

Abstract
Angiogenesis is essential during development and in pathological conditions such as chronic inflammation and cancer progression. Inhibition of angiogenesis by targeting vascular endothelial growth factor (VEGF) blocks disease progression, but most patients eventually develop resistance which may result from compensatory signalling pathways. In endothelial cells (ECs), expression of the pro-angiogenic chemokine CXCL12 is regulated by non-canonical nuclear factor (NF)-κB signalling. Here, we report that NF-κB-inducing kinase (NIK) and subsequent non-canonical NF-κB signalling regulate both inflammation-induced and tumour-associated angiogenesis. NIK is highly expressed in endothelial cells (ECs) in tumour tissues and inflamed rheumatoid arthritis synovial tissue. Furthermore, non-canonical NF-κB signalling in human microvascular ECs significantly enhanced vascular tube formation, which was completely blocked by siRNA targeting NIK. Interestingly, Nik−/− mice exhibited normal angiogenesis during development and unaltered TNFα- or VEGF-induced angiogenic responses, whereas angiogenesis induced by non-canonical NF-κB stimuli was significantly reduced. In addition, angiogenesis in experimental arthritis and a murine tumour model was severely impaired in these mice. These studies provide evidence for a role of non-canonical NF-κB signalling in pathological angiogenesis, and identify NIK as a potential therapeutic target in chronic inflammatory diseases and tumour neoangiogenesis. © 2014 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: NF-κB, endothelial cells, angiogenesis, inflammation, NIK
Introduction
Angiogenesis is defined as the formation of new blood vessels from the pre-existing vasculature. It is a highly coordinated process occurring both during normal development and in pathological conditions such as atherosclerosis, chronic inflammation, and cancer 1. Activation of endothelial cells (ECs) is pivotal in this process and can result in the formation of new blood vessels, most notably via vascular endothelial growth factor (VEGF) receptor stimulation 2. In chronic inflammation, including rheumatoid arthritis (RA), and in tumours, pathological angiogenesis occurs when activated stromal cells and inflammatory cells secrete pro-angiogenic factors such as VEGF 2–4 that trigger quiescent ECs to differentiate and form new blood vessels 1. Importantly, inhibition of angiogenesis by targeting VEGF blocks disease progression in (pre-)clinical models of arthritis 5,6 and various cancers 7. Anti-VEGF monoclonal antibodies are used in clinical practice for eye diseases with neovascularization and metastatic cancer. However, most patients develop resistance 8,9, which may result from compensatory signalling pathways 7. These pathways can, for instance, induce inflammatory cytokines and proteases that contribute to enhanced angiogenesis in chronic inflammation and tumours 1.
Nuclear factor-κB (NF-κB) transcription factors can be activated via two distinct signalling pathways. The canonical NF-κB pathway is activated by pro-inflammatory stimuli such as TNFα and requires inhibitor of κB (IκB) kinase (IKK) β, whereas IKKα is dispensable 10. In contrast, the non-canonical pathway is strictly dependent on NF-κB-inducing kinase (NIK) and IKKα, but does not require IKKβ 11. This pathway can be activated via TNF-receptor superfamily members such as the lymphotoxin β receptor (LTβR) and CD40 10. All known inducers of the non-canonical NF-κB pathway also stimulate canonical NF-κB activity 12.
In ECs, the canonical NF-κB pathway is critical for pro-inflammatory gene expression, including TNFα 13,14. Furthermore, VEGF-receptor signalling induces canonical NF-κB signalling in ECs, and VEGF gene expression is regulated in part via the canonical NF-κB pathway 15. In contrast, the function of non-canonical NF-κB signalling in ECs is largely unknown. CXCL12, an important chemokine for lymphocyte transendothelial migration with pro-angiogenic properties 16, is regulated by non-canonical NF-κB signalling in ECs 17 and is expressed both in RA synovial tissue ECs 18 and in cancer ECs (reviewed in ref 19). In lymphoid tissues, LTβR-dependent non-canonical NF-κB signalling is required for high endothelial venule (HEV) differentiation and function 20,21. Interestingly, HEVs are also present in chronically inflamed tissues such as RA synovial tissue 22, and are also induced in tumours after antibody–lymphotoxin fusion protein treatment 23. In addition, LTβ and LIGHT (homologous to Lymphotoxins, exhibits Inducible expression, and competes with HSV Glycoprotein D for HVEM, a receptor expressed by T-lymphocytes), both ligands of the LTβR, and CD40L are expressed in RA synovial tissue 24–26. Furthermore, LTβ and LIGHT are produced by host immune cells in tumours 27,28 and promote tumour growth 28,29. Non-canonical NF-κB signalling in tumour cells is involved in tumour progression 30, but its contribution to angiogenesis has not yet been investigated.
Therefore, we investigated the role of non-canonical NF-κB signalling in inflammation-induced and tumour-associated angiogenesis, and compared this with its requirement for angiogenesis during development. In our studies, we used human RA synovial tissue biopsies and various human cancer tissues, complemented with murine disease models in Nik−/− mice.
Materials and methods
Human tissues
Synovial tissue biopsies were taken from knee joints of active RA patients or from healthy individuals. Early RA was defined as a disease duration of less than 1 year and established RA as a disease duration of more than 2 years 31. Tumour tissues were obtained from the Department of Pathology of the Academic Medical Center (AMC). All tissue collection was carried out at the AMC and approved by the Medical Ethical Commission.
Mice
Nik−/− mice (C57/Bl6 background) and wild-type (Wt) littermate controls were kindly provided by DV Novack (Washington University, St Louis, MO, USA) 32. All experimental procedures were reviewed and approved by the Experimental Animal Committee of the AMC.
Immunohistochemical staining
Frozen human tissue sections (synovial tissues, tumour tissues, or normal healthy tissues) were cut (5 µm) and mounted on Star Frost adhesive glass slides (Knittelgläser, Braunschweig, Germany). Sections were fixed with acetone and endogenous peroxidase activity was blocked with 0.3% H2O2 in 0.1% sodium azide in PBS. Sections were stained with monoclonal primary antibodies against NIK (sc-8417; Santa Cruz Biotechnology, Santa Cruz, CA, USA), p52 (#4882; Cell Signaling Technology, Danvers, MA, USA), and CXCL12 (MAB350; R&D Systems, Abingdon, UK), and secondary antibodies goat anti-mouse (p0447; DAKO, Glostrup, Denmark) or swine anti-rabbit (p0399; DAKO), and streptavidin-labelled with horseradish peroxidase. Biotinylated tyramine was used for amplification, as previously described 33. As negative controls, sections were incubated with isotype control antibodies (all R&D Systems).
Immunofluorescence staining
Tissue sections were fixed with acetone and incubated with the following monoclonal mouse antibodies: anti-NIK (sc-8417; Santa Cruz Biotechnology); polyclonal goat antibodies: anti-CD31 (sc-1505; Santa Cruz Biotechnology), anti-CD34 (sc-7045; Santa Cruz Biotechnology); polyclonal rabbit antibodies: anti-IKKα (ab17943; Abcam, Cambridge, UK). For double fluorescence staining of NIK with CD31/34, slides were incubated with goat anti-mouse-HRP (p0447; DAKO) and subsequently incubated with streptavidin-Alexa 594 (S-32356; Molecular Probes Europe, Leiden, The Netherlands), Alexa 488-conjugated goat anti-rabbit (A-11008; Molecular Probes Europe), and Alexa 488-conjugated rabbit anti-goat (A-11034; Molecular Probes Europe). For double fluorescence staining of NIK with IKKα, slides were incubated with Alexa 594-conjugated goat anti-mouse (A-11005; Molecular Probes Europe) and Alexa 488-conjugated donkey anti-rabbit (A-21206; Molecular Probes Europe). Slides were mounted with Vectashield containing DAPI (VC-H-1500; Brunschwig Chemie, Amsterdam, The Netherlands). Isotype control antibodies were used as a negative control (all R&D Systems). Slides were analysed using a Leica DMRA fluorescence microscope (Leica, Wetzlar, Germany) coupled to a CCD camera and Image-Pro Plus software (Dutch Vision Components, Breda, The Netherlands). A ratio of NIK pixel values to CD31/CD34 pixel values was calculated for each image (n = 10) for determination of double-positive areas.
Human microvascular endothelial cell culture
Human microvascular endothelial cells (HMVECs) from foreskin were isolated and characterized as described previously 34. HMVECs were cultured on gelatin-coated culture plates in Medium 199 supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin (p/s), 2 mm l-glutamine (all Lonza, Verviers, Belgium), 5 U/ml heparin, 3.75 µg/ml EC growth factor (ECGF; crude extract from bovine brain), 10% heat-inactivated human serum (HSi; PAA Laboratories, Pasching, Austria), and 10% heat-inactivated newborn calf serum (NBCSi; Lonza). Confluent cells were trypsinized (0.05% trypsin in HBSS; both Lonza) and replated in a 1:3 density. Cells were used until passage 10.
In vitro tube formation assay
In vitro angiogenesis was determined with HMVECs in three-dimensional fibrin matrices, as described before 14. Fibrin matrices were prepared by addition of thrombin (0.1 U/ml) to a 2 mg/ml fibrinogen solution (Kordia, Leiden, The Netherlands) in M199 medium (Invitrogen, Carlsbad, CA, USA). After polymerization, thrombin was inactivated by adding M199 supplemented with p/s, 10% HSi, and 10% NBCSi. HMVECs were seeded in confluent density on the fibrin matrices and after 24 h, and subsequently at 48 h intervals, HMVECs were incubated with M199, p/s, 10% HSi, 10% NBCSi, 10 ng/ml TNFα (Sigma-Aldrich, St Louis, MO, USA), and 10 ng/ml fibroblast growth factor-2 (FGF-2; Preprotech, London, UK) in the presence or absence of 30 ng/ml lymphotoxin α1β2 (LT; R&D Systems), 30 ng/ml LIGHT (R&D Systems) or 30 ng/ml CD40L (Enzo Life Sciences, Antwerp, Belgium). The total length of formed tube-like structures from HMVECs into the fibrin matrices was analysed by phase contrast microscopy and Optimas image analysis software 35. Tube formation of HMVECs isolated from various donors was determined in quadruplicate wells. HMVECs were transfected with siRNAs targeting NIK, IKKα, IKKβ, or a non-targeting siRNA (siGENOME SMARTpool; Dharmacon, Lafayette, CO, USA) by using the AMAXA basic nucleofector kit for primary ECs (Lonza), according to the manufacturer's protocols. Transfection efficiency was determined by gene expression analysis with QPCR (see Supplementary methods).
Retinal whole-mounts
Mice were sacrificed on postnatal day 28 36. Mouse eyes were collected and fixed in 4% paraformaldehyde (PFA). Retinas were dissected, fixed in 4% PFA, dehydrated, and stored in methanol at −20 °C. Before analysis, retinal whole-mounts were rehydrated, permeabilized in PBS, 1% BSA, and 0.5% Triton X-100, and washed with PBS. Retinal whole-mounts were blocked in PBlec [PBS (pH 6.8), 1% Triton X-100, 0.1 mm CaCl2, 0.1 mm MgCl2, 0.1 mm MnCl2] and incubated in Alexa 488-labelled isolectin B4 (Sigma-Aldrich) in PBlec. After extensive washing in PBS, the retinas were flat mounted in Vectashield (Vector Laboratories, Burlingame, CA, USA). Images were taken using a wide-field fluorescence microscope (Leica) or confocal microscope (Leica). For vascular quantification, the total number of branch points in the superficial, intermediate, and outer plexus was counted per microscopic field 36, or the number of radial veins and arteries in the superficial plexus was counted per retina (see Supplementary methods).
Aortic ring assay
After sacrificing the mice, thoracic aortae were dissected and adipose tissue/fat was removed, washed, and cut into 1-mm rings 37. Next, the aortic rings were embedded in Matrigel (growth factor reduced; BD Biosciences, Oxford, UK) and cultured in M199 medium (Invitrogen) containing 2% FCS, 12% ECGS (Sigma-Aldrich), 200 µl/ml gentamycin (Invitrogen), and 2.5 µl/ml amphotericin (Lonza) in the presence or absence of LT (30 ng/ml), LIGHT (30 ng/ml), TNF (100 ng/ml) or VEGF (100 ng/ml) (all R&D Systems). After 8 days, microvessel outgrowth was analysed using Leica imaging software.
Antigen-induced arthritis
Antigen-induced arthritis was performed as described previously 38. In brief, Wt (n = 5) and Nik−/− (n = 5) mice were immunized with 100 µg of methylated (m)BSA (Sigma-Aldrich) emulsified in 0.1 ml of complete Freund's adjuvant (BD) intradermally at the base of the tail. Simultaneously, 1 µg of pertussis toxin (List Biological Laboratories Inc, Campbell, CA, USA) was injected intraperitoneally as an additional adjuvant. Arthritis was induced at day 21 by intra-articular injection of 100 µg of mBSA into the right knee joint, while the left knee joint was injected with PBS alone. Mice were sacrificed on day 32. For histological grading of the arthritis, hind paws were dissected and fixed in 10% buffered formalin. Fixed tissues were decalcified in EDTA, dehydrated, and embedded in paraffin. Sagittal sections (5 µm) were stained with H&E, anti-CD31 (sc-1505; Santa Cruz Biotechnology), or anti-CXCL12 (MAB350; R&D Systems). Next, slides were incubated with secondary antibody rabbit anti-goat (sc-2922; Santa Cruz Biotechnology) or goat anti-mouse (P044701; DAKO), and streptavidin-labelled with horseradish peroxidase. Biotinylated tyramine was used for amplification. Slides were scored blindly by two independent researchers. Inflammation was scored semi-quantitatively on a scale of 0–4, and synovial blood vessels were counted.
B16 melanoma bone metastasis model
The melanoma bone metastasis model was performed as described previously 39. In brief, Wt mice (n = 6) and Nik−/− mice (n = 6) at 38–46 days of age were anaesthetized and inoculated intra-arterially with 105 B16-FL murine melanoma cells in 50 µl of PBS. Mice were sacrificed on day 12 after tumour cell injection. For histological analysis, hind paws were dissected and fixed in 10% buffered formalin. Fixed tissues were decalcified in EDTA, dehydrated, and embedded in paraffin. Sagittal sections (5 µm) were stained with H&E, anti-CD31 (sc-1505; Santa Cruz Biotechnology), or anti-CXCL12 (MAB350; R&D Systems). Next, slides were incubated with secondary antibody rabbit anti-goat (sc-2922; Santa Cruz Biotechnology) or goat anti-mouse (P044701; DAKO), and streptavidin-labelled with horseradish peroxidase. Biotinylated tyramine was used for amplification. Tumour blood vessels were counted by two independent researchers.
Statistical analysis
All data are presented as mean ± standard error of the mean (SEM). Statistical analysis was carried out using Prism Software (GraphPad). For statistical comparison, mean values per group were compared by unpaired Mann–Whitney U-test. p values less than 0.05 were considered statistically significant (*p < 0.05, **p < 0.01).
Results
NIK is highly expressed in endothelial cells of inflamed RA synovial tissue
The non-canonical NF-κB stimuli CD40L, LT, and LIGHT are highly expressed in RA synovial tissue 24–26. Therefore, we investigated whether the non-canonical NF-κB pathway is activated in RA synovial tissue by studying the expression and distribution of NIK using immunohistochemistry (IHC). Interestingly, NIK was mainly expressed by vascular structures, both in early and in established RA (Figures 1A and 1B), whereas normal healthy synovial tissue did not contain NIK-positive vessels (Supplementary Figure 1A). Subsequent double-immunofluorescence microscopy analysis revealed that NIK strongly co-localized with the EC marker CD31/34 in small blood vessels (Figure 1D). Quantification of each NIK+ and CD31/34+ area in double-stained tissue sections showed that approximately three-quarters (74.4 ± 5.5%; n = 10) of ECs in RA synovial tissue were NIK-positive. These results fuel the hypothesis that NIK may be of importance in either activation of ECs or inflammation-induced neovascularization.
Figure 1.
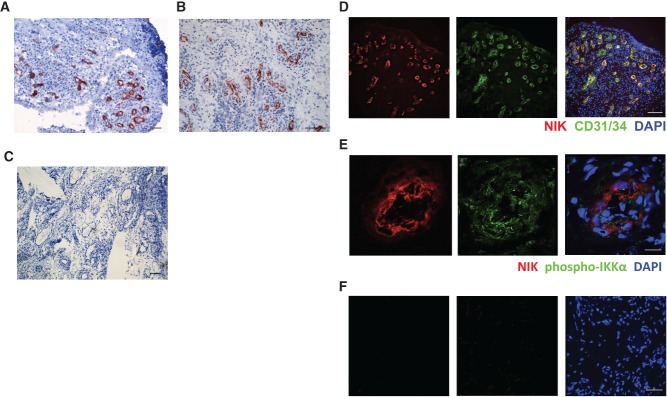
NIK expression in synovial tissue endothelial cells of rheumatoid arthritis patients. Synovial tissue sections from early and established RA patients showing (A, B) IHC staining of NIK and (C) the isotype control (scale bar in A, C = 100 µm; scale bar in B = 200 µm). (D) IF staining of NIK (red), CD31/34 (green), and nuclei (blue) (scale bar = 100 µm). (E) IF staining of NIK (red), phospho-IKKα (green), and nuclei (blue) (scale bar = 12.4 µm), and (F) the isotype control of E (scale bar = 27.9 µm). Representative pictures are shown (n = 10 different RA synovial tissues per staining).
Functional non-canonical NF-κB signalling in RA synovial tissue endothelial cells
Non-canonical NF-κB signalling in ECs induces the pro-angiogenic chemokine CXCL12 that also mediates lymphocyte transendothelial migration 16,17. To investigate whether non-canonical NF-κB signalling is functional in NIK-expressing ECs in RA synovial tissue, we performed additional staining for T-loop-activated phospho-IKKα, the non-canonical NF-κB subunit p52, and CXCL12. NIK expression (Figure1) co-localized with T-loop-activated phospho-IKKα (Figure1E), strongly suggesting that the non-canonical NF-κB pathway is activated in synovial ECs. In addition, both p52 (Supplementary Figure 1B) and CXCL12 (Supplementary Figure 1C) were expressed in blood vessels, further strengthening the hypothesis that the non-canonical NF-κB pathway is functionally active in ECs in RA synovial tissue. The NF-κB subunit RelB was also expressed in RA synovial tissue, but not exclusively in ECs (Supplementary Figure 1D), which is in line with previous reports 40.
NIK is also expressed by endothelial cells in tumour tissues
The non-canonical NF-κB pathway has been demonstrated to play an important role in tumour development 28,29,41. However, these studies did not focus on ECs or angiogenesis. To investigate whether NIK expression in newly formed blood vessels is specific for RA synovial tissue or also occurs in tumour neovascularization, we evaluated NIK expression in human melanoma, renal cell carcinoma, breast cancer, colorectal cancer, and pancreatic cancer. In these tissues, we detected NIK in vascular structures as well (Figures2A–2E). Double-immunofluorescence microscopy demonstrated that NIK and T-loop-activated phospho-IKKα were highly expressed by essentially all blood vessel ECs in cancer tissues, as depicted for breast cancer (Figures2F and 2G). Since both inflammation-induced angiogenesis and tumour-associated angiogenesis are in many aspects different from normal angiogenesis 1, we also studied the expression of NIK in blood vessels of healthy human tissues. Interestingly, ECs in blood vessels of normal human skin (Figure2H) and other normal healthy tissues did not exhibit NIK expression (Supplementary Figure 2). Collectively, these data show that both in chronic (synovial) inflammation and in tumours, NIK expression is exclusively vascular and increased selectively during neovascular growth.
Figure 2.

NIK is expressed in endothelial cells in tumour tissues. NIK expression in (A) renal carcinoma tissue, (B) breast cancer tissue, (C) pancreatic cancer tissue, (D) melanoma tissue, (E) colorectal cancer tissue, and (F) IF staining on NIK (red), CD31/CD34 (green), and nuclei (blue) in breast cancer tissue. (G) IF staining on NIK (red), phospho-IKKα (green), and nuclei (blue) in breast cancer tissue. (H) IF on NIK (red), CD31/CD34 (green), and nuclei (blue) in normal skin. Representative pictures are shown (n = 5–6 patients or healthy donors, two independent experiments). Scale bars: (A–F, H) 100 µm; (G) 11.2 µm.
Non-canonical NF-κB signalling induces in vitro sprouting of endothelial cells
Next, we evaluated the role of the non-canonical NF-κB pathway in vitro. HMVECs were seeded on fibrin matrices in the presence of bFGF/TNFα to facilitate outgrowth of tube-like structures, as a model for pathological sprout formation 14. Significantly increased tube formation was observed when LT, LIGHT or CD40L was added to bFGF/TNFα (Figures3A and 3B). This pro-angiogenic effect of the non-canonical NF-κB stimuli was dose-dependent, as shown for LT (Supplementary Figure 3A). To investigate whether the enhanced tube formation was dependent on canonical or non-canonical NF-κB signalling, we used siRNAs specifically targeting IKKβ, the essential kinase in canonical NF-κB signalling, or NIK or IKKα, the main kinases of the non-canonical NF-κB pathway (siIKKβ, siNIK or siIKKα). QPCR analysis demonstrated that target genes were all specifically knocked down 80–90% by the corresponding siRNAs (Supplementary Figures 3B–3D). Additional protein data confirmed that gene knockdown of NIK also prevented p100/p52 processing, as assessed by western blot (Supplementary Figure 3E). Specific siRNA-mediated gene silencing of NIK or IKKα blocked LT, LIGHT, and CD40L-enhanced tube formation significantly (Figures3C–3F). In contrast, siIKKβ blocked tube formation only partially. Furthermore, we also observed an effect of siNIK and siIKKα on basal tube formation, suggesting that the non-canonical pathway also contributed to basal sprouting in this model, although to a lesser extent. Altogether, it can be concluded that LT-, LIGHT-, and CD40L-induced tube formation relies mostly on non-canonical NF-κB signalling, a hitherto unknown function of this pathway in ECs. We confirmed that non-canonical NF-κB signalling in HMVECs results in nuclear translocation of p52 and DNA-binding activity, which significantly decreased in siNIK-treated cells (Supplementary Figure 3F). To further unravel the underlying mechanisms of NIK-induced angiogenesis in ECs, we performed PCR arrays of angiogenesis-related genes. Interestingly, knockdown of NIK in ECs resulted in significantly reduced expression levels of ANGPT2, BTG1, CD55, CSF3, CXCL4, CXCL5, CXCL6, CXCL9, CXCL11, CXCL12, FGF13, FN1, IFNB1, IL-6, IL-8, KITLG, PDGFB, PDGF, PGF, STAB1, and TGFA after LIGHT stimulation (Supplementary Table 1).
Figure 3.

Non-canonical NF-κB signalling induces in vitro sprouting of endothelial cells. (A) Representative pictures of tube formation by HMVECs under basal conditions or after LT, LIGHT, and CD40L stimulation (scale bar for all panels = 500 µm). (B) Quantification of LT-, LIGHT-, and CD40L-enhanced tube formation. n = 7 per group, three independent experiments. (C–F) HMVECs were transfected by the indicated siRNAs. Scrambled non-targeting siRNA (=siC) was used as a control. (C) Basal tube formation; (D) LT-enhanced tube formation; (E) LIGHT-enhanced tube formation; (F) CD40L-enhanced tube formation. n = 6 per group, three independent experiments. All panels: mean ± SEM. *p < 0.05; **p < 0.01. Specific knockdown of the genes targeted by the siRNAs is shown in Supplementary Figure 4.
Normal developmental angiogenesis, but reduced pathological angiogenic responses after genetic deletion of NIK
We made use of Nik−/− mice to further investigate the importance of NIK in angiogenesis in vivo. Nik−/− mice lack peripheral lymph nodes and have altered T- and B-cell responses 32. Despite these defects, Nik−/− mice are viable and appear to have a normal vasculature, suggesting that developmental angiogenesis is not impaired. To confirm this, we studied the vascular network in the retinas of Nik−/− mice compared with Wt mice on postnatal day 28, when all three vascular layers are fully mature 36. No differences were detected in the number of branch points in the superficial, intermediate, and outer plexuses between Nik−/− and Wt mice (Figures4A and 4B). Also, no abnormalities in the mean number of arteries and veins were observed (Supplementary Figure 4). To further study the importance of non-canonical NF-κB signalling in angiogenesis, we tested the angiogenic potential of Wt and Nik−/− mice in the aortic ring assay. Aortic rings from Wt and Nik−/− mice exhibited normal microvessel outgrowth after VEGF and TNFα stimulation, indicating that both VEGF- and TNFα-induced (canonical NF-κB-dependent) angiogenesis are unaffected (Figures4C and 4D), which is in line with the normal developmental angiogenesis of these mice. In contrast, whereas the non-canonical NF-κB stimuli LT and LIGHT significantly induced microvessel outgrowth in Wt mice, no microvessel outgrowth was observed in Nik−/− mice after LT or LIGHT stimulation (Figures4E and 4F). This strongly suggests that angiogenesis under pathological conditions, in the presence of stimuli such as LT and LIGHT, is dependent on NIK and subsequent non-canonical NF-κB signalling.
Figure 4.
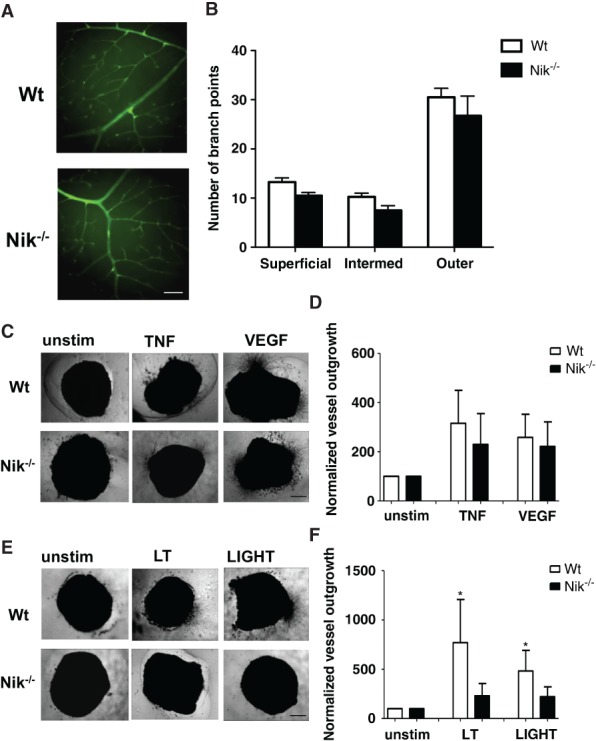
Genetic deletion of NIK results in reduced pathological angiogenic responses. (A) Representative retinas from Wt and Nik−/− mice, stained with isolectin B4 (green) (scale bar for both panels = 100 µm). (B) Quantification of vessel branching in the superficial, intermediate, and outer vascular plexuses. (C) Aortic rings of Wt and Nik−/− mice cultured in medium alone, TNF-α or VEGF. Representative pictures are shown (scale bar for all panels = 500 µm). (D) Quantification of TNF-α and VEGF-induced aortic vessel outgrowth. (E) Representative pictures of unstimulated, LT-, and LIGHT-stimulated aortic rings (scale bar for all panels = 500 µm). (F) Quantification of LT- and LIGHT-induced aortic vessel outgrowth. (B, C, D) n = 4 per group, two independent experiments; (A, E, F) n = 5 per group, two independent experiments. (B, D, F) Mean ± SEM. *p < 0.05.
Nik−/− mice exhibit reduced inflammation-induced and tumour-associated angiogenesis
Finally, we investigated whether pathological angiogenesis is dependent on non-canonical NF-κB signalling in vivo. To study inflammation-induced angiogenesis, we made use of an experimental arthritis model. Previously, we reported that NIK is important in the immune and bone-destructive components of inflammatory arthritis in antigen-induced arthritis 38. Histology of the arthritic hind paws of Nik−/− mice revealed less synovial inflammation (Figure5A). Quantification of the total number of synovial blood vessels demonstrated a 50% reduction in Nik−/− mice compared with Wt mice (Wt 20.00 ± 5.07 versus Nik−/− 10.20 ± 3.02; p = 0.1354) (Figure5B). In addition, we found that the number of CXCL12+ blood vessels was significantly decreased in the synovial tissue of Nik−/− mice (Wt 26.60 ± 1.63 versus Nik−/− 13.00 ± 1.23; p = 0.007) (Figure5C). This was accompanied by significantly reduced inflammation scores (Wt 3.40 ± 0.20 versus Nik−/− 1.67 ± 0.33; p = 0.008) (Figure5D). These findings suggest that non-canonical NF-κB signalling contributes to pathological, inflammation-induced angiogenesis.
Figure 5.

Nik−/− mice exhibit reduced inflammation-induced angiogenesis in arthritis and reduced tumour-associated angiogenesis in a melanoma model. (A) Representative picture of an mBSA-injected Wt mouse knee joint showing a severe inflammatory infiltrate (between arrows), in contrast to an mBSA-injected Nik−/− knee joint showing a reduced inflammatory infiltrate and intact cartilage (between arrows: scale bar = 500 µm). Sagittal sections of knee joint with H&E staining and CD31 or CXCL12 IHC staining (insets: scale bars = 100 µm). (B) Quantification of the total number of synovial blood vessels in Wt and Nik−/− mice. (C) Quantification of CXCL12+ blood vessels in synovial tissue of Wt and Nik−/− mice. (D) Quantification of synovial inflammation on a semi-quantitative scale of 0–4. (E) Representative pictures of tibial sections from Wt and Nik−/− mice, with B16 melanoma tumour tissue in trabecular bone. H&E-stained sections of tibias show marrow replacement by tumour cells beneath the growth plate (scale bar = 500 µm), and insets show CD31 and CXCL12 IHC staining on tumour tissue (scale bar in insets = 100 µm). (F) Quantification of the total number of blood vessels in melanoma tumour tissue in Wt and Nik−/− mice. (G) Quantification of CXCL12+ blood vessels in melanoma tumour tissue of Wt and Nik−/− mice. (A–D) n = 5 per group; (E–G) n = 6 per group. (B–D, F, G) Mean ± SEM. *p < 0.05; **p < 0.001.
Next, we investigated the contribution of NIK to tumour-associated angiogenesis using the B16 melanoma model. In this model, we have previously established that genetic deletion of NIK blocks tumour-induced loss of trabecular bone 39. Here, we demonstrate that Nik−/− mice have a significantly reduced total number of blood vessels inside the tumour compared with Wt mice (Wt 8.33 ± 6.36 versus Nik−/− 0.50 ± 0.34; p = 0.05) (Figures5E and 5F). In addition, the number of CXCL12+ blood vessels was significantly decreased in the tumour tissue of Nik−/− mice (Wt 12.5 ± 1.29 versus Nik−/− 2.33 ± 0.33; p = 0.005) (Figure5G), underlining the important role of non-canonical NF-κB signalling in pathological angiogenesis.
Discussion
Our data provide compelling evidence that NIK and subsequent non-canonical NF-κB signalling regulate inflammation-induced and tumour-associated angiogenesis, whereas NIK is dispensable for angiogenesis during development. Therefore, we propose that non-canonical NF-κB signalling specifically regulates pathological angiogenesis.
The non-canonical NF-κB pathway has been demonstrated to play an important role in tumour development 28,29,41. However, these studies mainly focused on the intrinsic role of non-canonical NF-κB signalling in tumour cells. Our study shows for the first time that NIK is expressed and active in ECs in inflamed RA synovial tissue as well as in a variety of tumour tissues, whereas the vasculature in normal healthy tissues does not express NIK. Importantly, our findings also identify NIK as a potential new therapeutic target in chronic inflammatory diseases, in particular arthritis, and cancer, since targeting components of the non-canonical NF-κB pathway efficiently blocked angiogenic responses. Previously, IKKβ-mediated canonical NF-κB signalling has been implicated in angiogenesis 42. However, knockdown of NIK or IKKα abrogated tube formation far more effectively than knockdown of IKKβ in our sprouting assay. Therefore, we conclude that LT-, LIGHT-, and CD40L-induced tube formation relies mostly on non-canonical NF-κB signalling, a hitherto unknown function of this pathway in ECs. Interestingly, knockdown of NIK or IKKα also blocked tube formation under basal conditions in the presence of low amounts of TNF and bFGF. This suggests that the non-canonical pathway is already somewhat activated in ECs in these conditions. A likely explanation for this is that ECs express both CD40 and CD40L 43, which could induce non-canonical NF-κB signalling in ECs due to cell–cell contact of adjacent ECs. The angiogenesis PCR array data point towards a pleiotropic role of non-canonical NF-κB signalling in chemokine expression and several other pro-angiogenic pathways in ECs, as subsequent gene ontology analysis did not reveal the involvement of one specific established angiogenesis pathway.
Our studies in Nik−/− mice demonstrate that both developmental retinal angiogenesis and VEGF-induced or TNFα-induced angiogenic responses are unaffected. In contrast, we observed significantly reduced non-canonical NF-κB-dependent microvessel outgrowth in the aortic ring assay and clearly impaired pathological, inflammation-induced angiogenesis in the antigen-induced arthritis model in Nik−/− mice. Of note, the observed reduction in arthritis and histological inflammation scores in Nik−/− mice may be at least in part due to impaired angiogenesis, since the number of CXCL12+ blood vessels was significantly reduced in Nik−/− mice.
Genetic ablation of NIK resulted in an almost complete block in pathological, tumour-associated angiogenesis in the B16 melanoma model. Scattered evidence from the literature underscores our findings that one of the signalling pathways of importance in tumour neoangiogenesis, and possibly also in anti-VEGF treatment resistance, may be the non-canonical NF-κB signalling pathway. Colorectal cancer ECs have been demonstrated to exhibit significantly increased IKKα expression compared with ECs in adjacent normal tissue 30. Furthermore, in ECs, expression of CXCL12, a chemokine that plays an important role in angiogenesis and the homing of CXCR4-expressing endothelial progenitor cells (EPCs) 16, is regulated by non-canonical NF-κB signalling 17. EPCs have been demonstrated to contribute to pathological angiogenesis both in developing tumours 44 and in lethal macrometastasis 45, as well as in the inflamed RA synovium 46,47. Therefore, non-canonical NF-κB-induced CXCL12 in cancer ECs 19 and synovial tissue ECs 18 may serve as a focal point modulating vasculogenesis, angiogenesis, and the attraction of immune cells.
Current strategies aimed at inhibition of angiogenesis in patients affect both pathological and physiological angiogenesis, resulting in unwanted side-effects. We hypothesize that targeting of NIK and subsequent non-canonical NF-κB signalling in ECs may overcome this problem. In addition, targeting of NIK could be combined with current anti-angiogenic cancer treatments to prevent or rescue resistance. In RA, pathological angiogenesis drives chronic inflammation and is therefore also an attractive therapeutic target 46. The recent description of the crystal structure of NIK may facilitate the development of new potent inhibitors 48,49. These inhibitors could be directed specifically to ECs using a multi-modular recombinant protein that specifically binds to cytokine-activated endothelium, which has been demonstrated to work very elegantly under inflammatory conditions in vivo 50.
Taken together, our studies point towards an important role of the non-canonical NF-κB pathway in pathological angiogenesis. Therefore, selective inhibition of non-canonical NF-κB signalling in ECs could be used as a novel therapeutic strategy, which may be beneficial not only in chronic inflammatory diseases such as RA and cancer, but potentially also in other diseases characterized by aberrant neovascularization, such as ocular diseases and atherosclerosis.
Acknowledgments
We thank the arthroscopy team of our department for obtaining the synovial tissue biopsies, Professor Dr Sandrine Florquin for providing tumour and healthy tissue specimens, Dr Marcel Teunissen for providing normal human skin biopsies, and Dr Rosalie Luiten and Esther Tjin for melanoma samples. We also acknowledge the invaluable technical support of Linda Hartkamp and Boy Helder, as well as the expert technical support of Dr Corine van der Horst with respect to digital imaging analysis. We thank Professor Dr Victor van Hinsbergh for critically reading the manuscript. This work was supported by a Veni grant of The Netherlands Organisation for Scientific Research (NWO) and a Clinical Fellowship (ZonMw) to SW Tas, and the National Institutes of Health (NIH) to DV Novack (grant No AR052705). P Koolwijk received a grant from The Netherlands Initiative for Regenerative Medicine (NIRM).
Author contribution statement
ARN contributed to the study design, all experiments, data analysis, and writing of the manuscript. KPMZ performed and collected data for in vitro and ex vivo experiments. EMW contributed to the tube formation experiments and drafting of the manuscript. PK contributed to and provided materials for tube formation experiments, discussion, and drafting the manuscript. CXM performed and collected data for in vitro and ex vivo experiments. MJS performed and analysed data of retina experiments. ROS provided materials, advice, and contributed to drafting of the manuscript. DVN performed and provided materials for in vivo experiments, discussion, and drafting of the manuscript. PPT contributed to study design, drafting of the manuscript and provided advice and discussion. SWT was responsible for project conceptualization, experimental design, writing of the manuscript, and funding of the project.
Supporting Information
The following supporting information may be found in the online version of this article
(related to Figure 1). NIK is not expressed in healthy synovial tissue, and downstream proteins of the non-canonical NF-κB pathway are expressed in RA synovial tissue.
(related to Figure 2). NIK is not expressed in related healthy tissues.
(related to Figure 3). The enhanced tube formation by HMVECs in 3D fibrin matrices is dose-dependent, and non-canonical NF-κB pathway-specific.
(related to Figure 4). We show a different quantification method of the retinal blood vessels of Wt and Nik−/− mice.
(related to Figure 3). This table shows a list of angiogenic genes that are significantly down-regulated by NIK siRNA in HMVECs.
References
- 1.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nature Rev Cancer. 2002;2:795–803. doi: 10.1038/nrc909. [DOI] [PubMed] [Google Scholar]
- 3.Kim KJ, Li B, Winer J, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 4.Fava RA, Olsen NJ, Spencer-Green G, et al. Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med. 1994;180:341–346. doi: 10.1084/jem.180.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi ST, Kim JH, Seok JY, et al. Therapeutic effect of anti-vascular endothelial growth factor receptor I antibody in the established collagen-induced arthritis mouse model. Clin Rheumatol. 2009;28:333–337. doi: 10.1007/s10067-008-1075-x. [DOI] [PubMed] [Google Scholar]
- 6.Sone H, Kawakami Y, Sakauchi M, et al. Neutralization of vascular endothelial growth factor prevents collagen-induced arthritis and ameliorates established disease in mice. Biochem Biophys Res Commun. 2001;281:562–568. doi: 10.1006/bbrc.2001.4395. [DOI] [PubMed] [Google Scholar]
- 7.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nature Rev Cancer. 2008;8:579–591. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 8.Welti J, Loges S, Dimmeler S, et al. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190–3200. doi: 10.1172/JCI70212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bakall B, Folk JC, Boldt HC, et al. Aflibercept therapy for exudative age-related macular degeneration resistant to bevacizumab and ranibizumab. Am J Ophthalmol. 2013;156:15–22 e11. doi: 10.1016/j.ajo.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 10.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nature Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 11.Senftleben U, Cao Y, Xiao G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 12.Tas SW, Vervoordeldonk MJ, Tak PP. Gene therapy targeting nuclear factor-kappaB: towards clinical application in inflammatory diseases and cancer. Curr Gene Ther. 2009;9:160–170. doi: 10.2174/156652309788488569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Martin MR, Hoeth M, Hofer-Warbinek R, et al. The transcription factor NF-kappa B and the regulation of vascular cell function. ArteriosclerThromb Vasc Biol. 2000;20:E83–E88. doi: 10.1161/01.atv.20.11.e83. [DOI] [PubMed] [Google Scholar]
- 14.Koolwijk P, van Erck MG, de Vree WJ, et al. Cooperative effect of TNFalpha, bFGF, and VEGF on the formation of tubular structures of human microvascular endothelial cells in a fibrin matrix. Role of urokinase activity 1. J Cell Biol. 1996;132:1177–1188. doi: 10.1083/jcb.132.6.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang S, Robinson JB, Deguzman A, et al. Blockade of nuclear factor-kappaB signaling inhibits angiogenesis and tumorigenicity of human ovarian cancer cells by suppressing expression of vascular endothelial growth factor and interleukin 8. Cancer Res. 2000;60:5334–5339. [PubMed] [Google Scholar]
- 16.Petit I, Jin D, Rafii S. The SDF-1–CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol. 2007;28:299–307. doi: 10.1016/j.it.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madge LA, Kluger MS, Orange JS, et al. Lymphotoxin-alpha 1 beta 2 and LIGHT induce classical and noncanonical NF-kappa B-dependent proinflammatory gene expression in vascular endothelial cells. J Immunol. 2008;180:3467–3477. doi: 10.4049/jimmunol.180.5.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santiago B, Baleux F, Palao G, et al. CXCL12 is displayed by rheumatoid endothelial cells through its basic amino-terminal motif on heparan sulfate proteoglycans. Arthritis Res Ther. 2006;8:R43. doi: 10.1186/ar1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–2931. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 20.Drayton DL, Bonizzi G, Ying X, et al. I kappa B kinase complex alpha kinase activity controls chemokine and high endothelial venule gene expression in lymph nodes and nasal-associated lymphoid tissue. J Immunol. 2004;173:6161–6168. doi: 10.4049/jimmunol.173.10.6161. [DOI] [PubMed] [Google Scholar]
- 21.Browning JL, Allaire N, Ngam-Ek A, et al. Lymphotoxin-beta receptor signaling is required for the homeostatic control of HEV differentiation and function. Immunity. 2005;23:539–550. doi: 10.1016/j.immuni.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Manzo A, Paoletti S, Carulli M, et al. Systematic microanatomical analysis of CXCL13 and CCL21 in situ production and progressive lymphoid organization in rheumatoid synovitis. Eur J Immunol. 2005;35:1347–1359. doi: 10.1002/eji.200425830. [DOI] [PubMed] [Google Scholar]
- 23.Schrama D, Thor SP, Fischer WH, et al. Targeting of lymphotoxin-alpha to the tumor elicits an efficient immune response associated with induction of peripheral lymphoid-like tissue. Immunity. 2001;14:111–121. doi: 10.1016/s1074-7613(01)00094-2. [DOI] [PubMed] [Google Scholar]
- 24.Takemura S, Braun A, Crowson C, et al. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167:1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 25.Pierer M, Brentano F, Rethage J, et al. The TNF superfamily member LIGHT contributes to survival and activation of synovial fibroblasts in rheumatoid arthritis. Rheumatology(Oxford) 2007;46:1063–1070. doi: 10.1093/rheumatology/kem063. [DOI] [PubMed] [Google Scholar]
- 26.Kang YM, Zhang X, Wagner UG, et al. CD8 T cells are required for the formation of ectopic germinal centers in rheumatoid synovitis. J Exp Med. 2002;195:1325–1336. doi: 10.1084/jem.20011565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hehlgans T, Stoelcker B, Stopfer P, et al. Lymphotoxin-beta receptor immune interaction promotes tumor growth by inducing angiogenesis. Cancer Res. 2002;62:4034–4040. [PubMed] [Google Scholar]
- 28.Daller B, Musch W, Rohrl J, et al. Lymphotoxin-β receptor activation by lymphotoxin-α(1)β(2) and LIGHT promotes tumor growth in an NFκB-dependent manner. Int J Cancer. 2011;128:1363–1370. doi: 10.1002/ijc.25456. [DOI] [PubMed] [Google Scholar]
- 29.Wolf MJ, Seleznik GM, Zeller N, et al. The unexpected role of lymphotoxin beta receptor signaling in carcinogenesis: from lymphoid tissue formation to liver and prostate cancer development. Oncogene. 2010;29:5006–5018. doi: 10.1038/onc.2010.260. [DOI] [PubMed] [Google Scholar]
- 30.Charalambous MP, Lightfoot T, Speirs V, et al. Expression of COX-2, NF-kappaB-p65, NF-kappaB-p50 and IKKalpha in malignant and adjacent normal human colorectal tissue. Br J Cancer. 2009;101:106–115. doi: 10.1038/sj.bjc.6605120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Hair MJ, Harty LC, Gerlag DM, et al. Synovial tissue analysis for the discovery of diagnostic and prognostic biomarkers in patients with early arthritis. J Rheumatol. 2011;38:2068–2072. doi: 10.3899/jrheum.110426. [DOI] [PubMed] [Google Scholar]
- 32.Yin L, Wu L, Wesche H, et al. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science. 2001;291:2162–2165. doi: 10.1126/science.1058453. [DOI] [PubMed] [Google Scholar]
- 33.Smeets TJ, Barg EC, Kraan MC, et al. Analysis of the cell infiltrate and expression of proinflammatory cytokines and matrix metalloproteinases in arthroscopic synovial biopsies: comparison with synovial samples from patients with end stage, destructive rheumatoid arthritis. Ann Rheum Dis. 2003;62:635–638. doi: 10.1136/ard.62.7.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Hinsbergh VW, Sprengers ED, Kooistra T. Effect of thrombin on the production of plasminogen activators and PA inhibitor-1 by human foreskin microvascular endothelial cells. Thromb Haemost. 1987;57:148–153. [PubMed] [Google Scholar]
- 35.van Hensbergen Y, Broxterman HJ, Peters E, et al. Aminopeptidase inhibitor bestatin stimulates microvascular endothelial cell invasion in a fibrin matrix. Thromb Haemost. 2003;90:921–929. doi: 10.1160/TH03-03-0144. [DOI] [PubMed] [Google Scholar]
- 36.Stahl A, Connor KM, Sapieha P, et al. The mouse retina as an angiogenesis model. Invest Ophthalmol Vis Sci. 2010;51:2813–2826. doi: 10.1167/iovs.10-5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baker M, Robinson SD, Lechertier T, et al. Use of the mouse aortic ring assay to study angiogenesis. Nature Protoc. 2012;7:89–104. doi: 10.1038/nprot.2011.435. [DOI] [PubMed] [Google Scholar]
- 38.Aya K, Alhawagri M, Hagen-Stapleton A, et al. NF-(kappa)B-inducing kinase controls lymphocyte and osteoclast activities in inflammatory arthritis. J Clin Invest. 2005;115:1848–1854. doi: 10.1172/JCI23763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vaira S, Johnson T, Hirbe AC, et al. RelB is the NF-kappaB subunit downstream of NIK responsible for osteoclast differentiation. Proc Natl Acad Sci U S A. 2008;105:3897–3902. doi: 10.1073/pnas.0708576105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pettit AR, MacDonald KP, O'Sullivan B, et al. Differentiated dendritic cells expressing nuclear RelB are predominantly located in rheumatoid synovial tissue perivascular mononuclear cell aggregates. Arthritis Rheum. 2000;43:791–800. doi: 10.1002/1529-0131(200004)43:4<791::AID-ANR9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 41.Keats JJ, Fonseca R, Chesi M, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu HG, Yu LL, Yang Y, et al. Increased expression of RelA/nuclear factor-kappa B protein correlates with colorectal tumorigenesis. Oncology. 2003;65:37–45. doi: 10.1159/000071203. [DOI] [PubMed] [Google Scholar]
- 43.Mach F, Schonbeck U, Sukhova GK, et al. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40–CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci U S A. 1997;94:1931–1936. doi: 10.1073/pnas.94.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lyden D, Hattori K, Dias S, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nature Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 45.Gao D, Nolan DJ, Mellick AS, et al. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319:195–198. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- 46.Szekanecz Z, Besenyei T, Szentpetery A, et al. Angiogenesis and vasculogenesis in rheumatoid arthritis. Curr Opin Rheumatol. 2010;22:299–306. doi: 10.1097/BOR.0b013e328337c95a. [DOI] [PubMed] [Google Scholar]
- 47.Distler JH, Beyer C, Schett G, et al. Endothelial progenitor cells: novel players in the pathogenesis of rheumatic diseases. Arthritis Rheum. 2009;60:3168–3179. doi: 10.1002/art.24921. [DOI] [PubMed] [Google Scholar]
- 48.de Leon-Boenig G, Bowman KK, Feng JA, et al. The crystal structure of the catalytic domain of the NF-kappaB inducing kinase reveals a narrow but flexible active site. Structure. 2012;20:1704–1714. doi: 10.1016/j.str.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 49.Li K, McGee LR, Fisher B, et al. Inhibiting NF-kappaB-inducing kinase (NIK): discovery, structure-based design, synthesis, structure–activity relationship, and co-crystal structures. Bioorg Med Chem Lett. 2013;23:1238–1244. doi: 10.1016/j.bmcl.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 50.Sehnert B, Burkhardt H, Wessels JT, et al. NF-kappaB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-kappaB in immune-mediated diseases. Proc Natl Acad Sci U S A. 2013;110:16556–16561. doi: 10.1073/pnas.1218219110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(related to Figure 1). NIK is not expressed in healthy synovial tissue, and downstream proteins of the non-canonical NF-κB pathway are expressed in RA synovial tissue.
(related to Figure 2). NIK is not expressed in related healthy tissues.
(related to Figure 3). The enhanced tube formation by HMVECs in 3D fibrin matrices is dose-dependent, and non-canonical NF-κB pathway-specific.
(related to Figure 4). We show a different quantification method of the retinal blood vessels of Wt and Nik−/− mice.
(related to Figure 3). This table shows a list of angiogenic genes that are significantly down-regulated by NIK siRNA in HMVECs.