affect oligodendrocytic lineage fate decisions in NSPCs? One possible explanation is that oligodendrocyte lineage cells are the most metabolically demanding (Sanchez-Abarca et al, 2001) and stress-sensitive central nervous system cell type (Husain & Juurlink, 1995). However, our results suggest that a more primary downstream effect of Nampt ablation on NSPC oligodendrocytic lineage fate decisions is the reduction in Sirt1 and Sirt2 activity (Fig8). Previous work has generated contradictory conclusions in the function of Sirt1 in NSPC differentiation (Hisahara et al, 2008; Prozorovski et al, 2008; Wang et al, 2011b; Zhang et al, 2011; Rafalski et al, 2013; Saharan et al, 2013) and in the function of Sirt2 in oligodendrocyte differentiation (Li et al, 2007; Ji et al, 2011). Yet, these contradictory findings could be explained by compensatory activity between Sirt1 and Sirt2. Since Sirt1 is highly expressed in NSPCs (Hisahara et al, 2008; Prozorovski et al, 2008; Saharan et al, 2013) and Sirt2 is highly expressed in oligodendrocytes (Li et al, 2007; Tyler et al, 2011), it is possible that the activities of these two proteins trade off during oligodendrocyte maturation.
Figure 8. Model for the role of Nampt-mediated NAD+ biosynthesis in NSPCs.
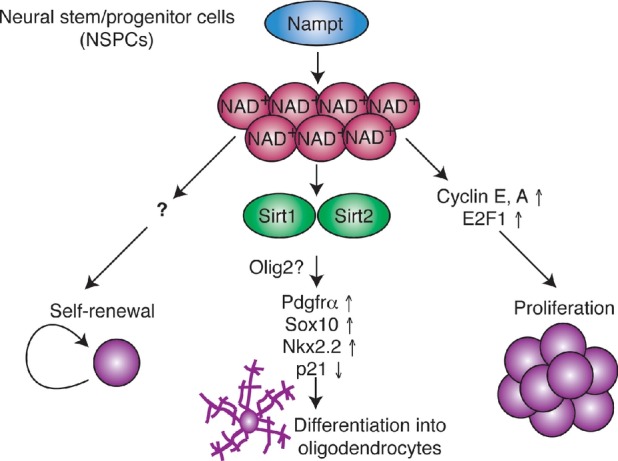
Nampt-mediated NAD+ biosynthesis promotes NSPC self-renewal, proliferation, and differentiation into oligodendrocytes. While the mechanism by which Nampt promotes NSPC self-renewal and proliferation remains unidentified, it involves transcriptional upregulation of cyclins E, A and their upstream regulator E2F1. Nampt-mediated NAD+ biosynthesis likely promotes NSPC oligodendrocyte lineage fate decisions by activating Sirt1 and Sirt2, which leads to transcriptional upregulation of Pdgfrα, Sox10, and Nkx2.2, as well as transcriptional downregulation of p21 (cdkn1a). Sirt1 and Sirt2 may act via an effect on Olig2 activity. See text for a detailed discussion.
The factor downstream of Sirt1/2, which promotes oligodendrocytic fate decisions, remains to be identified. Previous work has linked Sirt1 activity to Hes1 (Hisahara et al, 2008), Hes5 (Hisahara et al, 2008), and Mash1 expression (Prozorovski et al, 2008; Zhang et al, 2011). Given the importance of Hes5 in inhibiting oligodendrocyte differentiation (Liu et al, 2006), it will be of great importance to investigate the roles of these factors downstream of Nampt. Our results reveal that ablation of Nampt specifically reduced the proportion of NSPC-generated Pdgfrα+ OPCs as well as the transcription of Pdgfrα, Sox10, and Nkx2.2, but upregulated the expression of p21. One main regulator of these factors is the basic helix-loop-helix transcription factor Olig2 (Takebayashi et al, 2002; Zhou & Anderson, 2002; Ligon et al, 2007; Wegner, 2008). Olig2 is expressed in almost all (95–97%) of neurosphere cells and is necessary and sufficient to specify OPCs from the SVZ (Hack et al, 2004, 2005; Menn et al, 2006). Intriguingly, the number of Olig2+ cells in the SVZ decreases with age (Bouab et al, 2011). Recent work has shown that Olig2 competes with p53 for binding to an important coregulator that regulates p53 acetylation (Mehta et al, 2011; Sun et al, 2011). As Sirt1 and Sirt2 can deacetylate p53 (Polager & Ginsberg, 2009; van Leeuwen et al, 2013), it will be interesting to investigate the NAD+ dependence of Olig2 activity.
Identifying Nampt as a critical regulator of NSPC proliferation, self-renewal, and differentiation highlights the intimate link between stem cell metabolism and cell fate (Folmes et al, 2012; Zhang et al, 2012). Like loss of Nampt, inhibition of mitochondrial fatty acid oxidation impairs stem cell self-renewal decisions (Ito et al, 2012) and abrogation of de novo lipogenesis impairs NSPC proliferation (Knobloch et al, 2013). However, manipulations of other metabolic pathways can present very differently from loss of Nampt. For example, ablation of endogenous cholesterol production in NSPCs results in massive apoptosis of newborn neurons without affecting cell death or proliferation in the NSPCs themselves (Saito et al, 2009). And, mitochondrial inhibition alters NSPC neuronal lineage fate decisions without affecting the astrocytic or oligodendrocytic lineages (Voloboueva et al, 2010). Thus, our work supports the notion that cellular metabolism regulates NSPC functionality, but that alteration of each metabolic pathway generates specific effects.
Aging is an opposing factor for any type of regeneration (Jadasz et al, 2012). Consistent with the notion that altered metabolism plays a causal role in the age-related decline in NSPC function, NSPCs isolated from old mice had decreased regenerative capacity, fewer functional mitochondria, and less oxygen consumption relative to NSPCs isolated from young mice (Stoll et al, 2011). Rates of remyelination decline with age due to slower recruitment of OPCs into areas of demyelination and slower differentiation of OPCs into remyelinating oligodendrocytes (Decker et al, 2002; Sim et al, 2002; Doucette et al, 2010). Similarly, the lack of remyelination seen in multiple sclerosis, an autoimmune disease that leads to oligodendrocyte loss and demyelination (Jadasz et al, 2012), occurs with a failure of OPC differentiation (Franklin & Ffrench-Constant, 2008). Our results clearly showed that in neurospheres, treatment with NMN rescued defects in oligodendrogenesis caused by a reduction in NAD+ levels. Furthermore, systemic NMN administration was able to substantially augment hippocampal NAD+ levels and increase the NSPC pool. Thus, NMN administration could be an efficient intervention to enhance the NSPC pool and promote remyelination by activating endogenous NSPCs during the aging process and/or in neurodegenerative diseases that cause demyelination. In conclusion, our present study provides impetus to further delineate the therapeutic potential and signaling pathways downstream of Nampt-mediated NSPC self-renewal, proliferation, and differentiation into oligodendrocytes.
Materials and Methods
Mice
Mice were maintained on a regular chow ad libitum on a 12 h light/dark cycle (lights on from 6 am to 6 pm). Namptflox/flox mice (Rongvaux et al, 2008), in which exons 5 and 6 of the Nampt gene are flanked by loxP sites, were crossed to Nestin-CreERT2 mice (Lagace et al, 2007) to generate Nampt flox/+; Cre double heterozygous mice. Double heterozygous mice were bred to Namptflox/flox mice to obtain Namptflox/flox; Cre mutant mice (iNSPC-Nampt-KO mice) in the expected Mendelian ratio. To trace the progeny of adult NSPCs and to confirm the specificity and magnitude of the recombination induced by tamoxifen injection, iNSPC-Nampt-KO and Nestin-CreERT2 mice were crossed to a reporter mouse strain that expresses a loxP-flanked STOP cassette that prevents transcription of the downstream enhanced green fluorescent protein [ZsGreen1; Jackson laboratories #7906 (Madisen et al, 2010)]. Recombination PCR on hippocampal extracts of tamoxifen- or vehicle-treated mice showed successful deletion upon treatment with tamoxifen (Supplementary Fig S2C). All animal procedures were approved by the Washington University Animal Studies Committee and were in accordance with NIH guidelines.
Induction of Nampt deletion
Tamoxifen injections were performed as described previously (Lagace et al, 2007). Briefly, iNSPC-Nampt-KO mice (5–7 weeks old) were administered tamoxifen (TAM, Sigma T5648) at 180 mg/kg/d for 5 days (d, intraperitoneally; dissolved in 10% EtOH/90% sunflower oil), a protocol that produces maximal recombination with minimal lethality (5%) (Lagace et al, 2007).
BrdU incorporation
5′-Bromodeoxyuridine (BrdU, Sigma, B9285) was diluted in sterile saline and administered by intraperitoneal injections (100 mg/kg body weight). For analysis of the cumulative effects of loss of Nampt, mice were given BrdU twice a day for 2 days and sacrificed the following day or 28 days later. For analysis of the effect of loss of Nampt on adult NSC differentiation and postnatal oligodendrocyte differentiation, mice were given BrdU twice a day for 1 day and sacrificed 2 days later.
Cuprizone
Demyelination was induced by feeding 6- to 8-week-old mice a diet containing 0.2% cuprizone (bis-cyclohexanone oxaldihydrazone; Sigma C9012) mixed into a ground standard rodent chow for 4–5 weeks (Harlan Laboratories, TD.01453). To allow recovery from cuprizone treatment, food was replaced with standard chow for an additional 1 week. This protocol has been shown to successfully demyelinate and remyelinate the hippocampus (Skripuletz et al, 2011).
Reagents
The following primary and secondary antibodies were used:
Primary antibodies and their uses or cell type specificities (von Bohlen und Halbach, 2011):
Actin: normalization, WB 1:4,000 CPO1, Sigma; Gapdh: normalization, WB 1:4,000 6C5 Millipore CB1001; Nampt: IHC 1:1,000; WB 1:3,000 Alexis Biochemicals ALX-804-717-C100; Pdgfrα: oligodendrocyte precursor cells, IF 1:500 APA5 BD Biosciences; Olig2: all oligodendrocyte lineage cells, IHC 1:500, IF 1:1,000; Millipore; O4: immature oligodendrocytes, IF 1:1,000 Millipore, MAB345; APC: oligodendrocytes, IHC 1:1,000 Millipore CC-1 OP80; MBP: mature oligodendrocytes, IHC 1:1,000 Millipore MAB386; Ki67: proliferating cells, IHC, IF 1:500; WB 1:3,000 Abcam ab66155; Pcna: proliferating cells, WB 1:2,000; PC10 Cell signaling #2586; 5-bromo-2′-deoxyuridine (BrdU): a thymine analog that incorporates into the DNA of cells in S phase, IHC 1:500; OBT0030 Accurate; activated caspase-3: apoptosis, IHC, IF 1:500; Cell Signaling #9661; LC3B: autophagy, WB 1:1,000; Novus NB600-1384; TUNEL: cell death, Roche In Situ Cell Death Detection Kit 11 684 795 910; Dcx: newly born neurons, IHC 1:1,000; Cell Signaling #4604; NeuN: mature neurons, IHC, 1:500, Millipore, MAB377; Nestin: NSPCs, IHC, IF 1:1,000, Millipore MAB353; Sox2: NSPCs, IHC, IF 1:500; WB 1:2,000; Millipore AB5603; Gfap: NSPCs and astrocytes, IHC, IF 1:1,000; Millipore MAB360.
Secondary antibodies: Jackson ImmunoResearch anti-rat, anti-rabbit, anti-mouse Cy3 (1:400), Alexa Fluor488 (1:200), and Alexa Fluor647 (1:200). Anti-rabbit, anti-mouse horseradish peroxidase (Invitrogen).
FK866 (Hasmann & Schemainda, 2003) (Sigma F8557), EX527 (Peck et al, 2010) (Cayman Chemical 10009798), and AGK2 (Outeiro et al, 2007) (Sigma A8231) were dissolved in DMSO and used to inhibit Nampt, Sirt1, and Sirt2, respectively.
Neurosphere culture
Neurosphere cultures and culture media were prepared as described previously with minor modifications (Dasgupta & Gutmann, 2005; Lu & Ramanan, 2012). Briefly, postnatal hippocampi were dissected in Hibernate A (Invitrogen, A12475-01) and trypsinized at 37°C for 7 min. Cells were mechanically dissociated by pipetting and pelleted by centrifugation (213 g, 7 min). Dissociation medium (0.1% sodium bicarbonate, 15 mM HEPES, 0.5% glucose in HBSS) was used to wash the cells before they were resuspended in growth medium. Growth medium consisted of DMEM:F12 (1:1, Invitrogen 11966-025 and 21700-075, respectively), B27 (Invitrogen, 17504-044), N2 (Invitrogen, 17502-048), Pen/Strep (Invitrogen), epidermal growth factor (EGF, 20 ng/ml, Sigma, E4127), fibroblast growth factor (FGF, 10 ng/ml, R&D Systems, 233-fb), and heparin (Sigma). Cultures were maintained at 37°C with 5% CO2 and passaged twice before use in experiments. Three to nine independent samples, each in 1–3 replicates, from at least two different litters were used in all experiments. Neurospheres were cultured in the physiological glucose level of 5 mM (Dienel & Cruz, 2006), which has been previously shown to have no negative consequences on NSPC proliferation, differentiation, or death (Fu et al, 2006; Gao & Gao, 2007).
Neurosphere infection
Neurospheres derived from Namptflox/flox mice were infected with Ad5 Cre recombinase- or β-galactosidase-expressing (LacZ, control) adenoviruses at an MOI of 100. All assessments were performed at least 6 days post-infection.
Neurosphere proliferation analysis
Neurospheres derived from Namptflox/flox mice were dissociated by trypsin digestion and seeded at similar cell densities in 24-well plates with fresh growth medium. Every 24 h, neurospheres from triplicate wells were collected, dissociated, and counted on a hemocytometer using 0.2% trypan blue exclusion to distinguish viable cells. For analysis of neurosphere diameter, the largest neurosphere in each well was imaged (20× objective) and the diameter was calculated using ImageJ. For secondary neurosphere analysis, the total number of neurospheres in each well was counted at 7 days post-plating.
Neurosphere differentiation
Three to five days after their first passage, neurospheres were trypsinized, washed with dissociation medium, and plated at 150,000 cells per well in 24-well plates in differentiation medium [growth medium without FGF and EGF and with BDNF (5 ng/ml, Peprotech, 450-02) on glass coverslips coated with poly-D-lysine (50 μg/ml; Sigma) and laminin (20 μg/ml; BD Biosciences)]. Six-well plates were coated with poly-D-lysine (20 μg/ml) and laminin (10 μg/ml). To enrich for oligodendrocytes, PDGFαα (10 ng/ml, Peprotech 100-13A) was added to neurospheres at passage 2 and PDGFαα (2.5 ng/ml) and 3,3′,5-triiodo-L-thyronine (T3, 40 ng/ml, Sigma T4397) were added to differentiation medium. The percentage of oligodendrocyte precursor cells (OPCs) generated was analyzed after 2 days of differentiation, and the percentage of differentiated oligodendrocytes was analyzed after 6–7 days of differentiation.
Immunofluorescence
All tissue sections and cells were incubated in blocking/permeabilization solution containing 10% normal goat serum, 1% BSA, and 0.3% Triton X-100 in PBS for 45–60 min prior to 24 or 48 h of incubation with primary antibodies in 5% normal goat serum and 0.1% Triton X-100 in PBS at 4°C at the concentrations listed below. Alexa627-, Alexa488-, or Cy3-conjugated secondary antibodies diluted in 2% normal goat serum, 1% BSA, and 0.1% Triton X-100 in PBS were added for 2 h at room temperature. Nuclei were stained with 4,6-diamidino-2-phenylindole (Sigma) for 10 min at room temperature.
Cells were harvested by fixation with 4% paraformaldehyde in PBS (15 min). Mice were anesthetized by i.p. injection of ketamine and xylazine and perfused transcardially through left ventricle with cold 0.1 M phosphate buffer at pH 7.4 followed by a phosphate-buffered solution of 4% paraformaldehyde (PFA). Brains were post-fixed with 4% PFA overnight and placed into 15% sucrose followed by 30% sucrose, frozen, and stored at −80°C until use. Coronal sections (30 μm) were made by cryostat in a 1 in 8 series and stored at −30°C in cryoprotectant until use. To remove any endogenous peroxidase activity, all sections were incubated with 3% H2O2 for 10 min. Tissue sections used to assess BrdU incorporation were treated before the immunostaining procedure with 50% formamide in 2× saline/sodium citrate (SSC) at 65°C for 2 h, 2N HCl for 30 min at 37°C, 0.1 M borate pH 8.5, and then washed twice with PBS before proceeding with the staining protocol. Tissue sections not used to assess BrdU incorporation were either incubated in 50% formamide in 2× saline/sodium citrate (SSC) at 65°C for 2 h or 10 mM citrate buffer at 65°C for 1 h before proceeding with the staining protocol. Detection of Dcx, Nestin, Nampt, and APC was performed using the TSA-Plus fluorescein kit (PerkinElmer).
Quantification
For tissue sections, high-magnification (20×, 0.8DICII or 40× oil 1. 3DICII) microscopic imaging was performed using a Zeiss Axioimager.Z1. Images were taken in z-stacks of 1 μm steps through the range of tissue section immunoreactivity. For the dorsolateral corner of the SVZ, images were taken from bregma 1.10 to −0.10 mm. For the corpus callosum, images were taken from bregma −1.06 to −2.54 mm. For the dentate gyrus, images were taken from bregma −1.34 to −3.64 mm. Quantification was performed blinded to genotype on 3–8 tissue sections per animal. Cell densities were estimated by the number of immunoreactive cells divided by the area of the structure, measured with ImageJ. Verification of colocalization was achieved by importing stacks of Z images into ImageJ and performing 3D rendering. For cells, 10 or 20× microscopic imaging was performed using a Zeiss Axioimager.Z1. Quantification was performed blinded to genotype on 2–3 fields of view per sample and treatment, from 3 to 9 independent samples.
NAD+ measurement
NAD+ levels were determined using an HPLC system (Shimadzu) with a Supelco LC-18-T column (15 × 4.6 cm; Sigma), as described previously (Yoshino et al, 2011).
Microarrays and bioinformatic analyses
For individual genes, raw microarray data were subjected to Z score transformation, and Z ratios were calculated as described previously (Cheadle et al, 2003). Subsequent analysis, namely parametric analysis of gene set enrichment (PAGE), was performed as previously described (Yoshino et al, 2011). The microarray data used in this study have been deposited into the NCBI GEO database (GEO accession number GSE49784).
Western blotting
Protein extracts (15–50 μg) from mouse hippocampi or neurospheres were prepared as previously described (Yoshino et al, 2011).
Quantitative real-time RT–PCR
Total RNA was extracted from the hippocampus using the RNeasy kit (Qiagen) and reverse-transcribed into cDNA with the High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Quantitative real-time RT–PCR was conducted with the TaqMan Fast Universal PCR Master mix and appropriate TaqMan primers for each gene with the GeneAmp 7500 fast sequence detection system (Applied Biosystems). Relative expression levels were calculated for each gene by normalizing to Gapdh levels and then to a control.
Statistical analyses
Differences between the two groups were assessed using the Student's unpaired t-test. Comparisons among several groups were performed using one-way ANOVA with the Tukey–Kramer post hoc test except for Fig3J and Supplementary Fig S3D and E, in which the Games–Howell post hoc test and the Fisher LSD post hoc test were used, respectively. P-values < 0.05 were considered statistically significant.
Acknowledgments
We thank Jun Yoshino, Da Yong Lee, Paul P. Y. Lu, and members of the Imai laboratory for technical help and advice. We thank David Gutmann, Narendrakumar Ramanan, and Kelly Monk for making suggestions on and critically reading this manuscript. We thank the Oriental Yeast Co. (Tokyo, Japan) for providing the NMN used in the long-term NMN administration study. This work was supported in part by the National Institute on Aging (AG024150, AG037457) and the Ellison Medical Foundation to S.I, and Training Grant T32 GM007067 to L.R.S.
Author contributions
LRS and SI designed research, analyzed data, and wrote the paper. LRS performed research.
Conflict of interest
S.I. served as a scientific advisory board member for Sirtris, a GSK company, and had a sponsored research agreement with Oriental Yeast Co., Tokyo, Japan. S.I. is also a co-founder of Metro Midwest Biotech.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Arnett HA, Fancy SP, Alberta JA, Zhao C, Plant SR, Kaing S, Raine CS, Rowitch DH, Franklin RJ, Stiles CD. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science. 2004;306:2111–2115. doi: 10.1126/science.1103709. [DOI] [PubMed] [Google Scholar]
- Artegiani B, Calegari F. Age-related cognitive decline: can neural stem cells help us? Aging. 2012;4:176–186. doi: 10.18632/aging.100446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Abdallah NM, Slomianka L, Vyssotski AL, Lipp HP. Early age-related changes in adult hippocampal neurogenesis in C57 mice. Neurobiol Aging. 2010;31:151–161. doi: 10.1016/j.neurobiolaging.2008.03.002. [DOI] [PubMed] [Google Scholar]
- von Bohlen und Halbach O. Immunohistological markers for proliferative events, gliogenesis, and neurogenesis within the adult hippocampus. Cell Tissue Res. 2011;345:1–19. doi: 10.1007/s00441-011-1196-4. [DOI] [PubMed] [Google Scholar]
- Bouab M, Paliouras GN, Aumont A, Forest-Berard K, Fernandes KJ. Aging of the subventricular zone neural stem cell niche: evidence for quiescence-associated changes between early and mid-adulthood. Neuroscience. 2011;173:135–149. doi: 10.1016/j.neuroscience.2010.11.032. [DOI] [PubMed] [Google Scholar]
- Cheadle C, Vawter MP, Freed WJ, Becker KG. Analysis of microarray data using Z score transformation. J Mol Diagn. 2003;5:73–81. doi: 10.1016/S1525-1578(10)60455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D, Mori T, Brill MS, Pfeifer A, Falk S, Deng C, Monteiro R, Mummery C, Sommer L, Gotz M. Adult neurogenesis requires Smad4-mediated bone morphogenic protein signaling in stem cells. J Neurosci. 2008;28:434–446. doi: 10.1523/JNEUROSCI.4374-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Kuhn CM, Vroemen M, Brown J, Ye H, Thompson MA, Winkler J, Kuhn HG. Impaired adult neurogenesis in mice lacking the transcription factor E2F1. Mol Cell Neurosci. 2002;21:312–323. doi: 10.1006/mcne.2002.1176. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Gutmann DH. Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J Neurosci. 2005;25:5584–5594. doi: 10.1523/JNEUROSCI.4693-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker L, Picard-Riera N, Lachapelle F, Baron-Van Evercooren A. Growth factor treatment promotes mobilization of young but not aged adult subventricular zone precursors in response to demyelination. J Neurosci Res. 2002;69:763–771. doi: 10.1002/jnr.10411. [DOI] [PubMed] [Google Scholar]
- Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev. 2010;11:339–350. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA, Cruz NF. Astrocyte activation in working brain: energy supplied by minor substrates. Neurochem Int. 2006;48:586–595. doi: 10.1016/j.neuint.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Doucette JR, Jiao R, Nazarali AJ. Age-related and cuprizone-induced changes in myelin and transcription factor gene expression and in oligodendrocyte cell densities in the rostral corpus callosum of mice. Cell Mol Neurobiol. 2010;30:607–629. doi: 10.1007/s10571-009-9486-z. [DOI] [PubMed] [Google Scholar]
- Encinas JM, Michurina TV, Peunova N, Park JH, Tordo J, Peterson DA, Fishell G, Koulakov A, Enikolopov G. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell. 2011;8:566–579. doi: 10.1016/j.stem.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11:596–606. doi: 10.1016/j.stem.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- Friebe D, Loffler D, Schonberg M, Bernhard F, Buttner P, Landgraf K, Kiess W, Korner A. Impact of metabolic regulators on the expression of the obesity associated genes FTO and NAMPT in human preadipocytes and adipocytes. PLoS ONE. 2011;6:e19526. doi: 10.1371/journal.pone.0019526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Tay SS, Ling EA, Dheen ST. High glucose alters the expression of genes involved in proliferation and cell-fate specification of embryonic neural stem cells. Diabetologia. 2006;49:1027–1038. doi: 10.1007/s00125-006-0153-3. [DOI] [PubMed] [Google Scholar]
- Gao Q, Gao YM. Hyperglycemic condition disturbs the proliferation and cell death of neural progenitors in mouse embryonic spinal cord. Int J Dev Neurosci. 2007;25:349–357. doi: 10.1016/j.ijdevneu.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Gudi V, Moharregh-Khiabani D, Skripuletz T, Koutsoudaki PN, Kotsiari A, Skuljec J, Trebst C, Stangel M. Regional differences between grey and white matter in cuprizone induced demyelination. Brain Res. 2009;1283:127–138. doi: 10.1016/j.brainres.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Cullen KM, Lim CK, Smythe GA, Garner B, Kapoor V, Takikawa O, Brew BJ. Characterization of the kynurenine pathway in human neurons. J Neurosci. 2007;27:12884–12892. doi: 10.1523/JNEUROSCI.4101-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack MA, Sugimori M, Lundberg C, Nakafuku M, Gotz M. Regionalization and fate specification in neurospheres: the role of Olig2 and Pax6. Mol Cell Neurosci. 2004;25:664–678. doi: 10.1016/j.mcn.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Hack MA, Saghatelyan A, de Chevigny A, Pfeifer A, Ashery-Padan R, Lledo PM, Gotz M. Neuronal fate determinants of adult olfactory bulb neurogenesis. Nat Neurosci. 2005;8:865–872. doi: 10.1038/nn1479. [DOI] [PubMed] [Google Scholar]
- Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–7442. [PubMed] [Google Scholar]
- Hisahara S, Chiba S, Matsumoto H, Tanno M, Yagi H, Shimohama S, Sato M, Horio Y. Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc Natl Acad Sci USA. 2008;105:15599–15604. doi: 10.1073/pnas.0800612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain J, Juurlink BH. Oligodendroglial precursor cell susceptibility to hypoxia is related to poor ability to cope with reactive oxygen species. Brain Res. 1995;698:86–94. doi: 10.1016/0006-8993(95)00832-b. [DOI] [PubMed] [Google Scholar]
- Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, Schafer ZT, Evans RM, Suda T, Lee CH, Pandolfi PP. A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18:1350–1358. doi: 10.1038/nm.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonska B, Aguirre A, Raymond M, Szabo G, Kitabatake Y, Sailor KA, Ming GL, Song H, Gallo V. Chordin-induced lineage plasticity of adult SVZ neuroblasts after demyelination. Nat Neurosci. 2010;13:541–550. doi: 10.1038/nn.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, VandenBerg S, Alvarez-Buylla A. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006;51:187–199. doi: 10.1016/j.neuron.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Jadasz JJ, Aigner L, Rivera FJ, Kury P. The remyelination Philosopher's Stone: stem and progenitor cell therapies for multiple sclerosis. Cell Tissue Res. 2012;349:331–347. doi: 10.1007/s00441-012-1331-x. [DOI] [PubMed] [Google Scholar]
- Ji S, Doucette JR, Nazarali AJ. Sirt2 is a novel in vivo downstream target of Nkx2.2 and enhances oligodendroglial cell differentiation. J Mol Cell Biol. 2011;3:351–359. doi: 10.1093/jmcb/mjr009. [DOI] [PubMed] [Google Scholar]
- Jin K, Sun Y, Xie L, Batteur S, Mao XO, Smelick C, Logvinova A, Greenberg DA. Neurogenesis and aging: FGF-2 and HB-EGF restore neurogenesis in hippocampus and subventricular zone of aged mice. Aging Cell. 2003;2:175–183. doi: 10.1046/j.1474-9728.2003.00046.x. [DOI] [PubMed] [Google Scholar]
- Jin YH, Kim YJ, Kim DW, Baek KH, Kang BY, Yeo CY, Lee KY. Sirt2 interacts with 14-3-3 beta/gamma and down-regulates the activity of p53. Biochem Biophys Res Commun. 2008;368:690–695. doi: 10.1016/j.bbrc.2008.01.114. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Brandon EP, Gage FH. Environmental stimulation of 129/SvJ mice causes increased cell proliferation and neurogenesis in the adult dentate gyrus. Curr Biol. 1998;8:939–942. doi: 10.1016/s0960-9822(07)00377-6. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Gast D, Gage FH. Neuroplasticity in old age: sustained fivefold induction of hippocampal neurogenesis by long-term environmental enrichment. Ann Neurol. 2002;52:135–143. doi: 10.1002/ana.10262. [DOI] [PubMed] [Google Scholar]
- Knobloch M, Braun SM, Zurkirchen L, von Schoultz C, Zamboni N, Arauzo-Bravo MJ, Kovacs WJ, Karalay O, Suter U, Machado RA, Roccio M, Lutolf MP, Semenkovich CF, Jessberger S. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature. 2013;493:226–230. doi: 10.1038/nature11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg G, Bick-Sander A, Bunk E, Wolf C, Ehninger D, Kempermann G. Physical exercise prevents age-related decline in precursor cell activity in the mouse dentate gyrus. Neurobiol Aging. 2006;27:1505–1513. doi: 10.1016/j.neurobiolaging.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Lagace DC, Whitman MC, Noonan MA, Ables JL, DeCarolis NA, Arguello AA, Donovan MH, Fischer SJ, Farnbauch LA, Beech RD, DiLeone RJ, Greer CA, Mandyam CD, Eisch AJ. Dynamic contribution of nestin-expressing stem cells to adult neurogenesis. J Neurosci. 2007;27:12623–12629. doi: 10.1523/JNEUROSCI.3812-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen IM, Higgins M, Campbell J, McCarthy AR, Sachweh MC, Navarro AM, Lain S. Modulation of p53 C-terminal acetylation by mdm2, p14ARF, and cytoplasmic SirT2. Mol Cancer Ther. 2013;12:471–480. doi: 10.1158/1535-7163.MCT-12-0904. [DOI] [PubMed] [Google Scholar]
- Li W, Zhang B, Tang J, Cao Q, Wu Y, Wu C, Guo J, Ling EA, Liang F. Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha-tubulin. J Neurosci. 2007;27:2606–2616. doi: 10.1523/JNEUROSCI.4181-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, Anderson DJ, Stiles CD, Rowitch DH. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Li J, Marin-Husstege M, Kageyama R, Fan Y, Gelinas C, Casaccia-Bonnefil P. A molecular insight of Hes5-dependent inhibition of myelin gene expression: old partners and new players. EMBO J. 2006;25:4833–4842. doi: 10.1038/sj.emboj.7601352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/s0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- Lu PP, Ramanan N. A critical cell-intrinsic role for serum response factor in glial specification in the CNS. J Neurosci. 2012;32:8012–8023. doi: 10.1523/JNEUROSCI.5633-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugert S, Basak O, Knuckles P, Haussler U, Fabel K, Gotz M, Haas CA, Kempermann G, Taylor V, Giachino C. Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell. 2010;6:445–456. doi: 10.1016/j.stem.2010.03.017. [DOI] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S, Huillard E, Kesari S, Maire CL, Golebiowski D, Harrington EP, Alberta JA, Kane MF, Theisen M, Ligon KL, Rowitch DH, Stiles CD. The central nervous system-restricted transcription factor Olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer Cell. 2011;19:359–371. doi: 10.1016/j.ccr.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, Alvarez-Buylla A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci. 2006;26:7907–7918. doi: 10.1523/JNEUROSCI.1299-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nait-Oumesmar B, Decker L, Lachapelle F, Avellana-Adalid V, Bachelin C, Baron-Van Evercooren A. Progenitor cells of the adult mouse subventricular zone proliferate, migrate and differentiate into oligodendrocytes after demyelination. Eur J Neurosci. 1999;11:4357–4366. doi: 10.1046/j.1460-9568.1999.00873.x. [DOI] [PubMed] [Google Scholar]
- Norkute A, Hieble A, Braun A, Johann S, Clarner T, Baumgartner W, Beyer C, Kipp M. Cuprizone treatment induces demyelination and astrocytosis in the mouse hippocampus. J Neurosci Res. 2009;87:1343–1355. doi: 10.1002/jnr.21946. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- Peck B, Chen CY, Ho KK, Di Fruscia P, Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD, Lam EW. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther. 2010;9:844–855. doi: 10.1158/1535-7163.MCT-09-0971. [DOI] [PubMed] [Google Scholar]
- Picard-Riera N, Decker L, Delarasse C, Goude K, Nait-Oumesmar B, Liblau R, Pham-Dinh D, Evercooren AB. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci USA. 2002;99:13211–13216. doi: 10.1073/pnas.192314199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassman BL, Langa KM, Fisher GG, Heeringa SG, Weir DR, Ofstedal MB, Burke JR, Hurd MD, Potter GG, Rodgers WL, Steffens DC, McArdle JJ, Willis RJ, Wallace RB. Prevalence of cognitive impairment without dementia in the United States. Ann Intern Med. 2008;148:427–434. doi: 10.7326/0003-4819-148-6-200803180-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer. 2009;9:738–748. doi: 10.1038/nrc2718. [DOI] [PubMed] [Google Scholar]
- Prozorovski T, Schulze-Topphoff U, Glumm R, Baumgart J, Schroter F, Ninnemann O, Siegert E, Bendix I, Brustle O, Nitsch R, Zipp F, Aktas O. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat Cell Biol. 2008;10:385–394. doi: 10.1038/ncb1700. [DOI] [PubMed] [Google Scholar]
- Rafalski VA, Ho PP, Brett JO, Ucar D, Dugas JC, Pollina EA, Chow LM, Ibrahim A, Baker SJ, Barres BA, Steinman L, Brunet A. Expansion of oligodendrocyte progenitor cells following SIRT1 inactivation in the adult brain. Nat Cell Biol. 2013;15:614–624. doi: 10.1038/ncb2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, Milbrandt J, Kiess W, Imai S. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–375. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Galli M, Denanglaire S, Van Gool F, Dreze PL, Szpirer C, Bureau F, Andris F, Leo O. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J Immunol. 2008;181:4685–4695. doi: 10.4049/jimmunol.181.7.4685. [DOI] [PubMed] [Google Scholar]
- Rothgiesser KM, Erener S, Waibel S, Luscher B, Hottiger MO. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci. 2010;123:4251–4258. doi: 10.1242/jcs.073783. [DOI] [PubMed] [Google Scholar]
- Saharan S, Jhaveri DJ, Bartlett PF. SIRT1 regulates the neurogenic potential of neural precursors in the adult subventricular zone and hippocampus. J Neurosci Res. 2013;91:642–659. doi: 10.1002/jnr.23199. [DOI] [PubMed] [Google Scholar]
- Saito K, Dubreuil V, Arai Y, Wilsch-Brauninger M, Schwudke D, Saher G, Miyata T, Breier G, Thiele C, Shevchenko A, Nave KA, Huttner WB. Ablation of cholesterol biosynthesis in neural stem cells increases their VEGF expression and angiogenesis but causes neuron apoptosis. Proc Natl Acad Sci USA. 2009;106:8350–8355. doi: 10.1073/pnas.0903541106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Abarca LI, Tabernero A, Medina JM. Oligodendrocytes use lactate as a source of energy and as a precursor of lipids. Glia. 2001;36:321–329. doi: 10.1002/glia.1119. [DOI] [PubMed] [Google Scholar]
- Sim FJ, Zhao C, Penderis J, Franklin RJ. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci. 2002;22:2451–2459. doi: 10.1523/JNEUROSCI.22-07-02451.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skripuletz T, Gudi V, Hackstette D, Stangel M. De- and remyelination in the CNS white and grey matter induced by cuprizone: the old, the new, and the unexpected. Histol Histopathol. 2011;26:1585–1597. doi: 10.14670/HH-26.1585. [DOI] [PubMed] [Google Scholar]
- Soundarapandian MM, Selvaraj V, Lo UG, Golub MS, Feldman DH, Pleasure DE, Deng W. Zfp488 promotes oligodendrocyte differentiation of neural progenitor cells in adult mice after demyelination. Sci Rep. 2011;1:2. doi: 10.1038/srep00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab. 2012;23:420–428. doi: 10.1016/j.tem.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner B, Kronenberg G, Jessberger S, Brandt MD, Reuter K, Kempermann G. Differential regulation of gliogenesis in the context of adult hippocampal neurogenesis in mice. Glia. 2004;46:41–52. doi: 10.1002/glia.10337. [DOI] [PubMed] [Google Scholar]
- Stoll EA, Cheung W, Mikheev AM, Sweet IR, Bielas JH, Zhang J, Rostomily RC, Horner PJ. Aging neural progenitor cells have decreased mitochondrial content and lower oxidative metabolism. J Biol Chem. 2011;286:38592–38601. doi: 10.1074/jbc.M111.252171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Meijer DH, Alberta JA, Mehta S, Kane MF, Tien AC, Fu H, Petryniak MA, Potter GB, Liu Z, Powers JF, Runquist IS, Rowitch DH, Stiles CD. Phosphorylation state of Olig2 regulates proliferation of neural progenitors. Neuron. 2011;69:906–917. doi: 10.1016/j.neuron.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi H, Nabeshima Y, Yoshida S, Chisaka O, Ikenaka K, Nabeshima Y. The basic helix-loop-helix factor olig2 is essential for the development of motoneuron and oligodendrocyte lineages. Curr Biol. 2002;12:1157–1163. doi: 10.1016/s0960-9822(02)00926-0. [DOI] [PubMed] [Google Scholar]
- Tyler WA, Jain MR, Cifelli SE, Li Q, Ku L, Feng Y, Li H, Wood TL. Proteomic identification of novel targets regulated by the mammalian target of rapamycin pathway during oligodendrocyte differentiation. Glia. 2011;59:1754–1769. doi: 10.1002/glia.21221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Bruzzone S, Zocchi E, Fedele E, Schenk U, De Flora A, Matteoli M. Evidence of a role for cyclic ADP-ribose in calcium signalling and neurotransmitter release in cultured astrocytes. J Neurochem. 2001;78:646–657. doi: 10.1046/j.1471-4159.2001.00455.x. [DOI] [PubMed] [Google Scholar]
- Voloboueva LA, Lee SW, Emery JF, Palmer TD, Giffard RG. Mitochondrial protection attenuates inflammation-induced impairment of neurogenesis in vitro and in vivo. J Neurosci. 2010;30:12242–12251. doi: 10.1523/JNEUROSCI.1752-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Xu TY, Guan YF, Tian WW, Viollet B, Rui YC, Zhai QW, Su DF, Miao CY. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann Neurol. 2011a;69:360–374. doi: 10.1002/ana.22236. [DOI] [PubMed] [Google Scholar]
- Wang W, Esbensen Y, Kunke D, Suganthan R, Rachek L, Bjoras M, Eide L. Mitochondrial DNA damage level determines neural stem cell differentiation fate. J Neurosci. 2011b;31:9746–9751. doi: 10.1523/JNEUROSCI.0852-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner M. A matter of identity: transcriptional control in oligodendrocytes. J Mol Neurosci. 2008;35:3–12. doi: 10.1007/s12031-007-9008-8. [DOI] [PubMed] [Google Scholar]
- Wong JV, Dong P, Nevins JR, Mathey-Prevot B, You L. Network calisthenics: control of E2F dynamics in cell cycle entry. Cell Cycle. 2011;10:3086–3094. doi: 10.4161/cc.10.18.17350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CW, Chang YT, Yu L, Chen HI, Jen CJ, Wu SY, Lo CP, Kuo YM. Exercise enhances the proliferation of neural stem cells and neurite growth and survival of neuronal progenitor cells in dentate gyrus of middle-aged mice. J Appl Physiol. 2008;105:1585–1594. doi: 10.1152/japplphysiol.90775.2008. [DOI] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Xie Y, Wang T, Bi J, Li H, Zhang LQ, Ye SQ, Ding S. Neuronal protective role of PBEF in a mouse model of cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:1962–1971. doi: 10.1038/jcbfm.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang J, Chen G, Fan D, Deng M. Inhibition of Sirt1 promotes neural progenitors toward motoneuron differentiation from human embryonic stem cells. Biochem Biophys Res Commun. 2011;404:610–614. doi: 10.1016/j.bbrc.2010.12.014. [DOI] [PubMed] [Google Scholar]
- Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell. 2012;11:589–595. doi: 10.1016/j.stem.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109:61–73. doi: 10.1016/s0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.