

Abstract
Emily Cooley was a highly-regarded medical technologist and morphologist. The “Emily Cooley Lectureship and Award” was established to honor her, in particular, and medical technologists, in general. This article reviews some basic concepts about the “life of a red blood cell” and uses these to discuss the actual and potential consequences that occur in patients following clearance of transfused refrigerator storage-damaged red blood cells by extravascular hemolysis.
Keywords: red blood cells, transfusion, macrophages, cytokines, non-transferrin bound iron
Introduction
It is very meaningful for me to have received the honor of being the Emily Cooley Memorial Lecturer in 2013. I attended my first AABB meeting in 1980 when I was a resident, and that experience was critically important in my decision to pursue a career in academic Transfusion Medicine, combining clinical service with teaching and bench research. As will become clear at the end of this contribution, the AABB, as an institution and as a family of like-minded individuals, has been extremely important in my development; as such, I am eternally grateful to the support I have received through the years and am deeply honored by this award. Dr. Marion Reid was the 2012 Lecturer, and in her talk and accompanying paper,1 she provided an elegant and comprehensive history of Emily Cooley’s life and of the Lectureship; as such, I recommend the reader to refer to her outstanding paper.
The Life of a RBC
Our own research program mainly focuses on “the life of the RBC.” In both physiological and pathophysiological contexts, RBCs circulate, become damaged, and are then cleared from the circulation. Circulating RBCs are produced by normal or abnormal hematopoiesis, or introduced by transfusion. Circulating RBCs can already be damaged due to abnormal endogenous erythropoiesis in vivo (e.g., in patients with thalassemias) or as a result of refrigerated storage ex vivo (in the context of transfusion). Alternatively, RBCs can be targeted for clearance due to normal physiological senescence or from exogenous pathophysiological processes. The latter include immune mediated destruction (e.g. in various types of autoimmune hemolytic anemia, hemolytic transfusion reactions, or hemolytic disease of the fetus and newborn), oxidative damage (e.g., in glucose-6-phosphate dehydrogenase deficiency), hemolytic toxin-induced damage (e.g., after Black Recluse Spider bites2), radiation-induced damage (e.g., radiological exposure or burns), apoptosis (e.g., eryptosis), and infection (e.g., malaria). Once damaged, these RBCs can be cleared by sequestration (e.g. in hypersplenism), fragmentation (e.g., in thrombotic thrombocytopenic purpura), lysis (e.g., in IgM-mediated acute hemolytic transfusion reactions), and/or phagocytosis (e.g., in IgG-mediated delayed hemolytic transfusion reactions). These are summarized in Table 1. RBC clearance may have no downstream adverse effects (e.g., during normal RBC senescence), or can induce inflammation, circulatory disturbances, and/or coagulopathy, produce renal dysfunction, enhance infection, or lead to the death of the patient.
Table 1.
RBC Clearance: Types and Examples
| Clearance Type | Examples |
|---|---|
| Sequestration | |
| Hypersplenism | |
| Sickle Cell Anemia | |
| Cerebral Malaria | |
| Autoagglutination | |
| Fragmentation | |
| Disseminated Intravascular Coagulation | |
| Thrombotic Thrombocytopenic Purpura | |
| Prosthetic heart valves | |
| Pyropoikylocytosis | |
| Intravascular Lysis* | |
| IgM-mediated Acute Hemolytic Transfusion Reaction | |
| “Cold-type” Autoimmune Hemolytic Anemia | |
| Paroxysmal Cold Hemoglobinuria | |
| Paroxysmal Nocturnal Hemoglobinuria | |
| Black Recluse Spider Venom | |
| Phagocytosis† | |
| IgG-mediated Delayed Hemolytic Transfusion Reaction | |
| “Warm-type” Autoimmune Hemolytic Anemia | |
| Oxidative Damage (e.g., Glucose-6-phosphate Dehydrogenase Deficiency) | |
| Hemophagocytic Lymphohistiocytosis | |
| Physiological RBC Senescence |
Also termed “intravascular hemolysis.”
Also termed “extravascular hemolysis.”
Extravascular Hemolysis
Extravascular hemolysis, due to RBC ingestion by cells of the mononuclear phagocyte system (e.g., Kupffer cells in the liver and splenic macrophages), is a particularly important mechanism for clearing both normally senescent RBCs and pathologically-damaged RBCs. Macrophages become aware of the need to ingest nearby targets through “find me” signals and “eat me” signals. Interestingly, there are also “don’t eat me” signals that inhibit macrophage ingestion of potential targets.3,4 ATP is a classical “find me” signal, particularly for necrotic cells that release cytosolic ATP, thereby attracting and activating macrophages to clear cellular debris created during various pathological processes, such as infarction and trauma.5 Whether this phenomenon is important in RBC clearance is not yet known; nonetheless, circulating RBCs contain high cytosolic ATP levels.6 In contrast, a great deal of evidence suggests that various “eat me” signals on RBC surfaces, and their cognate receptors on macrophage membranes, are critically important in RBC clearance (see Figure 1). For example, in the context of IgG-mediated delayed hemolytic transfusion reactions, the RBC “eat me” signals are IgG molecules coating the RBC surface, and the cognate receptors on the macrophage surface are the Fc gamma receptors (FcγRs).7 In addition, depending on the specific macrophage receptor involved, differing signal transduction pathways are induced during receptor-mediated endocytosis, leading to various cellular effects in response to the ingested cargo. Interesting recent work suggests that, in addition to the roles played by multiple receptor-ligand pairs, macrophages, particularly in the spleen, may sense the degree of deformability of the RBCs with which they interact.8,9 Because damaged RBCs are typically less deformable than normal RBCs, macrophages may probe and interrogate the surfaces of passing RBCs, not only for the presence of molecular “eat me” signals, but also for the physical state of the entire cell. Finally, whether or not a RBC is cleared by extravascular hemolysis depends not only on the signals broadcast by the RBC itself, but also on the intrinsic activity of the phagocyte. Thus, under certain homeostatic or pathologic conditions (e.g., viral infection or hemophagocytic lymphohistiocytosis with increased interferon-γ levels), macrophages can become over-activated and even ingest apparently normal RBCs,10,11 whereas in other settings, down-regulated macrophages fail to ingest damaged or opsonized RBCs.12
Figure 1. Macrophage-RBC interactions between macrophage surface receptors and RBC ligands (“eat me” signals).

Cell-surface receptors on macrophages recognize various ligands expressed or newly-exposed on RBC surfaces, which signal macrophages to initiate erythrophagocytosis. All indicated RBC ligands function as “eat me” signals, except for CD47, which is both an “eat me” and a “don’t eat me” signal.3
Abbreviations: βGal: terminal β-Galactose residue; PS: phosphatidylserine; Gas6: growth-arrest 6 protein; MFG-E8: milk-fat globule protein-E8; SIRP-α: signal regulatory protein-α; ASGR: asialo-glycoprotein receptor; CR3: complement receptor 3; FcγR: Fc gamma receptor; PS-R; PS receptor; MER: c-mer proto-oncogene protein; αVβ5: an integrin subtype.
Red text: “don’t eat me” signal; black text: “eat me” signal
The Iron Hypothesis
Although multiple pathways lead to erythrophagocytosis in various settings, they all share one characteristic interaction; that is, the ingestion of RBCs, highly specialized cells in which ~98% of the cellular protein is composed of iron-rich hemoglobin.6 Iron is a critically important, but dangerous, element that readily undergoes oxidation-reduction reactions, converting between Fe+2 and Fe+3 (and even Fe+4). As such, it is an important constituent of the reactive center of multiple enzymes, functions in gas-carrying molecules (e.g., hemoglobin and myoglobin), and is absolutely required for life in virtually all known organisms. Unfortunately, given its highly reactive nature, iron also produces oxidative stress and tissue-damaging free radicals.13,14 Evolution addressed the latter issues by limiting the availability of “free” iron by tightly coupling iron to multiple types of “chaperones” to transport iron to its physiological destination and/or to store excess iron intracellularly; in mammals, these chaperones include transferrin, ferritin, lipocalin-2, and lactoferrin. However, in certain pathological settings, more iron is released than can be rapidly bound by the relevant chaperone; alternatively, more iron can be released than there are finite binding sites on the available chaperone. For the purposes of this review, the relevant chaperone is transferrin, which is the major physiological iron carrier in plasma; when iron production/release exceeds the binding capacity of transferrin, then “free,” non-transferrin bound iron (NTBI) is produced.15 Plasma/serum NTBI is undetectable in normal individuals, but is seen in various acute and chronic iron overload states, where it induces oxidative stress, produces cytotoxicity, and promotes the growth of iron-dependent (i.e., “ferrophilic”) pathogens. Indeed, the latter issue introduces the concept of “nutritional immunity”;16 in this case, there is ongoing competition for iron between the host and invading pathogens. The host sequesters iron from pathogens using, for example, the same chaperones described above (e.g., transferrin and lipocalin-2), thereby preventing pathogen proliferation. Nonetheless, various ferrophilic pathogens developed mechanisms for “stealing” iron from the host (e.g., transferrin receptors that ingest the iron-transferrin complex and microbial siderophores that extract iron from the host).17,18
Based on these concepts, we developed the “Iron Hypothesis,” which we believe is a unifying concept related to rapid extravascular hemolysis resulting from any underlying process (Figure 2; Ref. 19). Thus, under healthy steady-state conditions, the only RBCs cleared from the circulation are those undergoing normal senescence (~1% of all RBCs per day in humans). Under these conditions, clearance continues without any adverse consequences (e.g., inflammation, oxidative stress, or NTBI production). However, if a large bolus of damaged RBCs is rapidly delivered to the mononuclear phagocyte system, the metabolism of these ingested RBCs, along with their associated iron-rich hemoglobin, overwhelms the capacities of intracellular and extracellular iron chaperones (ferritin and transferrin, respectively) leading to increased levels of free intracellular iron (the “labile intracellular pool”) and increased circulating levels of NTBI.
Figure 2. Schematic representation of the “Iron Hypothesis”.
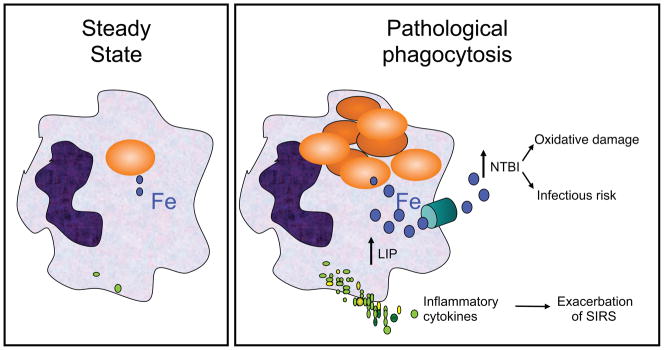
In pathological phagocytosis, metabolism of the hemoglobin in the large amount of ingested RBCs acutely increases the intracellular “free” iron levels (blue filled circles) in the labile intracellular pool (LIP), which, through a signal transduction pathway, enhances the production and secretion of pro-inflammatory cytokines (green filled circles); the latter can enhance the severity of the systemic inflammatory response syndrome (SIRS). In addition, excess “free” iron is exported from the cell through ferroportin (green cylinder), the physiological iron export channel. If the amount of exported iron exceeds the binding capacity of transferrin, then NTBI is produced, which can induce oxidative damage and enhance pathogen proliferation.
Is old blood bad? Mouse and human studies
The United States Food and Drug Administration (FDA) standards allow RBC units to be stored for up to 42 days before transfusion, which is deemed to be safe. Nonetheless, there is general agreement that one adverse consequence of this approach is that, on average, ~25% of these transfused RBCs are cleared from the circulation of healthy volunteers by 1–2 hours post-transfusion.20 In addition, there is general agreement that an even greater percentage of these transfused, refrigerator-stored RBCs are cleared from the circulation of ill patients (perhaps due to macrophage activation).21 There is also general agreement that this clearance results from RBC damage during refrigerated storage (i.e., the “RBC storage lesion”), which is, at least partly, due to oxidative stress.22 Finally, there is general agreement that some donors reproducibly appear to be “poor storers,” whereas some are “super storers,” although the underlying explanation of this phenomenon is not yet known.23,24
In contrast, a great deal of controversy exists concerning whether additional, more severe, adverse consequences occur following transfusions of older, stored RBCs. Thus, although there have been many, primarily observational, studies suggesting that older, stored RBC transfusions in hospitalized patients lead to increases in sepsis, pneumonia, multi-organ failure, myocardial infarction, thrombotic complications, renal dysfunction, and death, these studies have significant methodological problems. Therefore, this subject generates heated debate and is being intensely investigated by multiple groups around the world. Despite the absence of conclusive scientific evidence, particularly from well-designed prospective randomized trials, our group has taken the, admittedly unscientific, approach of “where there is smoke there is fire,” by studying the underlying mechanism(s) that could produce these adverse effects. Our decision is supported by a recent meta-analysis of 21 human studies involving 409,966 predominantly cardiac and surgical patients.25 The authors of this meta-analysis concluded that “based on available data, use of older stored blood is associated with a significantly increased risk of death” (odds ratio of 1.16; CI 1.07–1.24) and, that to save one life, 97 patients would need to be transfused with only “fresher” units (CI 63–199 patients).
Our group uses cell culture models, animal models, healthy human volunteers, and hospitalized patients to focus on the consequences induced by clearance of the storage-damaged RBCs themselves. In addition, other groups are focusing on the consequences of infusing components of the supernatant, including dissolved molecules (e.g., hemoglobin, eicosanoids, etc.)26,27 and cellular fragments (e.g., microparticles).26,28 Using the Iron Hypothesis as a guide, our approach predicts that transfusing one RBC unit at outdate into an adult human recipient would acutely deliver an iron load to the mononuclear phagocyte system that is ~60 times greater than it experiences during normal homeostatic conditions (i.e. ~60 mg of iron in the first hour post-transfusion due to clearance of 25% of the transfused RBCs present in one unit of 42-day old RBCs, as compared with ~1 mg of iron per hour due to normal clearance of senescent endogenous RBCs).
For example, in a mouse model of leukoreduced RBC storage, the murine equivalent of the FDA outdate standard is 14 days; that is, after 14 days of refrigerated storage in CPDA-1, the 24-hour post-transfusion recovery of murine RBCs in mouse recipients averages ~75%.29 The refrigerator storage-damaged transfused RBCs are cleared by extravascular hemolysis, mostly within 1–2 hours post-transfusion and predominantly by splenic macrophages and hepatic Kupffer cells; indeed, when these phagocytes are transiently depleted by liposomal clodronate, post-transfusion recovery approaches 100%.30 In this mouse model, transfusion of even one “mouse equivalent unit” of leukoreduced packed RBCs induces an acute inflammatory response with elevated levels of multiple pro-inflammatory cytokines (e.g., tumor necrosis factor (TNF)-alpha, monocyte chemoattractant protein (MCP)-1, interleukin (IL)-6, and IL-8).30 A dose-response relationship is also seen; that is, transfusions of larger volumes of RBCs lead to higher levels of circulating cytokines. In addition, only clearance of intact storage-damaged RBCs themselves induces this inflammatory response; no cytokine response is seen when supernatant, total hemolysate, or RBC “ghosts” are infused. In contrast, transfusions of washed 14-day old RBCs also induce a robust inflammatory response. Finally, the splenocytes responsible for cytokine synthesis and secretion in response to this erythrophagocytosis are tissue-resident macrophages (i.e., CD45+, CD11b+, F4/80+, Ly6C+, and CD19−, CD3−, Ly6G−).31,32 Taken together, these results are consistent with the Iron Hypothesis in that delivery of an acute bolus of a large amount of iron is required to increase the size of the “labile intracellular pool,” thereby initiating the intracellular signal transduction pathways required for cytokine synthesis and secretion.33
In addition to inducing inflammation, transfusions of older, stored RBCs produce increasing levels of circulating NTBI in this mouse model. Again, transfusions of washed RBCs, but not infusions of supernatant or RBC ghosts, lead to increased NTBI levels, suggesting that the major effects are due to metabolism in vivo of storage-damaged RBCs and not due to infusion of NTBI produced in the storage bag ex vivo.30,34,35 In addition, this NTBI enhances the growth in vitro of a human pathogen (i.e., Escherichia coli) in serum obtained from mice transfused with older, but not fresh, RBCs. Similarly, transfusions of older, stored RBCs enhance the pathogenicity in mice in vivo of both extracellular and intracellular pathogens (i.e., E. coli and Salmonella typhimurim, respectively).36 In summary, these results suggest that this mouse RBC storage model usefully mimics the human situation with regard to post-transfusion recovery, produces increased circulating “free” iron levels, and leads to two potentially-relevant adverse effects: inflammation and enhanced bacterial pathogenicity. Nonetheless, “mice are not human” and it is important to evaluate these findings, along with their potential mechanisms, in humans.37
To begin to study these issues, we performed a prospective study in 14 healthy human volunteers, each of whom consented to a two-unit RBC donation by apheresis, which was leukoreduced, divided in half, and stored in AS-1.38 These volunteers then received one autologous transfusion of an entire unit after 3–7 days of storage and the second autologous transfusion of an entire unit after 40–42 days of storage. The volunteers were carefully monitored for clinical signs and symptoms, and multiple samples were obtained pre- and post-transfusion for additional analysis. In brief, laboratory testing of these blood samples was consistent with rapid clearance of older, but not fresh, RBCs by extravascular hemolysis (i.e., statistically-significant acutely increased levels of serum bilirubin, with no detectable free hemoglobin or changes in circulating haptoglobin, lactate dehydrogenase, or potassium levels). In addition, older, stored, but not fresh, RBC transfusions led to statistically-significant acute elevations in circulating levels of various iron parameters (e.g., NTBI, serum iron, transferrin saturation, ferritin). Finally, serum samples obtained after transfusion of older, but not fresh, RBCs (i.e., with elevated NTBI levels) enhanced E. coli proliferation in vitro. Nonetheless, despite these direct similarities with the mouse model described above, there was no evidence of a pro-inflammatory response in healthy human volunteers receiving older, stored RBC transfusions, despite testing for multiple cytokines and related biomarkers.
The reasons underlying the differences between mice and humans with regard to inflammation following older, stored RBC transfusions remain unknown. For example, the murine inflammatory response may simply be more sensitive to this insult, although similar results were also seen in a canine model.39 Alternatively, the dose of refrigerator storage-damaged RBCs may be important; that is, mice exhibited a much more robust response to two, in contrast to one, “unit” transfusions;30 the human volunteers only received one unit in each instance.38 The sub-type of the phagocyte responsible for clearing the refrigerator-storage damaged RBCs may also be important. Thus, in the mouse model, the tissue resident splenic macrophages ingesting the storage-damaged RBCs also secrete the pro-inflammatory cytokines.32 However, the cell type responsible for this RBC clearance in humans is not yet known and other phagocyte types (e.g., monocyte-derived dendritic cells) actually produce an anti-inflammatory cytokine response following erythrophagocytosis.40 Finally, it is possible that robust transfusion-induced inflammation would be seen in humans with specific underlying conditions. For example, mice receiving sub-clinical doses of endotoxin exhibited much more robust inflammatory responses to older, stored RBC transfusions.30 In addition, older, stored RBC transfusions induced increased circulating levels of NTBI and cytokines (e.g., IL-8, TNF-alpha, and MCP-1) in premature infants;41 however, there was no correlation between NTBI levels and cytokine levels.42
Conclusions and future directions
In conclusion, results from the murine model conclusively show that older, stored RBCs and fresh RBCs yield markedly different (patho)physiological responses in transfusion recipients. In addition, some of the adverse consequences seen in mice following transfusions of older, stored RBCs are reminiscent of those described in observational studies of human patients (e.g., enhanced infection and sepsis). Future studies in mice will not only further investigate infectious complications and the inflammatory response, but will also attempt to model several types of thrombotic complications. In addition, based on the Iron Hypothesis, several therapeutic interventions will be studied to ameliorate these adverse effects. In addition, our studies with healthy human volunteers conclusively show that there is a clear physiological response, particularly regarding iron metabolism, to older, stored, but not fresh, RBC transfusions. To identify potentially “susceptible” human populations, particularly with respect to inflammation, studies are underway evaluating the effects of older, stored RBC transfusions in pediatric and neonatal intensive care unit patients, and in patients with sickle cell disease and β-thalassemia. Finally, to address the effects of oxidative stress on the RBC storage lesion, current studies in mice and humans are aimed at understanding whether individuals with RBCs less able to handle such stress (e.g., in glucose-6-phosphate dehyrdrogenase deficiency)43,44 are “poor storers.”
Teaching and mentoring
I am grateful that the Emily Cooley Award recognizes teaching ability and I am honored to have been able to teach and mentor so many outstanding students, residents, and fellows over the past 35 years. However, rather than discuss the individuals I have taught, I would prefer to provide a few thoughts about my mentors. I was initially attracted to specialize in Transfusion Medicine by two individuals: Ms. Mary Theresa (“Terry”) Cox and Professor Patrick L. Mollison. Terry Cox’s influence was “up close and personal.” Terry was the “teaching tech” in the Blood Bank at the University of Rochester, whose passion for all things immunohematological was infectious; her gentle prodding and enthusiastic encouragement led to my first abstract platform presentation and my first publication. In contrast, I cannot claim to have known Professor Mollison well personally, but I knew the Sixth Edition of his “Blood Transfusion in Clinical Medicine” extremely well, having read it cover-to-cover during my resident rotation. His lucid style, comprehensive knowledge, and elegant synthesis of the field ignited my interest. Fortunately, Dr. Neil Blumberg soon arrived on the scene in Rochester; his enthusiastic support and encouragement led to a fruitful collaborative relationship during the remainder of my residency training. The selfless mentorship of Dr. Henry (“Hank”) Martinique Sobell during a year-out post-doctoral research fellowship in Rochester was critically important in my decision to focus on a career in bench research. My year with Hank was an experiment in whether basic biomedical research was of interest to me and my project was a departure from the central theme of his experimental studies. Henry confidently gave me free reign in his laboratory, even though I had no idea what I was doing. Although these studies led to no publications (I disproved his hypothesis!), they provided me with a great deal of confidence that I could design appropriately controlled experiments and obtain robust and relevant data. This convinced me to pursue additional post-doctoral research training at the National Institutes of Health with Dr. Victor Ginsburg (1930–2003), one of the founding fathers of glycobiology.45 Vic was my most important research mentor, and a great friend, who taught me how ask appropriate scientific questions, how to evaluate data critically, how to function as a study section member, and, perhaps most importantly, how to write lucidly. Although no longer with us, his voice still reverberates in my head with advice and criticism when I write scientific papers (including this one right now!). In addition to the scientific mentoring described above, I benefited greatly from mentoring in the administrative, social, and political aspects of academic medicine by two outstanding, long-tenured pathology chairman: Drs. Leonard Jarett at the University of Pennsylvania (1985–1998) and Michael L. Shelanski at Columbia University (2003-present). They both provided incredible support and encouragement during this voyage. Over the years, I also came to realize that peer mentoring is extremely important. My most important peer mentor has been Dr. Leslie E. Silberstein, particularly when we were both Assistant Professors at the University of Pennsylvania. We met frequently and often strategized, regarding how to survive and thrive in an often difficult and hostile environment. Two phrases particularly remain with me from that time. “It’s a process…” is still useful when dealing with almost any difficult situation. “We’ll outlast and outlive them!” was often useful when dealing with particularly difficult senior faculty members; however, at this stage in my life, this phrase is more likely to be used by others about me than by me about others! Finally, although they are probably unaware of their role, there are ~50 AABB members in my age cohort (i.e., +/− 10 years of my age) who have been important and significant peer mentors and friends. In many ways, I have grown up in this organization and these individuals have been valuable advisors, supporters, and constructive critics throughout the years.
Acknowledgments
Some of the research described was supported in part by grants from the National Institutes of Health to S.L.S. (R01 HL098014 and R01 HD115557) and Dr. Eldad Hod (K08 HL103756), and from the Robert Wood Johnson Foundation to Dr. Richard O. Francis (Harold Amos Medical Faculty Development Program).
I would like to dedicate this paper to the memory of Dr. George Garratty, who began our relationship as an important mentor, critic, and colleague, and ended as a wonderful friend. I would also like to acknowledge the continuing support and friendship of the faculty members comprising the Laboratory of Transfusion Biology at Columbia University (Drs. Gary Brittenham, Eldad Hod, Richard Francis, Boguslaw Wojczyk, and Kevin Prestia) and of my key collaborator and friend, Dr. James Zimring at the Puget Sound Blood Center and University of Washington. Finally, I would like to acknowledge the support, patience, and understanding of my wife and son (Dr. Patrice Spitalnik and Joel Spitalnik) as they patiently indulged my obsession with research through the years.
Footnotes
Disclosure: The author declares no conflicts of interest relevant to the manuscript submitted to Transfusion.
Authorship contributions
S.L.S wrote the paper.
References
- 1.Reid ME. Emily Cooley Lecture 2012: Emily Cooley and techniques that have been applied to characterize DO and JR blood groups. Transfusion. 2013;53:1876–83. doi: 10.1111/trf.12207. [DOI] [PubMed] [Google Scholar]
- 2.Williams ST, Khare VK, Johnston GA, et al. Severe intravascular hemolysis associated with brown recluse spider envenomation. A report of two cases and review of the literature. American Journal of Clinical Pathology. 1995;104:463–67. doi: 10.1093/ajcp/104.4.463. [DOI] [PubMed] [Google Scholar]
- 3.Burger P, Hilarius-Stokman P, de Korte D, et al. CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood. 2012;119:5512–21. doi: 10.1182/blood-2011-10-386805. [DOI] [PubMed] [Google Scholar]
- 4.Oldenborg P-A, Zheleznyak A, Fang Y-F, et al. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–54. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 5.Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798–804. doi: 10.1016/j.cell.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 6.Gevi F, D’Alessandro A, Rinalducci S, et al. Alterations of red blood cell metabolome during cold liquid storage of erythrocyte concentrates in CPD–SAGM. Journal of Proteomics. 2012;76:168–80. doi: 10.1016/j.jprot.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 7.Ravetch JV, Boland S. IgG Fc receptors. Annual Review of Immunology. 2001;19:275–90. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 8.Discher D, Janmey P, Wang Y-l. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–43. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 9.Safeukui I, Buffet PA, Deplaine G, et al. Quantitative assessment of sensing and sequestration of spherocytic erythrocytes by the human spleen. Blood. 2012;120:424–30. doi: 10.1182/blood-2012-01-404103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jordan MB, Hildeman D, Kappler J, et al. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–43. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 11.Rosado FGN, Kim AS. Hemophagocytic Lymphohistiocytosis: An update on diagnosis and pathogenesis. American Journal of Clinical Pathology. 2013;139:713–27. doi: 10.1309/AJCP4ZDKJ4ICOUAT. [DOI] [PubMed] [Google Scholar]
- 12.Hod EA, Arinsburg SA, Francis RO, et al. Use of mouse models to study the mechanisms and consequences of RBC clearance. Vox Sanguinis. 2010;99:99–111. doi: 10.1111/j.1423-0410.2010.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aguirre JD, Culotta VC. Battles with iron: Manganese in oxidative stress protection. Journal of Biological Chemistry. 2012;287:13541–48. doi: 10.1074/jbc.R111.312181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arosio P, Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochimica et Biophysica Acta. 2010;1800:783–92. doi: 10.1016/j.bbagen.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Breuer W, Hershko C, Cabantchik ZI. The importance of non-transferrin bound iron in disorders of iron metabolism. Transfusion Science. 2000;23:185–92. doi: 10.1016/s0955-3886(00)00087-4. [DOI] [PubMed] [Google Scholar]
- 16.Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen–host interface. Nature Reviews Microbiology. 2012;10:525–37. doi: 10.1038/nrmicro2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cornelis P, Wei Q, Andrews SC, et al. Iron homeostasis and management of oxidative stress response in bacteria. Metallomics. 2011;3:540–49. doi: 10.1039/c1mt00022e. [DOI] [PubMed] [Google Scholar]
- 18.Johnson L. Iron and siderophores in fungal–host interactions. Mycological Research. 2008;112:170–83. doi: 10.1016/j.mycres.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 19.Hod EA, Spitalnik SL. Stored red blood cell transfusions: Iron, inflammation, immunity, and infection. Transfusion clinique et biologique. 2012;19:84–89. doi: 10.1016/j.tracli.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dumont LJ, AuBuchon JP BEST Collaborative. Evaluation of proposed FDA criteria for the evaluation of radiolabeled red cell recovery trials. Transfusion. 2008;48:1053–60. doi: 10.1111/j.1537-2995.2008.01642.x. [DOI] [PubMed] [Google Scholar]
- 21.Luten M, Roerdinkholder-Stoelwinder B, Schaap NPM, et al. Survival of red blood cells after transfusion: a comparison between red cells concentrates of different storage periods. Transfusion. 2008;48:1478–85. doi: 10.1111/j.1537-2995.2008.01734.x. [DOI] [PubMed] [Google Scholar]
- 22.Dumaswala UJ, Zhuo L, Jacobsen DW, et al. Protein and lipid oxidation of banked human erythrocytes: Role of glutathione. Free Radical Biology & Medicine. 1999;27:1041–49. doi: 10.1016/s0891-5849(99)00149-5. [DOI] [PubMed] [Google Scholar]
- 23.Dern RJ, Gwinn RP, Wiorkowski JJ. Studies on the preservation of human blood. I. Variability in erythrocyte storage characteristics among healthy donors. Journal of Laboratory and Clinical Medicine. 1966;67:955–65. [PubMed] [Google Scholar]
- 24.Zimring JC, Smith N, Stowell SR, et al. Strain-specific red blood cell storage, metabolism, and eicosanoid generation in a mouse model. Transfusion. 2013;54:137–48. doi: 10.1111/trf.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang D, Sun J, Solomon SB, et al. Transfusion of older stored blood and risk of death: a meta-analysis. Outcomes using old vs new stored blood. Transfusion. 2011;52:1184–95. doi: 10.1111/j.1537-2995.2011.03466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donadee C, Raat NJH, Kanias T, et al. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation. 2011;124:465–76. doi: 10.1161/CIRCULATIONAHA.110.008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spinelli SL, Lannan KL, Casey AE, et al. Isoprostane and isofuran lipid mediators accumulate in stored red blood cells and influence platelet function in vitro. Transfusion. 2013;54:1569–79. doi: 10.1111/trf.12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Danesh A, Inglis HC, Jackman RP, et al. Exosomes from red blood cell units bind to monocytes and induce proinflammatory cytokines, boosting T-cell responses in vitro. Blood. 2014;123:687–96. doi: 10.1182/blood-2013-10-530469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilson CR, Kraus TS, Hod EA, et al. A novel mouse model of red blood cell storage and posttransfusion in vivo survival. Transfusion. 2009;49:1546–53. doi: 10.1111/j.1537-2995.2009.02173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hod EA, Zhang N, Sokol SA, et al. Transfusion of red blood cells after prolonged storage produces harmful effects that are mediated by iron and inflammation. Blood. 2010;115:4284–92. doi: 10.1182/blood-2009-10-245001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davies LC, Jenkins SJ, Allen JE, et al. Tissue-resident macrophages. Nature Immunology. 2013;14:986–95. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wojczyk BS, Kim N, Bandyopadhyay S, et al. Macrophages clear refrigerator storage-damaged red blood cells and subsequently secrete cytokines in vivo, but not in vitro, in a murine model. Transfusion. 2014 doi: 10.1111/trf.12755. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.She H, Xiong S, Lin M, et al. Iron activates NF-kB in Kupffer cells. American Journal of Physiology. 2002;283:G719–G26. doi: 10.1152/ajpgi.00108.2002. [DOI] [PubMed] [Google Scholar]
- 34.Collard KJ, White DL. On the source of the non-transferrin-bound iron which accumulates in packed red blood cell units during storage. Blood Transfusion. 2014 doi: 10.2450/2014.0271-13. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marwah SS, Blann A, Harrison P, et al. Increased non-transferrin bound iron in plasma-depleted SAG-M red blood cell units. Vox Sanguinis. 2002;82:122–26. doi: 10.1046/j.1423-0410.2002.00153.x. [DOI] [PubMed] [Google Scholar]
- 36.Prestia K, Bandyopadhyay S, Slate A, et al. Transfusion of stored blood impairs host defenses against Gram-negative pathogens in mice. Transfusion. 2014 doi: 10.1111/trf.12712. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimring JC, Spitalnik SL. On the appropriate use and interpretation of animal models in transfusion medicine research. Transfusion. 2013;53:2334–39. doi: 10.1111/trf.12131. [DOI] [PubMed] [Google Scholar]
- 38.Hod EA, Brittenham GM, Billote GB, et al. Transfusion of human volunteers with older, stored red blood cells produces extravascular hemolysis and circulating non-transferrin-bound iron. Blood. 2011;118:6675–82. doi: 10.1182/blood-2011-08-371849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Callan MB, Patel RT, Rux AH, et al. Transfusion of 28-day-old leucoreduced or non-leucoreduced stored red blood cells induces an inflammatory response in healthy dogs. Vox Sanguinis. 2013;105:319–27. doi: 10.1111/vox.12058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohyagi H, Onai N, Sato T, et al. Monocyte-derived dendritic cells perform hemophagocytosis to fine-tune excessive immune responses. Immunity. 2013;39:584–98. doi: 10.1016/j.immuni.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 41.Keir AK, McPhee AJ, Andersen CC, et al. Plasma cytokines and markers of endothelial activation increase after packed red blood cell transfusion in the preterm infant. Pediatric Research. 2013;73:75–79. doi: 10.1038/pr.2012.144. [DOI] [PubMed] [Google Scholar]
- 42.Stark MJ, Keir AK, Andersen CC. Does non-transferrin bound iron contribute to transfusion related immune-modulation in preterms? Archives of Disease in Childhood - Fetal and Neonatal Edition. 2013;98:F424–F29. doi: 10.1136/archdischild-2012-303353. [DOI] [PubMed] [Google Scholar]
- 43.Francis RO, Jhang J, Hendrickson JE, et al. Frequency of glucose-6-phosphate dehydrogenase-deficient red blood cell units in a metropolitan transfusion service. Transfusion. 2012;53:606–11. doi: 10.1111/j.1537-2995.2012.03765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Francis RO, Jhang JS, Pham HP, et al. Glucose-6-phosphate dehydrogenase deficiency in transfusion medicine: The unknown risks. Vox Sanguinis. 2013;105:271–82. doi: 10.1111/vox.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kobata A, Zopf D, Magnani J. Victor Ginsburg (1930–2003): An unforgettable lab chief and mentor and a founder of glycobiology. Archives of Biochemistry and Biophysics. 2004;426:103–04. doi: 10.1016/j.abb.2004.04.005. [DOI] [PubMed] [Google Scholar]