

SUMMARY
Bone marrow (BM) metastasis remains one of the main causes of death associated with solid tumors as well as with Multiple Myeloma (MM). Targeting the BM niche to prevent or modulate metastasis has not been successful to date. Here we show that stromal cell derived factor-1 (SDF-1/CXCL12) is highly expressed in active MM, as well as in BM sites of tumor metastasis, and report on the discovery of the high affinity anti-SDF-1 PEGylated mirror-image l-oligonucleotide (olaptesed-pegol). In vivo confocal imaging showed that SDF-1 levels are increased within MM cell-colonized BM areas. Using in vivo murine and xenograft mouse models, we document that in vivo SDF-1 neutralization within BM niches leads to a microenvironment that is less receptive for MM cells and reduces MM cell homing and growth, thereby inhibiting MM disease progression. Targeting of SDF-1 represents a valid strategy for preventing or disrupting colonization of the BM by MM cells.
INTRODUCTION
Mechanisms that allow clonal multiple myeloma (MM) or solid tumor cells to home to and colonize bone marrow (BM) sites are not well defined. Seventy percent of patients with metastatic breast or prostate cancer present with involvement of the BM, and BM metastases also appear in about 15-30% of patients with colon, lung, stomach, thyroid, prostate and kidney cancer (Roodman, 2004). MM patients at diagnosis are characterized by the presence of multiple BM-lytic lesions, which indicate the perpetual dissemination of clonal plasma cells - originating in the birthplace of the MM clone - to multiple BM niches throughout the skeleton (Roodman, 2004); such lesions suggest that BM niches form an optimal environment for the lodgment and growth of MM cells. MM cells may also behave similarly to hematopoietic stem cells (HSCs), explaining why MM cells are selectively trafficked to BM sites (Karnoub and Weinberg, 2006; Scadden, 2006; Smith et al., 2004). In fact, it has been argued that clonal plasma cells - or other types of BM-metastasizing tumor cells - use the same signals and molecules that are critical for selection of the HSC niche: these include the chemoattractant stromal cell-derived factor-1 (SDF-1/CXCL12), which is a major retention factor for HSCs and mature immune cells in BM tissue (Scheiermann et al., 2013).
The biological effects of SDF-1 are related its ability to trigger motility, chemotactic responses, adhesion, and the secretion of matrix metalloproteinases and angiopoietic factors. The importance of SDF-1 is also reflected by its ability to activate integrins such as LFA-1, VLA-4, and VLA-5 (expressed on HSCs), and to thus promote adhesion and migration (Dar et al., 2006; Kucia et al., 2005; Peled et al., 1999; Peled et al., 2000; Wysoczynski et al., 2005). Other factors such as fibroblast growth factors, transforming growth factor β, insulin-like growth factors I and II, platelet-derived growth factors, and bone morphogenetic proteins also contribute to the preferential homing of tumor cells to the BM. And additionally, it is likely that areas of high blood flow in red marrow facilitate tumor cell metastasis to these regions of BM (Callander and Roodman, 2001; Chirgwin and Guise, 2000; Roodman, 2004). Moreover, MM cells are characterized by the presence of CXCR4 (one of the SDF-1 receptors), and SDF-1-dependent signaling via CXCR4 plays a critical role in regulating fundamental biological functions of MM cells, such as migration and adhesion in vitro, as well as their homing to the bone marrow in vivo (Alsayed et al., 2007; Azab et al., 2009). SDF-1 also activates the CXCR7 receptor (Tarnowski et al., 2010), which modulates trafficking and adhesion of human malignant hematopoietic cells.
Collectively, these findings point to the importance of targeting SDF-1, and of neutralizing CXCR4 and CXCR7, both of which are expressed on tumor cells (Burwick, 2008; Miao et al., 2007; Tarnowski et al., 2010; Xu et al., 2011). We reasoned that by neutralizing SDF-1, we can change the BM milieu, create a microenvironment that is less receptive for MM cells, diminish the homing of MM cells to the BM, and thereby inhibit MM disease progression. We report here on the discovery and characterization of olaptesed pegol (ola-PEG), a high affinity l-RNA Spiegelmer to SDF-1, and we demonstrate that ola-PEG-mediated neutralization of SDF-1 is a valid strategy for preventing MM progression in vivo.
RESULTS
Metastatic bone marrow niches reveal increased expression of SDF-1
Given the importance of SDF-1 in facilitating bone metastasis in a variety of clonal tumor cells, we compared the expression of this chemokine in the BM of patients with bone metastases of solid tumors or of multiple myeloma (MM), to that in the BM of healthy individuals or of patients with monoclonal gammopathy of undetermined significance (MGUS; the early precursor state of MM). Our findings indicate that BM that is affected by colonization of solid tumor cells or MM cells presents with higher expression of SDF-1, compared to samples from healthy controls or from MGUS patients which showed scattered, minimal SDF-1 expression (Figure 1A). These findings indicate the significance of SDF-1 in the BM niche of metastatic tumors, including MM as a model of BM metastasis.
Figure 1. SDF-1 expression in metastasized bone marrow niches.

(A) Bone marrow (BM) specimens were obtained from patients with multiple myeloma, MGUS, or solid tumors, and stained with anti-SDF-1 antibodies; MGUS samples were double-stained for SDF-1 (brown) and CD138 (blue). Normal BM was used as control. (B) SCID/Bg mice were injected (i.v.) with 5×106 MM.1S-GFP+ cells; after 3 weeks, AlexaFluor(AF)633-conjugated anti-SDF-1α antibody (Ab) was administered i.v., and mouse skull BM niches were imaged after 4 hours, using in vivo confocal microscopy. Evans Blue was used to visualize blood vessels (MM.1S-GFP+/Luc+ cells: green; vessels: red; AF633-anti-SDF-1α Ab: blue). (C) Levels of SDF-1α in primary BM mesenchymal stromal cells isolated from MM patients (n=10) and healthy subjects (n=5) were evaluated by ELISA.
We next focused on corroborating the role of SDF-1 in facilitating the homing, lodgment and growth of MM cells in vivo. We used in vivo live confocal imaging to determine the localization and spatial distribution of SDF-1 in BM niches that are occupied by MM cells. By injecting a fluorescently labeled anti-SDF-1 antibody in mice that harbor MM GFP+ tumors, we found that SDF-1 was enriched in BM areas that were colonized by MM cells, whereas SDF-1 was only observed within blood vessels in BM niches that were free of MM cells (Figure 1B). This observation is consistent with prior reports that transplanted hematopoietic stem and progenitor cell populations localize in subdomains of BM microvessels, where the SDF-1 chemokine is particularly abundant (Colmone et al., 2008; Sipkins et al., 2005). The finding was also supported by the significantly higher level of SDF-1 secreted from primary BM-mesenchymal stromal cells (MSCs) that were obtained from MM patients, relative to those that were harvested from healthy subjects (Figure 1C). Taken together, these results demonstrate that MM cells preferentially localized in SDF-1-enriched BM niche.
To better define the functional role of MM BM-MSC-secreted SDF-1 on MM cells, we used primary MM BM-MSCs to undertake SDF-1 loss-of-function studies. Transduction and knock-down efficiency of SDF-1 were evaluated with use of immunofluorescence and ELISA, respectively (Suppl. Fig. 1 A, B). Results showed that knocking down the secretion of SDF-1 from MM BM-MSCs inhibits the adhesion and also the migration of MM cells towards primary MM BM-MSCs (Suppl. Fig. 1C-D). Moreover, MM cells cultured in the presence of BM-MSC cells in which SDF-1 was knocked down, presented with reduced activation of pro-survival and migration-related pathways (Suppl. Fig. 1E).
Identification of the SDF-1-neutralizing Spiegelmer olaptesed pegol
We identified a high-affinity, structured l-RNA oligonucleotide (Spiegelmer (Klussmann et al., 1996) that binds and inhibits SDF-1. Initially, we applied an in vitro evolutionary screening technology based on the SELEX process (Tuerk and Gold, 1990) to select aptamers from a pool of randomized RNA oligonucleotides against d-SDF-1 (the enantiomer of natural l-SDF-1). This process led to the identification of aptamer sequences, which were synthesized as Spiegelmers by using non-natural l-ribonucleotides. The Spiegelmers were then able to bind to natural l-SDF-1.
For in vitro selection against d-SDF-1, we used reiterative rounds with increasing stringency and amplification steps that included mutagenic PCR (in rounds 15, 17) (Suppl. Fig. 2A). The enriched library from round 18 was cloned and sequenced. An alignment revealed sequences that differed by point mutations and by a non-related orphan sequence (Suppl. Fig. 2B). After truncating the primer binding sites, the best aptamer (193-G2), was trimmed to a 45mer (193-G2-001) without loss of affinity (Suppl. Fig. 2C). A pull-down assay showed that the binding of 193-G2-001 to d-SDF-1 resulted in a dissociation constant (Kd) of 268 pM at 37 °C (Figure 2A).
Figure 2. 193-G2-001/Olaptesed secondary structure and binding kinetics.

(A) Pull-down binding assay of aptamer d-193-G2-001, using the biotinylated selection target d-SDF-1. Fitting with a 3 parameter algorithm revealed a Kd of 268 pM. (B) Secondary structure prediction for olaptesed, with potential base-pairs forming three hydrogen bonds depicted in red, and those forming two hydrogen bonds with each other shown in blue. (C) Biacore analysis of olaptesed pegol binding to immobilized human SDF-1α, at 37 °C under physiological buffer conditions. Raw data: black; fitted data: red.
The RNA aptamer 193-G2-001 ola-PEG was converted into its corresponding Spiegelmer. A secondary structure prediction (mfold) is depicted in Figure 2B (Zuker, 2003). To slow down renal clearance of the Spiegelmer, we increased its molecular weight by conjugating a 40 kDa polyethylene glycol (PEG) to its 5′-end. This PEGylated l-RNA oligonucleotide was named 0p; all subsequent in vitro and in vivo data were generated with olaptesed pegol (ola-PEG). Dissociation constants of approximately 200 pM to human and mouse SDF-1α/β/γ were determined with use of surface plasmon resonance (Biacore) under physiological conditions (Figure 2C and Suppl. Tab. I). Since the three-dimensional structures of chemokines are conserved (Wells and Peitsch, 1997), we used a competitive assay format to test ola-PEG binding against a panel of 23 different chemokines. Other than soluble SDF-1, none of the other chemokines could compete with ola-PEG, demonstrating a high selectivity of ola-PEG to SDF-1 (Figure 3A).
Figure 3. In vitro characterization of ola-PEG.

(A) CC, CXC, CX3C and XC chemokines (2 μM) were checked for competing with the binding of ola-PEG (12.5 nM) to immobilized human SDF-1α. As expected, mixtures of ola-PEG with SDF-1α or SDF-1β competed fully, but none of the other chemokines did so. Data are double-referenced, and are plotted as mean response units ± SD of n=2 injections. (B) Inhibition of SDF-1α-induced CXCR4 receptor internalization by ola-PEG. Jurkat cells were incubated with 0.3 nM SDF-1, plus various concentrations of ola-PEG. CXCR4 surface expression was quantified by flow cytometry, using a CXCR4 specific PE-labeled antibody. Data points are means ± SD for triplicate measurements. ola-PEG inhibits CXCR4 internalization with an IC50 of approximately 200 pM. (C) Inhibition of SDF-1-induced chemotaxis. Ola-PEG inhibits chemotaxis of Jurkat cells, with an IC50 of approximately 200 pM. Baseline level (fluorescence measured without SDF-1) is reached at approximately 800 pM ola-PEG. Means ± SD for triplicate measurements are shown. (D) Inhibition of SDF-1-induced CXCR7 activation by ola-PEG. The mean IC50 value obtained from three independent experiments was 5.1 nM.
In vitro functional characterization of ola-PEG-dependent targeting of SDF-1
We evaluated the influence of SDF-1 on the expression of CXCR4 and assessed whether SDF-1-induced activity could be blocked by ola-PEG. Stimulation of Jurkat cells with human SDF-1 resulted in a dose-dependent internalization of CXCR4. The maximal effect was reached at around 3 nM, half-maximal internalization was achieved at around 0.3 nM. Increasing concentrations of ola-PEG inhibited SDF-1-mediated CXCR4 receptor internalization, with an IC50 of 200 pM (Figure 3B). Also, ola-PEG inhibited SDF-1-mediated chemotaxis of CXCR4-expressing Jurkat cells in a dose-dependent manner, displaying an IC50 of 200 pM (Figure 3C). Additionally, using a β-arrestin complementation assay with a CXCR7 reporter cell line, we confirmed that ola-PEG also blocks SDF-1-dependent activation of the second SDF-1 receptor CXCR7. At a fixed concentration of 10 nM SDF-1 (EC50), the half-maximal inhibition (IC50) was at 5.1 nM ola-PEG (Figure 3D). (note that for stoichiometric reasons, the assay does not allow measurement of IC50 values lower than 5 nM). A non-functional Spiegelmer with the reverse sequence showed no effects (data not shown).
Pharmacokinetics of olaptesed pegol
Following a single s.c. administration of 10 mg/kg ola-PEG in mice, maximum plasma concentrations (Cmax) of ola-PEG were observed at 12 and 6 h in male and female mice, respectively (tmax) (Suppl. Tab. II; Suppl. Fig. 2D). Thereafter, the plasma concentrations of ola-PEG declined, with a mean apparent half-life (t1/2) of 13 h, which is comparable to the value obtained after i.v. administration. The bioavailability (F) of ola-PEG in male and female mice was estimated to be approximately 29.8% and 39.8%, respectively, with no appreciable or consistent gender-related difference in systemic exposure (AUC and Cmax) to ola-PEG. Human pharmacokinetics and pharmacodynamics, along with safety evaluations, were performed in healthy volunteers in a clinical Phase I study (Vater, 2013): ola-PEG had a mean plasma half-life of 33-40 h, and was safe and well tolerated up to the highest dose of 10.8 mg/kg, when administered as a 15 min i.v. infusion. ola-PEG mobilized white blood cells and hematopoietic stem and progenitor cells, as shown by CD34+ cell counts and colony formation assays.
Ola-PEG-dependent neutralization of SDF-1 modulates BM niches, and inhibits bone engraftment and bone colonization by MM cells
To test the hypothesis that neutralization of SDF-1 in BM niches makes them less receptive to MM cell engraftment and colonization, we first examined whether ola-PEG-dependent SDF-1 inhibition prevents MM cell bone engraftment. MM cells were injected (i.v.) into control untreated mice, or into mice that were pre-treated with ola-PEG (20 mg/kg; every other day) or with AMD3100 (0.5mg/kg; daily; s.c.); treatment was continued after MM cell injection. Mice treated with ola-PEG showed significant reduction of MM cell tumor growth, relative to mice that were treated with AMD3100, or were untreated (Figure 4A), indicating that ola-PEG-dependent modulation of the BM milieu delays MM cell BM engraftment and tumor growth. Importantly, we detected differences in survival with ola-PEG treated mice displaying significantly improved survival compared to those treated with AMD3100, or to untreated mice (Figure 4B). We next investigated whether ola-PEG treatment could also reduce MM cell dissemination from a primary BM site to distant bones. MM cell lines (murine or human) were loaded into bone chips and implanted subcutaneously (s.c.) into recipient mice that were pretreated with either vehicle control, or with ola-PEG. After the implantation, ola-PEG treatment was continued in the mice that were ola-PEG-pre-treated. Results showed that, compared to control mice, SDF-1 neutralization by ola-PEG significantly inhibited the colonization of distant BM sites by MM cells (Figure 5A-D). Importantly, no ola-PEG-mediated cytotoxic effects were detected in MM cells in vitro (Suppl. Fig. 3A).
Figure 4. Ola-PEG-dependent neutralization of SDF-1 reduces MM tumor progression in vivo.

(A) Three weeks of pre-treatment with ola-PEG (20mg/kg injected every other day; s.c.) led to inhibition of MM tumor progression, shown with use of bioluminescence imaging. ola-treated mice are compared to mice that were pre-treated with AMD3100 (5,g/kg, daily; s.c.), or to untreated mice (n: 8/group). (B) Ola-PEG-pretreated mice presented with prolonged survival relative to AMD3100-pretreated or untreated mice (n: 10/group).
Figure 5. Ola-PEG-dependent neutralization of SDF-1 inhibits dissemination of MM cells from bone-to-bone.
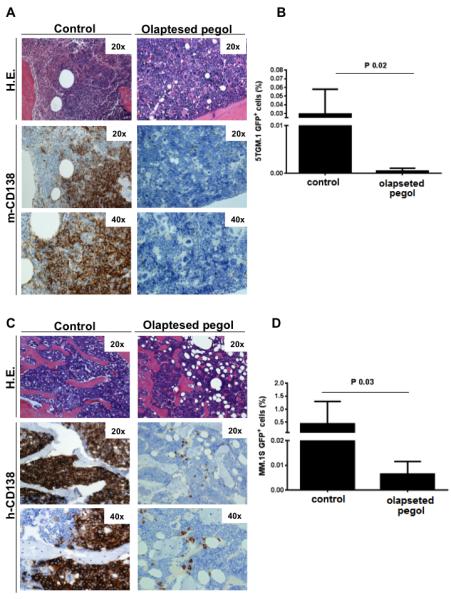
(A-D) ola-PEG-dependent inhibition of SDF-1 inhibited in vivo colonization of MM cells (GFP+5TGM1; GFP+MM.1S) - originating at a primary bone marrow site - to distant bone niches, as demonstrated by flow cytometry (GFP+) and immunostaining for murine (m) and human (h)-CD138. H.E. indicates hematoxylin-eosin staining (20×; 40×). Mice were euthanized when signs of paralysis were detected.
To better understand how BM niches are modulated by neutralizing SDF-1, we examined ola-PEG-mediated transcriptional changes in the BM microenvironment in vivo. SCID/Bg mice were treated with ola-PEG (s.c. injection, 20 mg/kg, given every other day) for three weeks. Untreated mice were used as controls. At the end of the 3rd week, BM was harvested, and gene expression profiling was performed. Ola-PEG induced transcriptional down-regulation of its target SDF-1, reduced expression of genes known to be regulators of angiogenesis, cell growth, cytokine production, vessel development and integrin-mediated cell adhesion, and down-regulated protein kinase activity; in contrast, heat-shock proteins were up-regulated (Suppl. Fig. 3B; P<0.05). Genes were categorized using dChip software; see supplemental data (Suppl. Gene List) for a detailed gene list.
To understand the in vivo distribution and localization of ola-PEG in areas of MM cell growth, we injected (i.v.) mice that harbor MM.1S-GFP+ tumors with AlexaFluor(AF)647-conjugated ola-PEG, and used live confocal imaging to determine the spatial localization of ola-PEG in BM niches that were colonized by MM cells; results documented that ola-PEG penetrated into BM areas that harbored MM cells (Suppl. Fig. 3C).
Ola-PEG mobilizes and chemosensitizes BM-colonizing MM cells in vivo
Because disruption of the SDF-1 gradient between BM and peripheral blood leads to the release of HSCs and lymphocytes into the circulation (Bleul et al., 1996; Dalakas et al., 2005; Dar et al., 2011; Kim and Broxmeyer, 1998; Kim et al., 1998), we argued that SDF-1 neutralization may lead to the release of MM cells from the BM into the peripheral circulation.
When mice with established MM were treated with ola-PEG for 5 weeks, we observed reduced numbers of MM cells within the BM niches, along with increased numbers of MM cells in the circulation (Suppl. Fig. 4A). These results indicate that ola-PEG has an impact on the interaction of MM cells with the BM milieu in vivo, leading to mobilization of MM cells. We therefore evaluated the effect of ola-PEG as monotherapy, or as a combination therapy with bortezomib, on tumor progression and MM cell dissemination in vivo. Control mice, and mice treated with ola-PEG as a single agent, showed similar growth rates for MM.1S-GFP+/Luc+, whereas mice treated with a combination of ola-PEG and bortezomib presented with a significant reduction of tumor burden; this reduction was more pronounced when compared to mice treated with bortezomib as a single agent (Figure 6A). These data were supported by the finding that ola-PEG and bortezomib work synergistically in inhibiting MM cell proliferation in vitro, when tested on a panel of MM cell lines (Suppl. Fig. 5A-D).
Figure 6. In vivo relevance of ola-PEG: effect on MM tumor growth.

(A) SCID/Bg mice were injected (i.v.) with 5×106 MM.1S-GFP+/Luc+ cells. Subsequently, mice were treated with vehicle (control), bortezomib (0.5 mg/kg, twice/week; i.p.) or ola-PEG (20 mg/kg, every other day; s.c.), alone or in combination (ola-PEG followed by Bortezomib) (n=5 per group). Tumor burden was detected by bioluminescence imaging, at different time points post-MM cell injection (t0: 2nd week; t1: 4th week; t2: 5th week). (B) Five weeks after MM cell inoculation, MM cell presence in BM structures of the skull was evaluated by intravital confocal microscopy (GFP+ MM cells: green; Evans Blue positive-blood vessels: red). Specific BM niches are highlighted, and relative magnification and 3D reconstruction are provided for each panel. (C) The presence of MM.1S-GFP+ cells ex vivo on femur tissues was detected by immunofluorescence. One representative image for one mouse from each group is shown (40×). For relative quantification, see Suppl. Figure 5E. (D) Primary MM BM-MSCs were treated with ola-PEG for 10 hours, and subsequently cultured in the presence of MM.1S cells for 8 hours. MM.1S cells were then harvested and cell lysates were subjected to Western blotting, with use of antibodies against p-ERK1/2, ERK1/2, p-cofilin, p-paxillin, p-Akt, p-Src, p-S6R, and p-GSK3. MM.1S cells cultured in the absence of MM BM-MSCs were used as control.
We next interrogated whether ola-PEG and bortezomib also act synergistically in modulating MM cell dissemination to distant BM niches in vivo, - and if so, whether this is due to the ability of ola-PEG to modulate cell adhesion. In vivo confocal microscopy revealed significant MM cell homing to the BM in control mice, while ola-PEG-treated mice presented with a lower number of MM cells within the whole skull-BM area that was imaged (Figure 6B). These observations point to two possibilities: i) that the BM of ola-PEG-treated mice is less receptive to MM cells, and ii) that the reduction in number of MM cells is due to an increase in mobilization of MM cells from the BM into the peripheral blood. Analysis of bortezomib-treated mice showed a significant reduction of MM cells, and this effect was even more significant with the combination of ola-PEG and bortezomib. These results were confirmed ex vivo, by quantifying the presence of MM cells in the femurs of treated mice (Figure 6C; Suppl. 5E), and were further supported by the demonstration of synergism between ola-PEG and bortezomib in inhibiting the adhesion of MM cells to primary MM BM-MSCs in vitro (Suppl. Fig. 6A-D).
The above findings were further corroborated at the protein level: primary MM BM-MSCs were pre-treated with ola-PEG and subsequently cultured in the presence of MM cells; both MM and BM-MSCs cells were then exposed to ola-PEG or bortezomib, used alone or as a combinatory regimen. Ola-PEG inhibited the BM-MSC-dependent up-regulation of p-ERK, p-Akt and p-Src in MM cells, and also similarly affected Akt-downstream targeted proteins (Figure 6D). Notably, ola-PEG treatment exerted a stronger effect when used in combination with bortezomib: in particular, ola-PEG was able to overcome the bortezomib-dependent activation of p-AKT, together with a more pronounced inhibition of Akt-downstream targeted proteins (p-S6R; p-GSK3). In addition, proteins such as cofilin and paxilin, which are involved in the modulation of cell adhesion, were also modulated by the ola-PEG/bortezomib treatment (Figure 6D).
DISCUSSION
In 1889, Stephan Paget presented the “seed-and soil” hypothesis for cancer metastasis, which states that certain malignant cells (seed) have a high and specific affinity for the milieu of specific organs (soil), and that the metastatic phenotype transpires only when the tumor cell and the surrounding microenvironment are compatible (Paget, 1889). In support of this hypothesis, a growing body of evidence now points to an essential role of the tumor microenvironment as a critical regulator of tumor cell metastasis, that facilitates the homing and expansion of the tumor clone from the primary neoplasm to distant organs (Valastyan and Weinberg, 2011). Along these lines, the receptivity of the BM milieu and its components is essential for tumor cells that preferentially metastasize to the bones, as reported for breast, prostate, and lung cancer (Bryden et al., 2002; Devys et al., 2001; Karnoub and Weinberg, 2006; Smith et al., 2004). A key factor in this cell trafficking network within the BM is the interaction of the chemokine SDF-1 (CXCL12) and its receptor CXCR4 (Alsayed et al., 2007; Azab et al., 2009; Dar et al., 2011).
The development of therapeutic agents that prevent or delay metastasis has been hampered by many limitations and failures; indeed, a number of anti-neoplastic agents that are effective on primary tumor sites do not display the same ability to inhibit the proliferation of disseminated cancer cells, and there is a pressing need to develop novel therapeutic strategies that disrupt the permissive metastatic niche and thus reduce the capacity of cancer cells to home to and engraft in these areas. Agents such as bisphosphonates, anti-RANK antibodies, and TGF-β inhibitors such as LY2157299 show promising activity, but none demonstrates significant clinical activity in preventing or delaying metastases. Novel anti-angiogenic molecules are implicated in inhibiting the process of metastasis, however recent evidence shows that such drugs may in fact paradoxically increase metastatic potential, due to the induction of hypoxia within the tumor mass (Ebos et al., 2009).
Here we use MM as a model of bone-to-bone cell dissemination. We first demonstrate that SDF-1 co-localizes in areas of the BM where metastasis occurs – not only in multiple epithelial tumors, but also in advanced stages of MM. Indeed, by comparing early precursor stages (such as MGUS) to late, disseminated stages of advanced MM, we were able to document a significant increase in SDF-1 levels in the BM with advanced disease. We also report on the identification and characterization of ola-PEG, a PEGylated l-RNA Spiegelmer that binds the chemokine SDF-1 with high affinity and selectivity. Our cell-based studies show that ola-PEG potently inhibits SDF-1 action, and thereby affects downstream signaling in tumor cells. Most interestingly, our data show that ola-PEG-dependent neutralization of SDF-1 modulates the pre-metastatic BM niche in vivo, and thus inhibits MM tumor progression and prolonged survival, compared to AMD3100-treated mice. SDF-1 inhibition also affects the BM gene expression signature by altering regulators of angiogenesis, adhesion and proliferation that collectively modify the BM niche; significantly reduces the homing of injected MM cells to the BM of SCID/Bg mice; and decreases MM cell bone metastases in vivo. Whether these effects are due to inhibition of SDF-1 signaling in the tumor cell itself, or due to changes in the stroma and its associated gene expression, has yet to be determined. Nevertheless, our findings clearly document a therapeutic effect of ola-PEG in a MM xenograft model of established disease. ola-PEG co-localizes in areas of high tumor burden within the BM, and disrupts the interaction of MM cells with the BM niches, as evidenced by its influence on diminishing tumor mobilization, homing, and growth within the BM niches. In addition, ola-PEG also chemosensitizes MM cells to bortezomib, despite the fact that it (ola-PEG) has no single-agent activity on the tumor cells, indicating that the activity of ola-PEG is specific to the niche and its interaction with SDF-1. Thus, combinations of ola-PEG with other chemotherapeutic agents are also likely to lead to synergistic effects.
In conclusion, our data suggest that the anti-SDF-1 Spiegelmer ola-PEG represents a first-in-class agent to successfully target the interaction between BM niches and tumor cells, thereby preventing or disrupting BM colonization by MM cells and potentially also by other bone-metastasizing tumor cells.
EXPERIMENTAL PROCEDURES
Cells
Primary BM-MSCs were obtained from MM patients: approval for these studies was obtained from the Institutional Review Board of the Dana-Farber Cancer Institute. Informed consent was obtained from all patients and healthy volunteers, in accordance with the Declaration of Helsinki protocol. BM-MSCs were devoid of hematopoietic cells (CD34−, CD138−, CD45−, CD14−), and were positive for markers (CD73+; CD90+; CD105+; CD106+) that indicate their multipotent MSC phenotype, as previously reported (Roccaro et al., 2013). Luciferase (luc)–expressing MM.1S-GFP/luc cell lines were generated by retroviral transduction of MM.1S with the pGC-gfp/luc vector (kind gift of Dr. A. Kung, Dana-Farber Cancer Institute, Boston, MA). The MM cell lines MM.1S, RPMI.8226, OPM2 and U266, were also used. Jurkat cells were obtained from DSMZ (Braunschweig, Germany). GFP+5TGM1 cells were kindly provided by Dr. G. D. Roodman (Indiana University).
In vitro selection and synthesis of ola-PEG
The oligonucleotide sequence of ola-PEG (5′-GCG UGG UGU GAU CUA GAU GUA UUG GCU GAU CCU AGU CAG GUA CGC-3′) was identified after 18 selection rounds, by screening a library of approx. 1015 different RNA oligonucleotides (carrying a 34 nt long random region) against biotinylated human SDF-1 in the all-d-configuration. The affinity of RNA libraries and aptamers to d-SDF-1 was determined by use of pull-down binding assays with radioactively labeled RNA. All oligonucleotides were synthesized at NOXXON Pharma AG, following standard synthesis protocols; and PEGylated Spiegelmers were synthesized as described (Hoffmann et al., 2011).
Use of surface plasmon resonance (SPR) to analyze the binding and selectivity of interaction between ola-PEG and SDF-1
SPR measurements were carried out at physiological conditions on a Biacore 2000 system, with use of CM4 sensor chips and immobilized SDF-1; a reference flow cell (activated and then blocked) on the same sensor chip served as a control. Data were analyzed, and dissociation constants (Kd) calculated with the BIAevaluation 3.1.1 software, using a modified Langmuir 1:1 stoichiometric fitting algorithm. Each measurement was done at least 3 times on different days. Selectivity data were plotted using the Prism 5.04 Software.
Cell-based assays for ola-PEG characterization
A detailed description of the methods is provided in the Supplemental Materials and Methods section. Briefly, CXCR4 internalization was determined in Jurkat cells using PE-labeled anti-CXCR4 antibodies and flow cytometry. CXCR7 signaling was analyzed with use of a β-arrestin complementation assay and a reporter cell line. MAP-kinase activation was studied in CHO-K1 cells transfected with a plasmid that codes for human CXCR4 (NM_003467.2); immunoblotting was performed with anti-MAP-K and anti-Phospho-MAP-K antibodies. Chemotaxis assays were carried out with Jurkat cells in Corning Transwell plates (5 μm pores) for 3 h. Prior to the addition of cells to the upper compartment, SDF-1 and various concentrations of ola-PEG were pre-incubated for 20-30 min in the lower compartment; migrated cells were quantified with resazurin.
Pharmacokinetics of ola-PEG
A detailed description of the pharamcokinetic data is provided in the Supplemental Materials and Methods section.
Reagents
Bortezomib was obtained from Hospital Pharmacy, diluted in DMSO, and stored at −20°C until use, then thawed and diluted in culture medium immediately before use. The maximum final concentration of DMSO (< 0.1%) did not affect cell proliferation and did not induce cytotoxicity on the cell lines tested (data not shown). AMD3100 was obtained from Sigma Aldrich (St. Louis, MO).
Immunoblotting
MM cells, cultured in presence or absence of SDF-1, were harvested and lysed using lysis buffer (Cell Signaling Technology, Beverly, MA) supplemented with 5 mM NaF, 2 mM Na3VO4, 1 mM PMSF (polymethilsulfonyl fluoride), 5 μg/mL leupeptine, and 5 μg/mL aprotinin. Whole-cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel eletrophoresis (SDS-PAGE) and transferred to polyvinyldene fluoride (PVDF) membrane (Bio-Rad Laboratories, Hercules, CA). Immunoblotting was done with antibodies against phospho(p)-ERK, ERK, p-cofilin, p-Akt, Akt, p-Src, p-S6R, p-GSK3, p-paxillin, p-cofilin, and α-tubulin(Cell Signaling, Danvers, MA).
ELISA
SDF-1 levels were determined with use of an SDF-1 ELISA (Human SDF-1 immunoassay, R&D Systems, Minneapolis, MN), according to the manufacturer’s protocol.
Gene expression profile
Gene expression profiling was performed on murine BM, using the Mouse Genome 430 2.0 Array. (GEO accession number: GSE42257). Untreated (n=3) and ola-PEG-treated (n=3) mice were compared, and differentially expressed genes were classified with use of dChip software (1.5 fold change; P <0.05): a detailed list of genes is provided as supplemental material.
In vivo studies
SCID/Bg mice were used for all in vivo studies. Six week-old female SCID-Bg mice (Charles River Labs, Wilmington, MA) were treated, monitored, and sacrificed in accordance with a protocol approved by the Dana-Farber Cancer Institute Animal Care and Use Committee.
Intravital confocal microscopy was performed as reported (Colmone et al., 2008; Roccaro et al., 2013), using a Zeiss 710 confocal system (Carl Zeiss Microimaging, Jena, Germany) on an upright Examiner stand with a custom stage. Briefly, a skin flap was made in the scalp to expose the underlying dorsal skull surface; high-resolution images with cellular detail were obtained through the intact skull, using a 10× 0.45NA Plan-Apo objective (Carl Zeiss Microimaging). Images were acquired at multiple imaging depths (up to 250 μm from the skull surface), and were merged with use of a maximum intensity z-projection. GFP was excited with the 488nm line on an Argon laser. Blood vessels were imaged using Evans Blue (Sigma-Aldrich, St. Louis, MO) excited with a 633 nm laser. Emission signals were collected with use of the Zeiss internal confocal Quasar detectors. To localize SDF-1 within the BM niches, SCID/Bg mice were injected (i.v.) with 5×106 MM.1S-GFP+ cells. After 3 weeks, AlexaFluor(AF)633-conjugated anti-SDF-1α antibody (Ab) (Santa Cruz Biotechnology, Dallas, TX) was administered i.v. (Colmone et al., 2008), and 4 hours later, intravital confocal microscopy was used to visualize mouse skull BM niches. Evans blue was injected i.v. to detect vessels.
To visualize tumor growth in vivo, mice were injected with 75 mg/kg of Luciferin (Xenogen, Hopkington, MA); 3 min later, tumor growth was detected by bioluminescence, using a Xenogen In Vivo Imaging System (Caliper Life Sciences, Hopkinton, MA), as reported (Roccaro et al., 2009).
For the in vivo metastasis model, bone chips were loaded with either murine (GFP+ 5TGM1) or human (GFP+ MM.1S) MM cells (2×106 cells/bone). Two bones were implanted subcutaneously into SCID/Bg mice, which were pre-treated with vehicle or ola-PEG (20 mg/kg; every other day; s.c.) for 3 weeks. Mice were euthanized once signs of limb paralysis became evident. MM cells were harvested from one host femur, and evaluated by flow cytometry for GFP positivity; the second host femur was used for immunostaining with murine and human anti-CD138 antibodies, to detect metastasized 5TGM1 and MM.1S cells, respectively.
To evaluate the effects of the bortezomib and ola-PEG combinatory regimen in vivo, SCID/Bg mice were injected (i.v.) with 5×106 MM.1S-GFP+/Luc+ cells; after 2 weeks, treatment was initiated with either vehicle (control), bortezomib (0.5 mg/kg, twice/week; i.p.), ola-PEG (20 mg/kg, every other day; s.c.), or a combination of ola-PEG and bortezomib (n=5 per group). Tumor burden was detected with use of bioluminescence imaging at different time points following the MM cell injection. Six weeks after MM cell inoculation, intravital confocal microscopy was used for a detailed evaluation of MM cell presence in BM structures of the skull. Evans Blue was injected i.v. to visualize vessels, and immunofluorescence was used to detect MM.1S-GFP+ cells ex vivo on femur tissues; nuclei were stained using DAPI.
Tissue immunofluorescence
Sections through the femurs of mice were immunostained as described (Hsieh et al., 2012), to detect MM.1S-GFP+ cells. Nuclei were stained with DAPI. Slides were analyzed on a fluorescence microscope (Nikon TE2000-E; objective 40× plan fluor 0.75NA). MM.1S-GFP+ cells were counted in 4 separate fields per slide. Images were taken on a Hamamatsu OrcaER camera, with NIS-Element software. Image J was used to merge channels.
In vitro studies
DNA synthesis was assessed by [3H]-thymidine ([3H]-TdR; Perkin Elmer, Boston, MA) uptake, as described (Roccaro et al., 2010; Sacco et al., 2011). Adhesion of MM cells to primary MM bone marrow MSCs was evaluated by an in vitro adhesion assay, using Calcein AM-labeling of MM cells, with degree of fluorescence measured on a spectrophotometer (485-520), as described (Roccaro et al., 2010). Migration of MM cells was evaluated as reported (Roccaro et al., 2010).
SDF-1 knock-down
SDF-1 knock-down in primary MM BM-MSCs was achieved with use of a tetracycline-inducible pTRIPz-Turbo-RFP vector (Thermo Scientific, Pittsburg, PA) containing the target sequence (clones 111678, 227310) or scramble control, according to manufacturer’s specifications. Transduction efficiency was determined by evaluating the proportion of cells expressing red fluorescent protein (RFP). SDF-1 knock down efficiency was validated by detecting levels of SDF-1 in the conditioned medium of infected cells.
Statistical analyses
P values for the in vitro assays were based on T-tests (two-tailed; α 0.05), and are provided for each figure. Drug synergism was analyzed by isobologram analysis, using the CalcuSyn software program (Biosoft, Ferguson, MO), as described (Roccaro et al., 2008; Roccaro et al., 2010; Sacco et al., 2011). The Kaplan-Meier curve was obtained using GraphPad Prism, and P values were calculated based on log-rank test.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Sonal Jhaveri for editing the manuscript; and Mrs. Ludmila Flores for technical assistance for paraffin-embedded tissue cutting.
Footnotes
CONFLICT OF INTEREST: Werner G. Purschke, Klaus Buchner, Christian Maasch, Dirk Zboralski, Stefan Zöllner, Stefan Vonhoff, Dirk Eulberg, Anna Kruschinski, Axel Vater, Sven Klussmann are employees of NOXXON Pharma AG.
All the remaining authors declare no competing financial interests.
AUTHORS’ CONTRIBUTION
AMR and IMG conceived of and designed the experiments, analyzed data and wrote the manuscript.
AMR and AS designed and performed the in vitro and in vivo functional experiments YM, MM, MRR carried out in vivo confocal microscopy.
GR, SL, MU, FF provided multiple myeloma bone marrow samples, and did the histology studies.
PM performed gene expression profiling analysis.
SK supervised the ola-PEG discovery program and revised the paper.
SV designed and performed manufacturing of ola-PEG.
AK advised on in vivo experiments and revised the paper.
AV designed the preclinical research, wrote the discovery section of the paper, and revised the paper.
DE designed concepts for SDF-1 as a target, as well as the designed experiments for in vitro selection.
WGP identified SDF-1-binding Spiegelmers, including ola-PEG, by in vitro selection, and characterized them by pull-down assays.
KB Designed a cell-based chemotaxis assay, and characterized selected SDF-1 Spiegelmers for inhibition of SDF-1alpha and beta of human origin and from other species. Also characterized inhibition of MAPK phosphorylation and CXCR7 activation. DZ designed and performed the receptor internalization assay with Jurkat cells.
CM characterized in vitro selected SDF-1 Spiegelmers for binding of SDF-1alpha and beta of human origin and from other species; also determined cross-reactivity of ola-PEG to other chemokines by surface plasmon resonance (Biacore).
SZ was responsible for the PK study, analysis of PK samples, and data interpretation.
ACCESSION NUMBER
GEO accession number: GSE42257.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alsayed Y, Ngo H, Runnels J, Leleu X, Singha UK, Pitsillides CM, Spencer JA, Kimlinger T, Ghobrial JM, Jia X, et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood. 2007;109:2708–2717. doi: 10.1182/blood-2006-07-035857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azab AK, Runnels JM, Pitsillides C, Moreau AS, Azab F, Leleu X, Jia X, Wright R, Ospina B, Carlson AL, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113:4341–4351. doi: 10.1182/blood-2008-10-186668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1) J Exp Med. 1996;184:1101–1109. doi: 10.1084/jem.184.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryden AA, Islam S, Freemont AJ, Shanks JH, George NJ, Clarke NW. Parathyroid hormone-related peptide: expression in prostate cancer bone metastases. Prostate Cancer Prostatic Dis. 2002;5:59–62. doi: 10.1038/sj.pcan.4500553. [DOI] [PubMed] [Google Scholar]
- Burwick N, Moreau AS, Jia X, Leleu X, Runnels J, Ngo HT, Melhem MR, Roccaro AM, Azab AK, Ghobrial IM. CXCR7 Regulates SDF-1 Induced Adhesion and Homing in Multiple Myeloma Blood. 2008;112 doi: 10.1182/blood-2007-12-129395. Abstract 1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callander NS, Roodman GD. Myeloma bone disease. Semin Hematol. 2001;38:276–285. doi: 10.1016/s0037-1963(01)90020-4. [DOI] [PubMed] [Google Scholar]
- Chirgwin JM, Guise TA. Molecular mechanisms of tumor-bone interactions in osteolytic metastases. Crit Rev Eukaryot Gene Expr. 2000;10:159–178. [PubMed] [Google Scholar]
- Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322:1861–1865. doi: 10.1126/science.1164390. [DOI] [PubMed] [Google Scholar]
- Dalakas E, Newsome PN, Harrison DJ, Plevris JN. Hematopoietic stem cell trafficking in liver injury. FASEB J. 2005;19:1225–1231. doi: 10.1096/fj.04-2604rev. [DOI] [PubMed] [Google Scholar]
- Dar A, Kollet O, Lapidot T. Mutual, reciprocal SDF-1/CXCR4 interactions between hematopoietic and bone marrow stromal cells regulate human stem cell migration and development in NOD/SCID chimeric mice. Exp Hematol. 2006;34:967–975. doi: 10.1016/j.exphem.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Dar A, Schajnovitz A, Lapid K, Kalinkovich A, Itkin T, Ludin A, Kao WM, Battista M, Tesio M, Kollet O, et al. Rapid mobilization of hematopoietic progenitors by AMD3100 and catecholamines is mediated by CXCR4-dependent SDF-1 release from bone marrow stromal cells. Leukemia. 2011;25:1286–1296. doi: 10.1038/leu.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devys A, Lortholary A, Audran M. [PTHrP and breast cancer] Bull Cancer. 2001;88:1075–1080. [PubMed] [Google Scholar]
- Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S, Hoos J, Klussmann S, Vonhoff S. RNA aptamers and spiegelmers: synthesis, purification, and post-synthetic PEG conjugation. Curr Protoc Nucleic Acid Chem. 2011;46:41–30. doi: 10.1002/0471142700.nc0446s46. Chapter 4, Unit 4. [DOI] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnoub AE, Weinberg RA. Chemokine networks and breast cancer metastasis. Breast Dis. 2006;26:75–85. doi: 10.3233/bd-2007-26107. [DOI] [PubMed] [Google Scholar]
- Kim CH, Broxmeyer HE. In vitro behavior of hematopoietic progenitor cells under the influence of chemoattractants: stromal cell-derived factor-1, steel factor, and the bone marrow environment. Blood. 1998;91:100–110. [PubMed] [Google Scholar]
- Kim CH, Pelus LM, White JR, Broxmeyer HE. Differential chemotactic behavior of developing T cells in response to thymic chemokines. Blood. 1998;91:4434–4443. [PubMed] [Google Scholar]
- Klussmann S, Nolte A, Bald R, Erdmann VA, Furste JP. Mirror-image RNA that binds D-adenosine. Nat Biotechnol. 1996;14:1112–1115. doi: 10.1038/nbt0996-1112. [DOI] [PubMed] [Google Scholar]
- Kucia M, Reca R, Miekus K, Wanzeck J, Wojakowski W, Janowska-Wieczorek A, Ratajczak J, Ratajczak MZ. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4 axis. Stem Cells. 2005;23:879–894. doi: 10.1634/stemcells.2004-0342. [DOI] [PubMed] [Google Scholar]
- Miao Z, Luker KE, Summers BC, Berahovich R, Bhojani MS, Rehemtulla A, Kleer CG, Essner JJ, Nasevicius A, Luker GD, et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci U S A. 2007;104:15735–15740. doi: 10.1073/pnas.0610444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paget S. The distribution of secondary growths in cancer of breast. Lancet. 1889;1 [PubMed] [Google Scholar]
- Peled A, Grabovsky V, Habler L, Sandbank J, Arenzana-Seisdedos F, Petit I, Ben-Hur H, Lapidot T, Alon R. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34(+) cells on vascular endothelium under shear flow. J Clin Invest. 1999;104:1199–1211. doi: 10.1172/JCI7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled A, Kollet O, Ponomaryov T, Petit I, Franitza S, Grabovsky V, Slav MM, Nagler A, Lider O, Alon R, et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood. 2000;95:3289–3296. [PubMed] [Google Scholar]
- Roccaro AM, Leleu X, Sacco A, Jia X, Melhem M, Moreau AS, Ngo HT, Runnels J, Azab A, Azab F, et al. Dual targeting of the proteasome regulates survival and homing in Waldenstrom macroglobulinemia. Blood. 2008;111:4752–4763. doi: 10.1182/blood-2007-11-120972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roccaro AM, Sacco A, Husu EN, Pitsillides C, Vesole S, Azab AK, Azab F, Melhem M, Ngo HT, Quang P, et al. Dual targeting of the PI3K/Akt/mTOR pathway as an antitumor strategy in Waldenstrom macroglobulinemia. Blood. 2010;115:559–569. doi: 10.1182/blood-2009-07-235747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, Azab F, Flores LM, Campigotto F, Weller E, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123:1542–1555. doi: 10.1172/JCI66517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roccaro AM, Sacco A, Thompson B, Leleu X, Azab AK, Azab F, Runnels J, Jia X, Ngo HT, Melhem MR, et al. MicroRNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood. 2009;113:6669–6680. doi: 10.1182/blood-2009-01-198408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- Sacco A, Aujay M, Morgan B, Azab AK, Maiso P, Liu Y, Zhang Y, Azab F, Ngo HT, Issa GC, et al. Carfilzomib-dependent selective inhibition of the chymotrypsin-like activity of the proteasome leads to antitumor activity in Waldenstrom’s Macroglobulinemia. Clin Cancer Res. 2011;17:1753–1764. doi: 10.1158/1078-0432.CCR-10-2130. [DOI] [PubMed] [Google Scholar]
- Scadden DT. The stem-cell niche as an entity of action. Nature. 2006;441:1075–1079. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- Scheiermann C, Kunisaki Y, Frenette PS. Circadian control of the immune system. Nat Rev Immunol. 2013;13:190–198. doi: 10.1038/nri3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipkins DA, Wei X, Wu JW, Runnels JM, Cote D, Means TK, Luster AD, Scadden DT, Lin CP. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435:969–973. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MC, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, Luker GD. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64:8604–8612. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- Tarnowski M, Liu R, Wysoczynski M, Ratajczak J, Kucia M, Ratajczak MZ. CXCR7: a new SDF-1-binding receptor in contrast to normal CD34(+) progenitors is functional and is expressed at higher level in human malignant hematopoietic cells. Eur J Haematol. 2010;85:472–483. doi: 10.1111/j.1600-0609.2010.01531.x. [DOI] [PubMed] [Google Scholar]
- Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vater A. Hematopoietic stem and progenitor cell mobilization in mouse and man by a first-in-class mirror-image oligonucleotide inhibitor of CXCL12. Clinical Pharmacology & Therapeutics. 2013 doi: 10.1038/clpt.2013.58. [DOI] [PubMed] [Google Scholar]
- Wells TN, Peitsch MC. The chemokine information source: identification and characterization of novel chemokines using the WorldWideWeb and expressed sequence tag databases. J Leukoc Biol. 1997;61:545–550. doi: 10.1002/jlb.61.5.545. [DOI] [PubMed] [Google Scholar]
- Wysoczynski M, Reca R, Ratajczak J, Kucia M, Shirvaikar N, Honczarenko M, Mills M, Wanzeck J, Janowska-Wieczorek A, Ratajczak MZ. Incorporation of CXCR4 into membrane lipid rafts primes homing-related responses of hematopoietic stem/progenitor cells to an SDF-1 gradient. Blood. 2005;105:40–48. doi: 10.1182/blood-2004-04-1430. [DOI] [PubMed] [Google Scholar]
- Xu H, Wu Q, Dang S, Jin M, Xu J, Cheng Y, Pan M, Wu Y, Zhang C, Zhang Y. Alteration of CXCR7 expression mediated by TLR4 promotes tumor cell proliferation and migration in human colorectal carcinoma. PLoS One. 2011;6:e27399. doi: 10.1371/journal.pone.0027399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.