

Abstract
Bile salt export pump (BSEP) is responsible for biliary secretion of bile acids, a rate limiting step in the enterohepatic circulation of bile acids and transactivated by nuclear receptor farnesoid x receptor (FXR). Intrahepatic cholestasis of pregnancy (ICP) is the most prevalent disorder among diseases unique to pregnancy and primarily occurs in the third trimester of pregnancy with a hallmark of elevated serum bile acids. Currently, the transcriptional regulation of BSEP during pregnancy and its underlying mechanisms and involvement in ICP are not fully understood. In this study, the dynamics of BSEP transcription in vivo in the same group of pregnant mice before, during and after gestation were established with in vivo imaging system (IVIS). BSEP transcription was markedly repressed in the later stages of pregnancy and immediately recovered after parturition, resembling the clinical course of ICP in human. The transcriptional dynamics of BSEP was inversely correlated with serum 17β-estradiol (E2) levels before, during and after gestation. Further studies showed that E2 repressed BSEP expression in human primary hepatocytes, Huh 7 cells and in vivo in mice. Such transrepression of BSEP by E2 in vitro and in vivo required estrogen receptor α (ERα). Mechanistic studies with chromatin immunoprecipitation (ChIP), protein co-immunoprecipitation (Co-IP) and bimolecular fluorescence complementation (BiFC) assays demonstrated that ERα directly interacted with FXR in living cells and in vivo in mice. In conclusion, BSEP expression was repressed by E2 in the late stages of pregnancy through a non-classical E2/ERα transrepressive pathway, directly interacting with FXR. E2-mediated repression of BSEP expression represents an etiological contributing factor to ICP and therapies targeting the ERα/FXR interaction may be developed for prevention and treatment of ICP.
Keywords: BSEP, Bile acids, 17β-estradiol, FXR, ERα
INTRODUCTION
Pregnancy causes profound maternal changes in physiology and metabolism (1–4). One such change is the serum lipid profile with elevated bile acids, cholesterol and triglycerides, especially in the third trimester of gestation (1–3, 5–7). Most of those changes are either directly or secondarily resulted from elevated pregnancy-related hormones, mainly estrogens, such as 17β-estradiol (E2), and progesterone. In a subgroup of pregnant women, when their bodies cannot adequately adapt to elevated reproductive hormones, pregnancy-related diseases occur (8, 9).
Intrahepatic cholestasis of pregnancy (ICP) is the most prevalent disorder among diseases unique to pregnancy (8–10). The incidence of ICP varies widely among ethnic groups ranging from less than 1% to 14% (11, 12). ICP occurs predominantly during the third trimester of pregnancy and spontaneously disappears after labor (10). Although ICP has a mild impact on the mother, it poses significant risks to the fetus for complications, such as preterm delivery, respiratory stress and prenatal mortality (8–10, 13). ICP shares the characteristic manifestations of intrahepatic cholestasis with a hallmark of markedly elevated levels of serum bile acids, which has become the most accurate diagnosis for ICP (6, 7, 14).
Bile acid homeostasis is achieved through a tightly regulated enterohepatic circulation and canalicular secretion through bile salt export pump (BSEP, ABCB11) is the rate-limiting step (15, 16). BSEP expression is coordinately regulated by distinct but related transactivation pathways, notably the bile acid/farnesoid x receptor (FXR, NR1H4) signaling pathway (17–21). Activation of FXR by bile acids strongly induces BSEP expression in vitro and in vivo (17, 18). Such feed-forward regulation of BSEP by bile acid/FXR is considered as a major mechanism to prevent excessive accumulation of toxic bile acids in hepatocytes.
It has been recognized that the etiology of ICP is complex with genetic and endocrine contributing factors (10). Indeed, genetic variants of BSEP and FXR have been associated with an increased risk for ICP (22–24). On the other hand, steroid hormones and their metabolites have been implicated in the development of ICP (25–29). Currently, the transcriptional regulation of BSEP during pregnancy and its underlying mechanisms and involvement in ICP are not fully understood.
In this study, the transcriptional dynamics of BSEP in vivo in the same group of pregnant mice before, during and after gestation were established, resembling the clinical course of ICP in human. Further studies showed that BSEP transcription was inversely correlated with serum E2 levels during pregnancy and E2 repressed BSEP expression in vitro and in vivo through a non-classical E2/ERα transrepressive pathway, directly interacting with FXR.
MATERIALS AND METHODS
Plasmid constructs
Human and mouse BSEP promoter reporters phBSEP(−2.6kb) and pmBSEP(−2.6kb) were described elsewhere (20, 30). Human FXR, Flag-FXR, ERα and ERβ were provided by Drs. David Mangelsdorf and Matthew Stoner. Construct eGFPn-FXR was made by fusing the N-terminal 172 residues of enhanced green fluorescence protein (eGFP) to human FXR while ERα-eGFPc was generated by fusing the human ERα to the C-terminal fragment of eGFP with a linker (RSIATGS) in between. Promoter reporters phBSEP(−805b), phBSEP(−405b), phBSEP(−205b), phBSEP(−160b) and phBSEP(−120b) were described previously (19). The estrogen response element (ERE) reporter pTK-2xERE was made by cloning two copies of the ERE consensus sequences into the pTK-Luc vector. FXR response element (FXRE) reporters pGL-2xFXREcon and pGL-2xhIR1 were constructed by cloning two copies of the FXRE consensus (5’-AGGTCA T TGACCT-3’) or IR1a (inverted repeat spaced by one nucleotide, 5’-GGGACA T TGATCC-3’) in human BSEP promoter into the pGL3/promoter vector.
Treatments of human primary hepatocytes and Huh 7 cells
Human primary hepatocytes obtained through Liver Tissues Procurement and Distribution System and Huh 7 cells were treated with chenodeoxycholic acid (CDCA) (5 or 10µM) or a combination of CDCA and various concentration of E2 (0, 1, 10 or 100nM) for 30h in a phenol red-free DMEM medium containing 1% charcoal-stripped FBS.
Reporter luciferase assays
Transient transfection and dual luciferase assays were carried out as described elsewhere (31).
Quantitative real-time PCR
Total RNA isolation from human primary hepatocytes, Huh 7 cells or liver tissues and subsequent TaqMan real-time PCR assays were performed as described previously (20, 30).
Living imaging with in vivo imaging system (IVIS)
Before mating, thirty female CD-1 mice were hydrodynamically injected with mouse BSEP promoter reporter pmBSEP(−2.6kb) via tail-vein (0.5µg/g). Hepatic luciferase expressions were monitored by IVIS (30) before, during and after the gestation in both pregnant and non-pregnant mice. In the study with E2 treatment, twenty female CD-1 mice were randomly divided into E2 (5mg/kg daily for 5 days subcutaneously) and vehicle ethanol (EtOH) group. All mice were hydrodynamically injected with pmBSEP(−2.6kb) plasmid prior to E2 treatment. Luciferase levels were detected by IVIS before and 7 days post-treatment. All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Rhode Island.
ERα(−/−) mice and treatments
ERα(−/−) mice were obtained through in-house breeding with ERα(−/+) heterozygous male and female mice (Jackson Laboratories). Eleven ERα(−/−) mice were randomly divided into E2 treatment (5mg/kg daily for 5 days subcutaneously) and control EtOH group. ERα wt mice from the same colony were included as controls. Liver tissues were harvested and processed for BSEP mRNA and protein detection.
Bimolecular fluorescence complementation (BiFC) assays
Huh 7 cells were cotransfected with 1µg eGFPn-FXR and 1µg ERα-eGFPc, ERα-LBD-Mut-eGFPc or pcDNA5. Twenty four hours post-transfection, the cells were treated with 100nM E2 for 24h, followed by detection of fluorescence under a confocal microscope. Eighteen CD-1 female mice were randomly divided into three groups. Each group of mice were hydrodynamically co-injected 5µg eGFPn-FXR with 5µg ERα-eGFPc, ERα-LBD-Mut-eGFPc or pcDNA5 via tail vein. Twenty four hours post-injection, mouse primary hepatocytes were collected through liver perfusion. Hepatocytes were either cultured on slide chambers for florescence detection or subjected to FACS analysis.
Western blotting
Cell lysates were prepared from Huh 7 cells or mouse liver tissues for detecting BSEP, ERα and small heterodimer partner (SHP) by regular or capillary electrophoresis-based Western as described previously (30).
Serum E2 detection
Serum samples were collected from mice through tail vein before, during and after gestation. Serum E2 levels were detected with an E2 ELISA kit from Calbiotech.
Site-directed mutagenesis
Mutagenesis was performed using QuickChange mutagenesis kit (Stratagene). ERα DNA binding domain mutant (ERα-DBD-Mut) was made by substituting two amino acids at E207A and G208A in ERα wt. The ERα ligand binding domain mutants (ERα-LBD-Mut) and ERα-LBD-Mut-eGFPc were generated by replacing the glycine at 521 with arginine in ERα wt or ERα-eGFPc.
Co-immunoprecipitation (Co-IP) assays
HEK 293T or Huh 7 cells were transfected with 2µg Flag-FXR or FXR and ERα, followed by treatment with 100nM E2 and 10μM CDCA for 24h and subsequently lysed. Liver tissues from pregnant mice at gestation day 18 were homogenized and lysed. Cell lysates (500 µg total protein) underwent Co-IP with the Dynabeads Antibody Coupling kit (Life Technologies) using ERα (sc-542, Santa Cruz Biotechnology), custom made FXR antibodies or rabbit IgG, followed by Western blotting with anti-Flag (Sigma-Aldrich), FXR or ERα antibodies.
Chromatin immunoprecipitation (ChIP) assays
Chromatins were prepared from Huh 7 cells transfected with 2μg ERα and FXR plasmids, followed by treatment of transfected cells with 10μM CDCA and 100nM E2 or 1% EtOH for 1 or 24h. ChIP assays were performed with the ChIP-IT Express Kit (Active Motif) following the procedure described elsewhere (20, 32).
Electrophoretic mobility shift assays (EMSA)
Nuclear extracts of Huh 7 cells transfected with FXR and ERα were prepared and EMSAs were performed using the LightShift Chemiluminescent EMSA kit (Pierce) as described previously (32)
Statistical analysis
Student’s t-test was applied to pair-wise comparison for normally distributed data. One-way ANOVA was applied to analyze data with multiple groups, followed by Tukey post-hoc test for multiple comparisons. Non-parametric Mann-Whitney test was used for pair-wise comparison for non-normally distributed data. Pearson correlation test was used to determine the correlation coefficient. The P value of 0.05 or lower was considered statistically significant.
RESULTS
BSEP transcription was repressed in the late stages of pregnancy
Serum bile acids are mildly elevated in normal pregnancy and markedly increased in ICP, especially in the third trimester of pregnancy (6, 7, 14). BSEP is responsible for biliary secretion of bile acids, a rate-limiting step in the enterohepatic circulation of bile acids (15, 16). To determine whether BSEP expression is transcriptionally altered during pregnancy, especially at the late stages of pregnancy, the dynamics of BSEP transcription in the same group of mice hydrodynamically injected with BSEP promoter reporter was monitored by IVIS before, during and after gestation. As shown in Fig. 1A and 1B, significant decreases in BSEP transactivation were detected on days 2 and 1 prior to parturition and on the day of parturition with a p value of 0.005, <0.001 and 0.003, respectively. The lowest levels were detected on the day of parturition and only 3.4 % BSEP transactivation levels remained with one mouse below the detection level. One day after parturition, BSEP transactivation was immediately recovered to approximately 50% of the premating levels. By day 6 and 11 post-parturition, the BSEP transactivation levels were completely recovered. The data for individual mice were presented in Supplement Table 1. No significant changes were detected in non-pregnant mice during the same monitoring period (Fig. 1C and 1D). Taken together, for the first time, the transcriptional dynamics of BSEP was established in the same group of mice during the entire pregnancy. Such dynamics resemble the clinical course of ICP in human, which occurs in the late stages of pregnancy and spontaneously disappears within a week after delivery (10).
Fig. 1. The dynamics of BSEP transactivation before, during and after gestation in the same group of pregnant mice.


(A) mouse BSEP promoter reporter pmBSEP(-2.6kb) were hydrodynamically injected (0.5µg/g) before mating. Hepatic luciferase expression levels as luminescent signals were monitored by IVIS before, during and after gestation in the same group of pregnant mice (n=15). (B) quantification of the luciferase activities as luminescent signals (photons) at various time points. (C) hepatic luciferase expression levels were monitored in a group of non-pregnant mice (n=10) in parallel with the pregnant mice showed in (A). (D) quantification of the luciferase activities in non-pregnant mice. Median value of each group was indicated by a short line. Mann-Whitney’s nonparametric test was applied for pair-wise comparison. * p<0.05 and ** p<0.01.
Serum E2 levels were increased in the late stages of pregnancy and inversely correlated with BSEP transactivation
Among estrogens, E2 is the predominant hormone during pregnancy. As shown in Fig. 2A, serum E2 levels steadily increased throughout the pregnancy and peaked right before delivery. Significant increases in E2 levels were detected on days 3, 2, 1 prior to parturition and on the day of delivery with a p value of 0.023, 0.005, 0.001 and 0.015, respectively. The E2 levels were then quickly dropped to the premating levels on days 1, 2 and 7 post-delivery. The data for individual mice were presented in Supplement Table 2.
Fig. 2. Serum E2 levels were increased in the late stages of pregnancy and inversely correlated with BSEP transactivation.



(A) serum samples were collected before, during and after gestation and E2 levels were determined in the same group of pregnant (n=14) and (B) non-pregnant mice (n=14). Median value of each group was indicated by a short line. Mann-Whitney’s nonparametric test was used for pair-wise comparison. * p<0.05 and ** p<0.01. (C) Pearson correlation test was performed between BSEP transactivation and serum E2 levels in the same groups of pregnant mice. Group means for BSEP transactivation and serum E2 levels were used for the calculation on the following time points: premating, day-5/6, day −2, day −1, day 0, day +1, and day +6/7.
No significant alterations in E2 serum levels were detected in a group of non-pregnant mice in all the time points except for one (Fig. 2B), in which the E2 level is statistically higher than the premating concentration (p=0.021). Such significant elevation in E2 might reflect the fluctuation of E2 levels during normal estrus cycle.
To determine whether BSEP transactivation dynamics are correlated with the changes in serum E2 levels, Pearson correlation test was performed. As shown in Fig. 2C, BSEP transactivation was negatively correlated with serum E2 concentrations (r = −0.902) with a p value of 0.005. It was thus concluded that elevation of E2 concentrations in the late stages of pregnancy was a possible cause for the suppression of BSEP transactivation.
E2 suppressed BSEP expression in vivo
To determine whether E2 directly suppresses BSEP transactivation, the effects of E2 on BSEP transactivation were investigated. As shown in Fig. 3A, BSEP transactivation levels were comparable among all the mice prior to E2 treatment. However, it was markedly suppressed by E2 (75% decrease) (p<0.05) (Fig. 3A). To confirm the results, endogenous BSEP mRNA and protein levels were quantified with real-time PCR and Western blot. Consistent with the IVIS data, E2 significantly decreased BSEP expression by approximately 50% (p<0.05) (Fig. 3B and 3C). It was thus concluded that E2 directly suppressed BSEP expression in mice.
Fig. 3. E2 suppressed BSEP expression in vivo in mice and in vitro in human primary hepatocytes and hepatoma Huh 7 cells.




(A) twenty CD-1 mice hydrodynamically injected with pmBSEP(-2.6kb) (0.5µg/g) via tail-vein prior to treatment were randomly divided into treatment (E2, 5mg/kg daily for 5 days) and control group (vehicle EtOH). Luciferase activities were quantified by IVIS before and after treatment. (B) endogenous BSEP mRNA and (C) protein levels in E2 or EtOH treated mice were detected by real-time PCR or Western blot with GAPDH as internal standard. (D) human primary hepatocytes or (E) Huh 7 cells were treated with CDCA (5µM) or a combination of CDCA and various concentrations of E2, followed by detection of BSEP mRNA by real-time PCR and (F) BSEP protein levels by Western blotting. The Student’s t-test was applied to pair-wise comparison in (A), (B) and (C). One-way ANOVA was applied to analyze data in (D) and (E), followed by Tukey post-hoc test for multiple comparisons. * p<0.05 and ** p<0.01.
E2 suppressed BSEP expression in human primary hepatocytes and Huh 7 cells
As expected, treatment with CDCA significantly induced BSEP mRNA expression in both human primary hepatocytes and Huh 7 cells. However, such inductions were significantly suppressed by E2 (Fig. 3D and 3E). Consistently, BSEP protein levels were dose-dependently suppressed by E2 (Fig. 3F). Taken together, it was concluded that E2 suppressed BSEP expression in both human primary hepatocytes and Huh 7 cells primarily through transcription.
E2 repressed BSEP promoter transactivation through ERα in vitro
To determine whether E2-mediated repression of BSEP expression is mediated through ERα or ERβ, the effects of E2 on BSEP promoter transactivation were evaluated in the presence of ERα or ERβ. As shown in Fig. 4A, co-transfection of ERα significantly decreased basal as well as CDCA-mediated BSEP transactivation. More importantly, E2 further significantly decreased BSEP transactivation. In contrast, ERβ had minimal effects on BSEP transactivation in the presence or absence of E2. In the dose response studies, both ERα and E2 dose-dependently repressed BSEP transactivation (Fig. 4B and 4C). The results thus established that E2 repressed BSEP transactivation through ERα but not ERβ, and such repression was dose-dependent of ERα and E2.
Fig. 4. E2 repressed BSEP promoter transactivation in vitro through ERα.


(A) Huh 7 cells were transfected with phBSEP(−2.6kb), FXR and ERα or ERβ, followed by treatment with CDCA (10µM) or CDCA plus E2 (10nM). Luciferase activities were detected by the dual luciferase assays. (B) dose-response studies were performed with ERα and (C) E2. Data were presented as means ± SD of at least three individual experiments. One-way ANOVA was applied to analyze data in (A) and (C), followed by Tukey post-hoc test for multiple comparisons. The Student’s t-test was applied to pair-wise comparison in (B). * p<0.05 and ** p<0.01
ERα was required for E2 to repress BSEP expression in vivo
After demonstrating that E2 repressed BSEP transactivation through ERα in vitro, we next carried out experiments to confirm such findings in ERα−/− mice in vivo. As shown in Fig. 5A and 5B, BSEP mRNA levels were significantly decreased in wt mice treated with E2 (p<0.01) but the decrease was not observed in ERα(−/−) mice. Consistently, BSEP protein levels were markedly reduced by E2 in wt mice whereas such repression was not detected in ERα(−/−) mice (Fig. 5C and 5D). The data thus firmly established that ERα was required for E2 to repress BSEP expression.
Fig. 5. ERα was required for E2 to repress BSEP expression in vivo.


(A) ERα(−/−) and (B) ERα wt mice from the same colony were treated with E2 (5mg/kg) or vehicle daily for 5 days, followed by detection of BSEP mRNA levels with real-time PCR. The mean and SD were indicated in each group. (C) BSEP protein expression was detected by capillary electrophoresis-based Western blotting with GAPDH as internal standard. (D) quantification of BSEP protein levels. Mann-Whitney’s nonparametric test was used for pair-wise comparison. * p<0.05 and ** p<0.01.
ERα expression in Huh 7 cells and mice liver before, during and after pregnancy
Both ERα mRNA and protein were readily detected in Huh 7 cells and markedly increased in ERα-transfected cells (Supplement Fig 1A and 1B). E2 had no significant effects on ERα mRNA levels but decreased ERα protein levels, consistent with the notion that ligand-binding to ERα promotes ERα protein turnover. ERα expression was increased during pregnancy at both mRNA and protein levels (Supplement Fig. 2A and 2B) but the increases were not statistically significant.
Mapping of the cis-acting element mediating E2/ERα transrepression to FXRE
To localize the cis-element, a series of human BSEP promoter constructs with various lengths (Fig. 6A) were used to evaluate their ability to respond to E2. As shown in Fig. 6B, all the promoter constructs exhibited similar patterns in responding to E2. The minimal promoter which retained its ability to respond to E2 is phBSEP(−120b). It should be mentioned that further deletion of the minimal promoter resulted in a total loss of the promoter activity.
Fig. 6. Mapping of the cis-acting element mediating E2/ERα transrepression to FXRE.



(A) human BSEP promoter reporters with various lengths. (B) transrepression of various BSEP promoter reporters and (C) FXRE consensus pGL-2xFXREcon and human BSEP IR1a element pGL-2xhIR1 reporters by E2 (10nM) in the presence of CDCA (10µM). (D) chromatins were prepared from Huh 7 cells transfected with ERα and FXR, and subsequently treated with CDCA and E2. Chromatins were precipitated with anti-ERα antibodies or IgG, followed by PCR detection with a primer set flanking or upstream (negative control) the FXRE in the human BSEP promoter. (E) Huh 7 cells were transfected with phBSEP(−2.6kb), FXR and pcDNA5, ERα, ERα-LBD-Mut or ERα-DBD-Mut, followed by treatment with CDCA or CDCA and E2. Luciferase activities were determined. Data were presented as means ± SD of at least three individual experiments. The Student’s t-test was applied to pair-wise comparison in (B) and (C). One-way ANOVA was applied to analyze data in (E), followed by Tukey post-hoc test for multiple comparisons. * p<0.05 and ** p<0.01.
Bioinformatic analysis of the minimal promoter revealed a number of potential cis-elements including FXRE. Given the fact that E2/ERα repressed basal as well as CDCA-induced BSEP transactivation, the FXRE was the first target to be investigated. Two FXRE reporters, pGL-2xFXREcon and pGL-2xhIR1a, were tested. E2 exhibited a similar transrepressive pattern on the two FXRE reporters (Fig. 6C) as to BSEP promoter reporters (Fig. 6B). Thus, the data strongly suggested that E2/ERα repressed BSEP transactivation through the FXRE in the BSEP promoter. Indeed, ERα was specifically recruited to the FXRE in the BSEP promoter in the ChIP assays (Fig. 6D).
Ligand but not DNA binding was required for ERα to mediate transrepression on BSEP
To investigate whether ERα directly binds to the FXRE, an ERα DNA-binding mutant (ERα-DBD-Mut) was tested for its ability to transrepress BSEP. As shown in Fig. 6E, ERα-DBD-Mut exhibited similar transrepressive activity as ERα wt while ERα ligand binding domain mutant ERα-LBD-Mut totally lost its ability to repress BSEP transactivation. As expected, both ERα-DBD-Mut and ERα-LBD-Mut diminished their ability to respond to E2 with an ERE reporter pTK-2xERE (Supplement Fig.3). The data thus concluded that DNA-binding is not required for ERα to mediate the transrepression. Therefore, it is unlikely that ERα directly binds to the FXRE to interfere in the FXR signaling pathway. More logically, ERα may interrupt FXR signaling through protein-protein interaction with FXR.
ERα and FXR were co-immunoprecipitated in cells and livers from pregnant mice
To determine whether ERα forms a protein complex with FXR in the cells, Flag-tagged FXR or FXR and ERα were over-expressed in 293T or Huh 7 cells, followed by detection of ERα-FXR protein complex. As shown in Fig. 7A and 7B, FXR and ERα formed a protein complex, which was immunoprecipitated with anti-Flag or anti-ERα antibodies, followed by detection with anti-ERα or anti-FXR antibodies.
Fig. 7. Association of ERα with FXR in celllines and livers from pregnant mice.


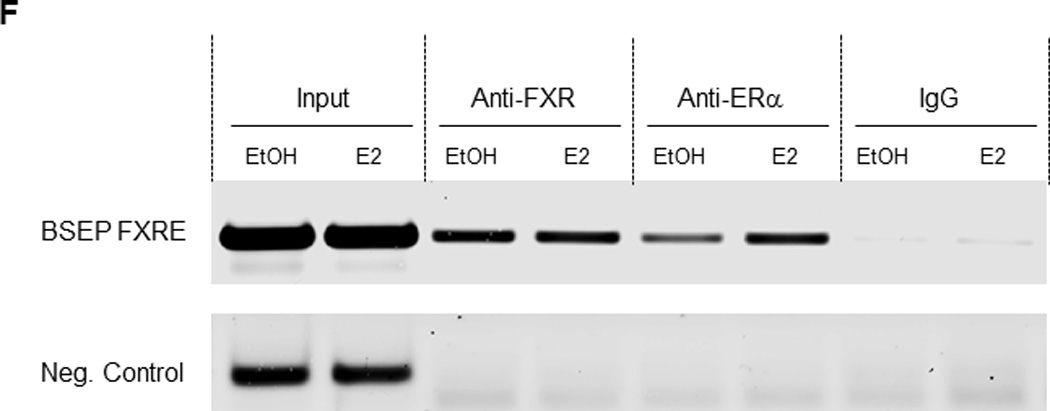
(A) HEK 293T cells were transfected with Flag-FXR and ERα, followed by treatment with CDCA (10µM) and E2 (100nM). Whole cell lysates were precipitated with rabbit anti-ERα polyclonal antibodies or rabbit IgG as negative control. The precipitated proteins were analyzed by Western blotting with anti-Flag antibodies. (B) Huh 7 cells were transfected with FXR and ERα. Cell lysates were immunoprecipitated with anti-FXR, followed by detection by Western blotting with anti-ERα antibodies. (C) Liver tissues were collected from pregnant mice at gestation day 18, homogenized and lysed. Cell lysates were immunoprecipitated with anti-ERα antibiodies, followed by Western blotting with anti-FXR antibodies. (D) cell lysates from the mouse liver tissues were precipitated with anti-FXR antibodies, followed by detection with anti-ERα antibodies on Western blot. (E) nuclear extracts were prepared from Huh 7 cells transfected with FXR and ERα. EMSA assays were performed using the FXRE of human BSEP as probe in the presence of DMSO (0.1%), EtOH (0.1%), E2 (100nM), CDCA (10µM) or a combination of E2 and CDCA. (F) chromatins were prepared from Huh 7 cells transfected with FXR and ERα and treated with E2 (100nM) or EtOH (0.1%), followed by immunoprecipitation with rabbit anti-FXR, ERα or control rabbit IgG. ChIP assays were performed with the ChIP-Express Kit from Active Motif.
To determine whether such FXR-ERα protein complex forms in vivo in pregnant mice, liver tissues were collected from mice at gestation day 18 and similar Co-IP assays were performed. Consistent with the data from transfected cells, protein complexes of FXR-ERα were readily detected in the liver of pregnant mice (Fig. 7C and 7D).
Effects of E2 on FXR and ERα recruitment to FXRE
In the EMSA assays, E2 decreased FXR binding to the FXRE. However, such decrease largely disappeared when CDCA was present (Fig. 7E). In the ChIP assays, E2 increased ERα recruitment to the FXRE of BSEP promoter without affecting FXR recruitment (Fig. 7F).
Direct interaction between ERα and FXR in living cells and in vivo
To determine whether ERα directly interacts with FXR, BiFC assays were performed (33). After construction and confirming the activities of the fusion proteins eGFPn-FXR and ERα-eGFPc (Fig. 8A, Supplement Fig. 4A and 4B), direct physical interaction between the two fusion proteins was evaluated in vitro in Huh 7 cells and in vivo in mice. Minimal fluorescent signals were detected in cells transfected with eGFPn-FXR and vector pcDNA5. However, significant fluorescence was observed in the cells cotransfected with eGFPn-FXR and ERα-eGFPc (Fig. 8B and 8C), indicating a direct interaction between ERα and FXR. Mutation of the LBD in ERα-LBD-Mut-eGFPc markedly decreased the fluorescent signals, indicating that ligand binding is required for ERα to achieve maximal interaction with FXR. Similar results were obtained with in vivo mouse study (Fig. 8D and 8E). Minimal fluorescent signals were detected in hepatocytes derived from mice injected with eGFPn-FXR and pcDNA5. However, strong fluorescent signals were detected in hepatocytes from the mice injected with eGFPn-FXR and ERα-eGFPc. Much weaker signals were detected in hepatocytes from mice injected with eGFPn-FXR and ERα-LBD-Mut-eGFPc. It was thus concluded that ERα directly interacted with FXR in vitro in living cells and in vivo in mice, and maximal interaction required ligand binding to ERα.
Fig. 8. Direct interaction between ERα and FXR in vitro in Huh 7 cells and in vivo in mice.






(A) construction of fusion proteins eGFPn-FXR, ERα-eGFPc and ERα-LBD-Mut-eGFPc. (B) Huh 7 cells were transfected with eGFPn-FXR and either pcDNA5 vector, ERα-eGFPc or ERα-LBD-Mut-eGFPc, followed by treatment with E2 (100nM). Fluorescent signals were detected under a confocal microscope. Images were captured at a magnification of 40X with fluorescent and differential interference contrast (DIC) settings. (C) quantification of fluorescent signals in (B). (D) three groups of mice were hydrodynamically co-injected eGFPn-FXR with either ERα-eGFPc, ERα-LBD-Mut-eGFPc or pcDNA5 vector via tail vein. Twenty four hours post-injection, primary hepatocytes collected through liver perfusion were cultured on slide chambers for fluorescence detection under confocal microscope or directly subjected to FACS analysis. (E) quantification of the cells gated in M2 window showed in (D). (F) cellular localization of ERα and FXR interaction as fluorescent signals in Huh 7 cells and primary hepatocytes. One-way ANOVA was applied to analyze data in (C) and (E), followed by Tukey post-hoc test for multiple comparisons. * p<0.05 and ** p<0.01.
It is our expectation that the interaction between ERα and FXR should occur in the nuclei. Indeed, fluorescent signals were almost exclusively observed in the nuclei of transfected Huh 7 cells (upper panel, Fig. 8F). However, it was noticed that fluorescent signals were observed in various cellular compartments in mouse hepatocytes in vivo with predominant presence around the nuclear membrane likely associated with endoplasmic reticulum and to a less extent in the cytoplasm possibly associated with vesicle-like structures and nucleus (lower panel, Fig. 8F).
DISCUSSION
Pregnancy is associated with dramatic physiological changes in the mother. One of the changes during pregnancy is elevated levels of serum bile acid (3, 5–7). When such elevation reaches in an excess of 10 µM, ICP develops (6, 7, 14). Canalicular effluxers including BSEP and multidrug resistance protein 3 (MDR3) have long been suspected as the targets for causing ICP (22, 23, 27). Indeed, genetic variants of BSEP are associated with ICP (22, 23). Also, estrogen and its metabolites have been reported to inhibit BSEP function or cell surface expression (27, 28). In this study for the first time, the dynamics of BSEP transactivation during pregnancy was established in the same group of mice and it was demonstrated that BSEP expression was significantly transrepressed during the late stages of pregnancy. Furthermore, such transrepression was inversely correlated with elevated E2 levels during pregnancy and E2 repressed BSEP expression in vitro and in vivo. These findings support the notion that down-regulation of BSEP expression by elevated E2 during late stages of pregnancy is a contributing factor to the pathogenesis of ICP. Consistent with a previous report (26), SHP mRNA levels were significantly decreased during and after pregnancy (Supplement Fig. 5A). However, the decreases were not statistically significant at the protein levels (Supplement Fig. 5B).
Since most women do not experience ICP during their normal pregnancy, it becomes apparent that repression of BSEP expression by E2 alone is not sufficient to cause ICP. Other factors, notably genetics, are also risk factors for ICP. The prevalence of ICP varies markedly among ethnic groups ranging from less than 1% to 14% (11, 12) with the highest in South America. Such variations in ICP incidence among ethnic groups strongly indicate that certain genetic traits predispose women to ICP. It is therefore reasonable to speculate that women carrying those genetic changes increase their sensitivity to elevated E2 during the late stages of pregnancy, leading to ICP.
The question remains regarding the physiological significance of BSEP transrepression by E2/ERα through attenuating the FXR signaling during the pregnancy, especially at the later stages of the pregnancy. Transrepression of BSEP leads to decreased canalicular disposition of bile acids thus causing retention of bile acids in the liver. Such increase in bile acid levels in turn slows down the conversion of cholesterol into bile acids through feedback mechanisms (34), subsequently leading to conservation of cholesterol. Cholesterol is an essential building block for cell membranes and a precursor for steroid hormone production. A growing fetus requires cholesterol for rapid growth and steroid hormone production, especially in the late stages of pregnancy. Therefore, down-regulation of BSEP, as a result of attenuation of the FXR signaling by E2/ERα, may represent an adaptive mechanism by the mother to meet the increasing demand of the growing fetus for cholesterol. Consistent with such speculation, serum cholesterol levels are elevated in pregnant women (1–3).
In the classical signaling pathway estrogens, including E2, regulate their target genes through activating ERα or ERβ (35), which directly binds to ERE in the promoter of the target genes and subsequently results in the transactivation of the target genes (35). In this study, we demonstrated that E2 transrepressed BSEP expression through ERα, which directly interacts with FXR. Such transrepression through protein-protein interaction represents another non-classical estrogen signaling pathway (36). In support of our findings, it has been shown that ERα co-immunoprecipitated with FXR in a breast cancer cell line MCF-7 (37) and ERα interacted FXR in a GST-pulldown assay (26).
As FXR and ERα are both nuclear receptors, we expected that their interaction occurs in the nucleus. Indeed, the interaction in Huh 7 cells was almost exclusively in the nucleus. However, the interaction was more widely distributed in mice in vivo, with a predominant presence around the nuclear membrane, especially outside the membrane. The precise reasons for such discrepancy are not clear. The predominant presence of FXR and ERα complexes outside the nucleus in mouse liver raised an interesting question of whether retention of FXR in the cytoplasm as an ERα/FXR complex represents a possible mechanism for E2/ERα to attenuate the FXR signaling pathway.
We consistently demonstrated that ERα markedly decreased basal and CDCA-induced transactivation of BSEP promoter in the absence of exogenously added E2 (Fig. 4A, 4B and 6E). One possible explanation is that there are endogenous estrogenic compounds in cells or cell medium that bind to ERα to mediate the transrepression. Another possible explanation is that ERα has both ligand-dependent and independent transrepressive activities on BSEP.
Given the fact that in addition to bile acid homeostasis, FXR signaling pathway is involved in many cellular functions including cholesterol, lipids, glucose, energy and vascular regulations (38–40), it is reasonable to assume that many other physiological and metabolic changes during pregnancy are direct results of estrogen’s effects through the ERα/FXR interaction.
Supplementary Material
ACKNOWLEDGEMENTS
Financial Support:
This work was supported by the National Institutes of Health Grant R01DK087755. B.Y. is supported by the National Institutes of Health Grants R01GM61988, R01ES07965 and R15AT007705.
Instrumental supports from the RI-INBRE Core Facility in the College are greatly appreciated, which is supported by an Institutional Development Award (IDeA) from the National Institutes of General Medical Sciences of the National Institutes of Health under grant P20 GM103430-13. We thank Dr. David Mangelsdorf for providing human FXRα1 expression plasmid, Dr. Matthew Stoner for providing expression plasmids for Flag-FXR, ERα and ERβ, and Dr. Chang-Deng Hu for providing the expression constructs of the N- and C-terminal fragments of eGFP, pBiFC-VN173 (pFLAG-Venus 1-172) and pBiFC-VC155 (pHA-Venus 155-238).
Abbreviations
- BSEP
bile salt export pump
- ABCB11
ATP-binding cassette subfamily B member 11
- BiFC
bimolecular fluorescence complementation
- CDCA
chenodeoxycholic acid
- ChIP
chromatin immunoprecipitation
- Co-IP
co-immunoprecipitation
- DBD
DNA binding domain
- LBD
ligand binding domain
- DMSO
dimethyl sulfoxide
- E2
17β-estradiol
- eGFP
enhanced green fluorescence protein
- ERα
estrogen receptor-α
- ERβ
estrogen receptor-β
- ERE
estrogen response element
- EtOH
ethanol
- FACS
fluorescence activated cell sorting
- FXR
farnesoid x receptor
- NR1H4
nuclear receptor subfamily 1 group H member 4
- FXRE
FXR response element
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- IACUC
institutional animal care and use committee
- ICP
intrahepatic cholestasis of pregnancy
- IR1
inverted repeat spaced by one nucleotide
- IVIS
in vivo imaging system
- MDR3
multidrug resistance protein 3
- SHP
small heterodimer partner
- WT
wild-type
Contributor Information
Xiulong Song, Email: songxiulong@hotmail.com.
Alexander Vasilenko, Email: alex_vasilenko@yahoo.com.
Yuan Chen, Email: chenyuan@ymail.com.
Leila Valanejad, Email: leilavalanejad@gmail.com.
Ruchi Verma, Email: ruchi9991@gmail.com.
Bingfang Yan, Email: byan@uri.edu.
Ruitang Deng, Email: dengr@mail.uri.edu.
REFERENCES
- 1.Herrera E. Metabolic adaptations in pregnancy and their implications for the availability of substrates to the fetus. Eur J Clin Nutr. 2000;54(Suppl 1):S47–S51. doi: 10.1038/sj.ejcn.1600984. [DOI] [PubMed] [Google Scholar]
- 2.Lippi G, Albiero A, Montagnana M, Salvagno GL, Scevarolli S, Franchi M, et al. Lipid and lipoprotein profile in physiological pregnancy. Clin Lab. 2007;53(3–4):173–177. [PubMed] [Google Scholar]
- 3.Abu-Hayyeh S, Papacleovoulou G, Williamson C. Nuclear receptors, bile acids and cholesterol homeostasis series - bile acids and pregnancy. Mol Cell Endocrinol. 2013;368(1–2):120–128. doi: 10.1016/j.mce.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 4.Aleksunes LM, Yeager RL, Wen X, Cui JY, Klaassen CD. Repression of hepatobiliary transporters and differential regulation of classic and alternative bile acid pathways in mice during pregnancy. Toxicol Sci. 2012;130:257–268. doi: 10.1093/toxsci/kfs248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fulton IC, Douglas JG, Hutchon DJ, Beckett GJ. Is normal pregnancy cholestatic? Clin Chim Acta. 1983;130(2):171–176. doi: 10.1016/0009-8981(83)90114-6. [DOI] [PubMed] [Google Scholar]
- 6.Lunzer M, Barnes P, Byth K, O'Halloran M. Serum bile acid concentrations during pregnancy and their relationship to obstetric cholestasis. Gastroenterology. 1986;91(4):825–829. doi: 10.1016/0016-5085(86)90682-7. [DOI] [PubMed] [Google Scholar]
- 7.Heikkinen J, Maentausta O, Ylostalo P, Janne O. Changes in serum bile acid concentrations during normal pregnancy, in patients with intrahepatic cholestasis of pregnancy and in pregnant women with itching. Br J Obstet Gynaecol. 1981;88(3):240–245. [PubMed] [Google Scholar]
- 8.Hay JE. Liver disease in pregnancy. Hepatology. 2008;47(3):1067–1076. doi: 10.1002/hep.22130. [DOI] [PubMed] [Google Scholar]
- 9.Joshi D, James A, Quaglia A, Westbrook RH, Heneghan MA. Liver disease in pregnancy. Lancet. 2010;375(9714):594–605. doi: 10.1016/S0140-6736(09)61495-1. [DOI] [PubMed] [Google Scholar]
- 10.Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15(17):2049–2066. doi: 10.3748/wjg.15.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reyes H, Gonzalez MC, Ribalta J, Aburto H, Matus C, Schramm G, et al. Prevalence of intrahepatic cholestasis of pregnancy in Chile. Ann Intern Med. 1978;88(4):487–493. doi: 10.7326/0003-4819-88-4-487. [DOI] [PubMed] [Google Scholar]
- 12.Abedin P, Weaver JB, Egginton E. Intrahepatic cholestasis of pregnancy: prevalence and ethnic distribution. Ethn Health. 1999;4(1–2):35–37. doi: 10.1080/13557859998173. [DOI] [PubMed] [Google Scholar]
- 13.Rook M, Vargas J, Caughey A, et al. Fetal outcomes in pregnancies complicated by intrahepatic cholestasis of pregnancy in a Northern California cohort. PLoS One. 2012;7(3):e28343. doi: 10.1371/journal.pone.0028343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glantz A, Marschall HU, Mattsson LA. Intrahepatic cholestasis of pregnancy: Relationships between bile acid levels and fetal complication rates. Hepatology. 2004;40(2):467–474. doi: 10.1002/hep.20336. [DOI] [PubMed] [Google Scholar]
- 15.Meier PJ, Stieger B. Bile salt transporters. Annu Rev Physiol. 2002;64:635–661. doi: 10.1146/annurev.physiol.64.082201.100300. [DOI] [PubMed] [Google Scholar]
- 16.Kullak-Ublick GA, Stieger B, Meier PJ. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology. 2004;126(1):322–342. doi: 10.1053/j.gastro.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001;276(31):28857–28865. doi: 10.1074/jbc.M011610200. [DOI] [PubMed] [Google Scholar]
- 18.Plass JR, Mol O, Heegsma J, Geuken M, Faber KN, Jansen PL, et al. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology. 2002;35(3):589–596. doi: 10.1053/jhep.2002.31724. [DOI] [PubMed] [Google Scholar]
- 19.Deng R, Yang D, Radke A, Yang J, Yan B. The hypolipidemic agent guggulsterone regulates the expression of human bile salt export pump: dominance of transactivation over farsenoid X receptor-mediated antagonism. J Pharmacol Exp Ther. 2007;320(3):1153–1162. doi: 10.1124/jpet.106.113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song X, Kaimal R, Yan B, Deng R. Liver receptor homolog 1 transcriptionally regulates human bile salt export pump expression. J Lipid Res. 2008;49(5):973–984. doi: 10.1194/jlr.M700417-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weerachayaphorn J, Cai SY, Soroka CJ, Boyer JL. Nuclear factor erythroid 2-related factor 2 is a positive regulator of human bile salt export pump expression. Hepatology. 2009;50(5):1588–1596. doi: 10.1002/hep.23151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lam P, Soroka CJ, Boyer JL. The bile salt export pump: clinical and experimental aspects of genetic and acquired cholestatic liver disease. Semin Liver Dis. 2010;30(2):125–133. doi: 10.1055/s-0030-1253222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stieger B, Geier A. Genetic variations of bile salt transporters as predisposing factors for drug-induced cholestasis, intrahepatic cholestasis of pregnancy and therapeutic response of viral hepatitis. Expert Opin Drug Metab Toxicol. 2011;7(4):411–425. doi: 10.1517/17425255.2011.557067. [DOI] [PubMed] [Google Scholar]
- 24.Van Mil SW, Milona A, Dixon PH, Mullenbach R, Geenes VL, Chambers J, et al. Functional variants of the central bile acid sensor FXR identified in intrahepatic cholestasis of pregnancy. Gastroenterology. 2007;133(2):507–516. doi: 10.1053/j.gastro.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 25.Abu-Hayyeh S, Papacleovoulou G, Lovgren-Sandblom A, Tahir M, Oduwole O, Jamaludin NA, et al. Intrahepatic cholestasis of pregnancy levels of sulfated progesterone metabolites inhibit farnesoid X receptor resulting in a cholestatic phenotype. Hepatology. 2013;57(2):716–726. doi: 10.1002/hep.26055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milona A, Owen BM, Cobbold JF, Willemsen EC, Cox IJ, Boudjelal M, et al. Raised hepatic bile acid concentrations during pregnancy in mice are associated with reduced farnesoid X receptor function. Hepatology. 2010;52(4):1341–1349. doi: 10.1002/hep.23849. [DOI] [PubMed] [Google Scholar]
- 27.Stieger B, Fattinger K, Madon J, Kullak-Ublick GA, Meier PJ. Drug- and estrogen-induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology. 2000;118(2):422–430. doi: 10.1016/s0016-5085(00)70224-1. [DOI] [PubMed] [Google Scholar]
- 28.Leslie KK, Reznikov L, Simon FR, Fennessey PV, Reyes H, Ribalta J. Estrogens in intrahepatic cholestasis of pregnancy. Obstet Gynecol. 2000;95(3):372–376. doi: 10.1016/s0029-7844(99)00533-5. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto Y, Moore R, Hess HA, Guo GL, Gonzalez FJ, Korach KS, et al. Estrogen receptor alpha mediates 17alpha-ethynylestradiol causing hepatotoxicity. J Biol Chem. 2006;281(24):16625–16631. doi: 10.1074/jbc.M602723200. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, Song X, Valanejad L, et al. Bile salt export pump is dysregulated with altered farnesoid X receptor isoform expression in patients with hepatocellular carcinoma. Hepatology. 2013;57(4):1530–1541. doi: 10.1002/hep.26187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng R, Yang D, Yang J, et al. Oxysterol 22(R)-hydroxycholesterol induces the expression of the bile salt export pump through nuclear receptor farsenoid X receptor but not liver X receptor. J Pharmacol Exp Ther. 2006;317(1):317–325. doi: 10.1124/jpet.105.097758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song X, Chen Y, Valanejad L, Kaimal R, Yan B, Stoner M, et al. Mechanistic insights into isoform-dependent and species-specific regulation of bile salt export pump by farnesoid X receptor. J Lipid Res. 2013;54(11):3030–3044. doi: 10.1194/jlr.M038323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerppola TK. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys. 2008;37:465–487. doi: 10.1146/annurev.biophys.37.032807.125842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50(10):1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, et al. Mechanisms of estrogen action. Physiol Rev. 2001;81(4):1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 36.Safe S, Kim K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol. 2008;41(5):263–275. doi: 10.1677/JME-08-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Journe F, Laurent G, Chaboteaux C, Nonclercq D, Durbecq V, Larsimont D, et al. Farnesol, a mevalonate pathway intermediate, stimulates MCF-7 breast cancer cell growth through farnesoid-X-receptor-mediated estrogen receptor activation. Breast Cancer Res Treat. 2008;107(1):49–61. doi: 10.1007/s10549-007-9535-6. [DOI] [PubMed] [Google Scholar]
- 38.Trauner M, Claudel T, Fickert P, Moustafa T, Wagner M. Bile acids as regulators of hepatic lipid and glucose metabolism. Dig Dis. 2010;28(1):220–224. doi: 10.1159/000282091. [DOI] [PubMed] [Google Scholar]
- 39.Teodoro JS, Rolo AP, Palmeira CM. Hepatic FXR: key regulator of whole-body energy metabolism. Trends Endocrinol Metab. 2011;22(11):458–466. doi: 10.1016/j.tem.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 40.Stanimirov B, Stankov K, Mikov M. Pleiotropic functions of bile acids mediated by the farnesoid X receptor. Acta Gastroenterol Belg. 2012;75(4):389–398. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.