

Abstract
The past two decades have brought dramatic progress in the neuroscience of anxiety due, in no small part, to animal findings specifying the neurobiology of Pavlovian fear-conditioning. Fortuitously, this neurally mapped process of fear learning is widely expressed in humans, and has been centrally implicated in the etiology of clinical anxiety. Fear-conditioning experiments in anxiety patients thus represent a unique opportunity to bring recent advances in animal neuroscience to bear on working, brain-based models of clinical anxiety. The current presentation details the neural basis and clinical relevance of fear conditioning, and highlights generalization of conditioned fear to stimuli resembling the conditioned danger cue as one of the more robust conditioning markers of clinical anxiety. Studies testing such generalization across a variety of anxiety disorders (panic, generalized anxiety disorder, and social anxiety disorder) with systematic methods developed in animals will next be presented. Finally, neural accounts of over-generalization deriving from animal and human data will be described with emphasis given to implications for the neurobiology and treatment of clinical anxiety.
Keywords: anxiety disorders, Pavlovian fear-conditioning, stimulus generalization, neurobiology, fMRI, fear-potentiated startle
I. Introduction
Central to etiological accounts of clinical anxiety are abnormalities in Pavlovian fear conditioning1–3: the associative learning process by which an initially benign conditioned stimulus (CS) assumes anxiety eliciting properties by way of its co-occurrence with a naturally aversive unconditioned stimulus (US). Though fear-conditioning generally serves an adaptive, self-preserving function, it becomes a source of pathology when anxious reactivity persists to CSs no longer accompanied by, or predictive of, the aversive US. Different anxiety disorders are distinguished primarily by the nature of the US and CS. For example, in social anxiety disorder, panic disorder, and PTSD the respective USs are “time limited” embarrassing events, panic attacks, and traumatic encounters that elicit unconditioned anxiety, and the CSs are those people, places, and things associated with the relevant US. Conditioning processes are similarly thought to contribute to all these disorders by conferring anxiogenic valence to CSs, and stimuli resembling the CSs, that are then capable of eliciting and maintaining (conditioned) anxiety responses well after the termination of the time-limited US.
As alluded above, fear conditioning is more than a phenomenon that goes awry in clinical anxiety but is a fundamental learning process expressed in all people. Indeed, fear conditioning can be thought of as a basic building block of the fear network4, or web of fear-related memories, through which memories of neutral stimuli are incorporated as a function of their association with an aversive outcome. Fear-conditioning is not just basic to the human experience but is ubiquitously expressed in numerous species from snails to non-human primates—a cross-species feature that has led to the elegant elucidation of conditioned fear in the brains of lower mammals5. Such animal data provide important insights on human fear-conditioning, generally, as well as neural substrates of conditioning aberrancies in clinical anxiety, specifically. The current paper will detail the neurobiology of fear conditioning derived from work in animals, link these findings to clinical anxiety through meta-analytic results implicating over-generalization of conditioned fear as one of the more promising conditioning correlates of anxiety pathology, describe a program of work designed to further investigate the clinical relevance and brain basis of human generalization, and propose candidate neural mechanisms through which clinical abnormalities in generalization may be explained and, in the future, treated.
II. Neurocircuitry of Unconditioned and Conditioned Fear
Unconditioned fear
Evidence from animal studies demonstrates the centrality of the amygdala for processing fear-relevant stimuli. As shown in Figure 1 (Pathway A), when a naturally aversive US (e.g., electric shock) is encountered, the thalamus relays sensory information to the input node of the amygdala: the lateral nucleus (La), resulting in activation of the amygdala’s output node: the central nucleus (Ce), which then excites subcortical and brainstem structures (e.g., lateral hypothalamus, central grey, bed nucleus of the stria terminalis) responsible for the autonomic, behavioral, and neuroendocrine constituents of the fear response)6.
Figure 1.

Schematic of fear circuitry subserving unconditioned and conditioned fear derived from animal data. (A) Solid black arrows mark the processing path for an inherently aversive unconditioned stimulus (US: e.g., electric shock) which activates the input and output nodes of the amygdala, culminating in the behavioral, endocrine, and autonomic constituents of the fear response. (B) The light grey processing path subserves acquisition of conditioned fear, ensuing when a neutral conditioned stimulus (CS+) causes the release of glutamate in LA neurons in close temporal proximity to US-induced depolarization of these same neurons. The co-occurrence of this glutamate release and depolarization results in a cascade of intracellular processes in the La (release of Ca2+ which binds to NMDA receptors → excitation of protein kinases [e.g., MAPK] → activation of transcription factors [CREB] → synthesis of RNA and protein) which then makes new proteins used, in part, to increase the amygdala’s sensitivity to the CS+. (C) Broken arrows delineate extinction of conditioned fear, whereby repeated presentations of the CS+ in the absence of the US weakens the conditioned fear response. This weakening is thought to ensue when an mPFC-ITC mediated inhibition of the central nucleus of the amygdala outcompetes the excitatory pathway formed during acquisition. (D) Denoted by compound arrows is the processing path for discrimination of conditioned fear to a conditioned safety cue (CS-) with perceptual resemblance to the CS+. Sensory and affective discrimination of CS− from CS+ begins with higher order processing of CS− in the sensory cortex. Next the hippocampus may perform a schematic match between the cortical representation of the previously encountered CS+ and that of the currently presented CS−. With sufficient mismatch, the hippocampus initiates pattern separation resulting in activation of the mPFC, which then inhibits fear by activating a group of inhibitory, intercalated (ITC) neurons in the amygdala. La = lateral nucleus of the amygdala; Ce = central nucleus of the amygdala; Ca2+ = calcium; NMDAR = N-Methyl-D-aspartic acid receptor; MAPK = Mitogen-activated protein kinase; CREB = cAMP response element-binding transcription factor; BST = bed nucleus of the stria terminalis; PAG = periaqueductal gray; RPC = reticularis pontis caudalis; hypo = hypothalamus;
 = excitatory connections;
= excitatory connections;
 = inhibitory connections.
= inhibitory connections.
Acquisition
The amygdala-based circuit mediating the formation, or acquisition, of fear to a conditioned danger cue (CS+) following its pairing with an aversive US is illustrated in Figure 1, (Pathway B). Unlike exposure to the US, pre-conditioning exposures to the CS+ do not lead to activation of the input (La) or output (Ce) nodes of the amygdala, but do—by way of the thalamus—evoke the release of glutamate in the La which then binds to glutamate receptors of La neurons7. During acquisition of conditioned fear, when the US follows the CS+ closely in time, activation of La neurons by the US occurs in the presence of CS+ provoked glutamate, triggering a cascade of intracellular processes (e.g., release of Ca2+ which binds to NMDA receptors → excitation of protein kinases [e.g., MAPK] → activation of transcription factors [CREB] → synthesis of RNA and protein)—the end result of which is the availability of new proteins used, in part, to increase the amygdala’s sensitivity to the CS+7. For example, some new proteins are used to form additional glutamate receptors in the La, rendering the La more sensitive to CS+ induced glutamate. Through repeated parings of the CS+ and US (i.e., repeated acquisition trials) these intracellular changes accumulate, and the amygdala becomes increasingly reactive to the CS+—a stimulus previously incapable of activating the amygdala.
Extinction
Figure 1 (Pathway C) denotes the circuit engaged during extinction when the CS+ is repeatedly presented in the absence of the US. At the outset of extinction, the CS+ continues to excite the Ce of the amygdala (via the thalamo-LACe pathway) resulting in the fear response. However, with repeated presentations of the CS+ in the absence of the US, the thalamus increasingly activates the medial prefrontal cortex which relays excitation to intercalated cells of the amygdala, a group of GABAergic amygdaloid neurons that inhibit activation of the Ce and its fear-related projections. Extinction, or neutralization, of fear to the CS+ results when the inhibitory influence of medial prefrontal cortex on the Ce, outcompetes excitatory communications from the La to the Ce, resulting in the inactivation of Ce projections underlying the fear response8.
In addition to acquisition and extinction, a variety of other learning processes (e.g., extinction retention, consolidation, reconsolidation, contextual conditioning, reversal, discrimination/generalization) have been linked to brain mechanisms with lab-based Pavlovian conditioning experiments in animals (for a review see9,10). Though a detailed description of this array of findings is beyond the purview of the current paper, they illustrate the wealth of animal data available on the neurobiology of a number of different processes of conditioned fear. Importantly, the clinical relevance of brain circuits subserving these different conditioning processes depends on human data, from comparable experiments, specifying conditioning aberrancies associated with anxiety disorders.
III. The Road to Generalization
Existing conditioning experiments in anxiety disorders generally apply discriminative conditioning, whereby the degree to which a subject’s fear response is differentially larger to a CS paired with an aversive US (CS+: learned danger cue) versus a CS presented in the absence of the US (CS−: learned safety cue) is assessed. A meta-analysis of such studies reflecting psychophysiologically indexed conditioning scores across 453 anxiety patients and 455 healthy comparisons failed to show the expected elevation in differential responding (i.e., CS+ > CS−) among those with, versus without, an anxiety disorder, but rather implicated enhanced fear-responding to the CS− as one of the more robust conditioning correlates of clinical anxiety2. Given that CS+ and CS− employed by this literature, invariably share many stimulus properties (e.g., size, shape, duration), such findings implicate an enhanced tendency among anxiety patients to generalize conditioned fear from the danger cue to resembling safety cues. This “generalization” interpretation of data is consistent with clinical conceptualizations of anxiety pathology in which over-generalization is thought to contribute etiologically by proliferating anxiety cues in the individual’s environment that then increase and/or sustain anxiety symptoms.
Though both lab-based data and clinical observation implicate over-generalization of conditioned fear as an important marker of anxiety pathology, no psychobiological studies prior to studies by our group11, 12 systematically examined the conditioned generalization process in any of the anxiety disorders. Indeed, past conditioning studies in clinical anxiety were not explicitly interested in assessing generalization, and reactions to CS− were included only as control conditions with which to contrast levels of CS+ reactivity. Because this lack of work in the clinical realm was accompanied by a virtual absence of such work in humans, we designed and psychophysiologically validated a novel paradigm13 employing systematic tests of generalization developed in animals referred to as generalization gradients: the gradual weakening of the conditioned response as the target (generalization) stimulus becomes increasingly dissimilar to the CS+14. The steepness of this gradient indexes generalization, with less steep downward gradients indicating more generalization. Our generalization paradigm, as displayed in Figure 2, employs 6 classes of rings of gradually increasing size. The extreme ring sizes serve as CS+ (paired with electric shock) and CS− (unpaired with shock), with the CS+ being the smallest for half of participants and the largest for remaining participants. Generalization stimuli (GSs) consist of the 4 classes of intermediately sized rings (GS1,GS2,GS3,GS4) and create a continuum-of-similarity from CS+ to CS−. Conditioned fear as well as its generalization was assessed by fear-potentiated startle: the reliable enhancement of the startle reflex when an organism is in a state of fear15. Data in Figure 2A demonstrate continuous decreases in generalization of conditioned fear in healthy controls, with the highest level of fear-potentiated startle to the CS+, and gradually subsiding levels to stimuli of decreasing similarity to the CS+.
Figure 2.
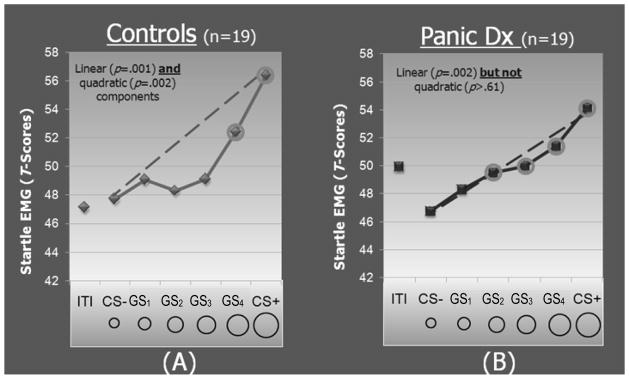
Average standardized startle-blink EMG magnitudes at generalization test, by group, for conditioned stimuli paired (CS+) and unpaired (CS−) with shock, generalization stimuli (GS1, GS2, GS3, GS4), and intertrial intervals (ITIs). The dotted lines reflect linear decreases in startle from CS+ to CS− with which to visualize the deviation of gradients from linearity. Such deviations reflect a significant quadratic component in the generalization gradient of healthy comparison subjects (p=0.001) but not patients with panic disorder (p=0.62). The circled data points indicate stimulus classes for which startle is potentiated relative to the CS− (at the Hochberg-adjusted p value) for each group.
IV. Clinical Testing of the Human Generalization-Gradient Paradigm
Panic disorder and generalization
Panic disorder (PD) was the first clinical group to which this paradigm was applied11. In the context of PD, the US is the panic attack itself and the CSs are the people, places, and sensations that co-occur with the attack. Conditioning is thought to contribute to the onset and maintenance of PD by conferring panicogenic valence to the CSs that are then capable of eliciting anticipatory anxiety for, or actual occurrence of, additional attacks. However, CSs contributing to PD are not thought limited to the original CSs but proliferate to resembling stimuli through the generalization process16–18. For example, learned fear of a particular public place where a panic attack occurred tends to generalize to other public places (malls, cafeterias, or subways). Additionally, acquired fear of autonomic sensations experienced during panic is known to generalize to benign activities of everyday life that elicit resembling sensations (e.g., exercise, climbing stairs, or running for a bus). These clinically observed proclivities to generalize among panic patients led to the hypothesis that panic patients would over-generalize conditioned fear-potentiated startle to stimuli resembling the CS+. Figures 2A and 2B display results for healthy comparisons versus panic patients. Group differences in the shape of gradients emerged with more shallow linear decreases in patients and steeper quadratic decreases in healthy comparisons, indicating more generalization among those with panic disorder. Further evidence of over-generalization in panic patients derives from follow-up contrasts, the results of which are presented in Figure 2 as circled data points reflecting stimulus categories eliciting fear-potentiated startle relative to CS−. Such analyses revealed generalization of conditioned fear to stimuli with up to three units of CS+ differentiation in panic patients, but only one unit of differentiation in healthy comparisons.
GAD, SAD, and generalization
In subsequent applications of this paradigm, patients with generalized anxiety disorder (GAD) and social anxiety disorder (SAD) were tested with opposite predictions. In GAD, a multiplicity of situations lead to anxiety and part of this multiplicity may well stem from the overgeneralization of fear to resembling situations. Supporting this prediction of over-generalization in GAD, are findings of increased sensitivity to uncertainty19 as well as a tendency to interpret ambiguous stimuli as threatening among GAD patients20. Because generalization stimuli in this paradigm are intermediary sized rings constituting threat of a more uncertain and ambiguous nature, we expected over responding in GAD patients to our generalization stimuli. In contrast to GAD, SAD is characterized by circumscribed anxiety responses associated with the fear of social scrutiny and humiliation. Threat of electric shock used in this program of work is a more physical threat that falls outside the circumscribed content of their phobic fear and should elicit normative levels of conditioning and generalization. Thus SAD patients were expected to show normal levels of generalization and served as a psychiatric control group. Predictions were largely confirmed with evidence of over-generalization in conditioned fear-potentiated startle, akin to that found in panic disorder, among those with GAD but not SAD21.
V. The Neurobiology of Generalization
Our group next turned to the neural characterization of conditioned generalization in an effort to identify candidate brain processes subserving the over-generalization found in clinical anxiety. Owing to the cross-species relevance of this conditioning process, hypotheses for human generalization were informed by neurobiological findings in animals. Among them are findings suggesting a critical role for the hippocampus, with increased generalization of conditioned responses to stimuli resembling the CS+ in lower mammals with lesions of the hippocampus22, 23. Of note, generalization constitutes a failure to discriminate CS+ from CS− and, as such, previously reported deficits evoked by hippocampal lesions can also be interpreted as impairments in discriminative conditioning. Figure 1 (Pathway D) displays a neural model of generalization/discrimination centered on the hippocampus. Following acquisition of conditioned fear to the CS+, when presented with the CS−, the thalamus is thought to relay sensory information to the cortex for higher order sensory processing, resulting in activation of the cortical representations of CS− in sensory cortex24, 25. The hippocampus is next thought to complete a schematic match (same-different appraisal) between the cortical representation of the previously encountered CS+ and the currently experienced CS−. With sufficient overlap in CS+ and CS− representations, the hippocampus may initiate pattern completion, whereby a subset of cues from a previous experience (i.e., the CS+) activates the whole of the stored pattern representing the currently experienced stimulus (i.e., the CS−)26. Because part of the pattern of brain activity subserving the CS+ includes fear related brain areas, completion of this pattern, via CS− exposure, will result in activation of such structures as the amygdala and anterior insula culminating in the amygdala-mediated fear response (i.e., fear generalization to the CS−). In the event of insufficient overlap, the hippocampus is thought to initiate pattern separation resulting in spreading of activation to the medial-prefrontal-cortex which projects to the intercalated cells of the amygdala that then inhibit the Ce and its anxiety related projections—an inhibitory cascade resulting in little generalization of fear to the CS−.
To what degree do these animal findings relate to human generalization? Preliminary findings from an fMRI version of the generalization gradient paradigm displayed in Figure 2 reveal brain areas engaged by human generalization, that overlap with animal findings, including the hippocampus, medial prefrontal cortex, and amygdala21. Specifically, activations in the hippocampus and ventro-medial prefrontal-cortex (vmPFC) were weakest to generalization stimuli (GS) with the best “schematic match” to the CS+ (i.e., stimuli most similar to the CS+) and strengthened in a curve-linear manner as the schematic match decreased. Inversely, amygdala (and anterior insula) activations decreased curve-linearly as the match between GS and CS+ decreased. This pattern of results, informed by inferences from animal findings, suggests that GSs with decreasing schematic match to the CS+ elicited increasing, hippocampally-mediated, pattern separation of GS from CS+ representations, resulting in increasingly strong vmPFC activations and a correspondingly strong inhibition of such fear-related areas as the amygdala and anterior insula.
VI. Candidate Neural Loci of Over-Generalization in the Anxiety Disorders
Clinical inferences from preliminary fMRI data
The neural correlates of generalization described above afford several predictions regarding potential brain abnormalities subserving overgeneralization of the kind generated psychophysiologically in clinical anxiety. One such prediction is reduced hippocampally-mediated pattern separation in response to GSs among those with clinical anxiety. That is, anxiety patients may require greater dissimilarity between the CS+ and GS before the hippocampus initiates pattern separation leading to activation of the fear inhibiting vmPFC. Additional clinical aberrancies may lie in the vmPFC and amygdala, whereby fear fails to be inhibited following hippocampally-mediated pattern separation due to insufficient vmPFC activity, overly strong amygdala activity that eludes vmPFC control, or both.
Further clinical inferences from animal work
During mammalian fear-conditioning, the amygdala activates the nucleus basalis of Meynert27 which then projects to the visual, auditory, and somatosensory cortices—providing acetylcholine to the representation of the CS+ in sensory cortex28. This influx of acetylcholine has been shown to retune receptive fields in sensory cortex toward CS+ attributes, such that an increased number of sensory neurons become responsive to the CS+ (for a review, see29). Importantly, neurons that retune to the CS+ following conditioning have also been shown to retune to stimuli resembling the CS+30 with decreasing levels of retuning as the stimulus becomes less similar to the CS+29. Thus the memorial representation of the post-acquisition CS+ includes an enlarged population of cortical neurons that extends beyond those engaged by the pre-acquisition CS+ to neurons comprising the cortical representation of sensory stimuli similar to the CS+. This conditioning-induced neural overlap between the CS+ representation and its approximations increases the likelihood that post-acquisition exposure to stimuli similar to the CS+ will activate the CS+ representation, via hippocampally-mediated pattern completion, culminating in the neural and behavioral constituents of the fear response (i.e., fear generalization to the CS+ approximate).
As mentioned previously, the nucleus-basalis mediated cholinergic influx into sensory cortex during aversive conditioning follows from fear-related amygdala excitation of the nucleus basalis27. Given findings of increased amygdala activity during fear conditioning in clinical anxiety31, it logically follows that anxiety patients undergoing fear-conditioning may incur greater influx of acetylcholine into sensory cortex, increased neural overlap between the memorial representations of the CS+ and its approximations, and a heightened likelihood of activating the CS+ representation—and the associatively linked fear circuitry—upon exposure to stimuli resembling the CS+. Given the cholinergic nature of this conditioning-dependent plasticity, medications with anticholinergic properties (e.g., scopolamine) may facilitate improved sensory discrimination of the conditioned danger cue and its approximations and may thereby treat overgeneralization of the kind associated with panic disorder.
A second candidate neural mechanism for conditioned generalization gleaned from animal work is the NMDA-dependent rise in intracellular Ca2+ in lateral amygdala neurons, thought essential for the formation of associative links between the CS+ and US7. Consistent with this idea, the partial NMDA agonist D-cycloserine (DCS) has been found to enhance acquisition32 and retention33 of conditioned fear – learning critically reliant on the CS+/US connection. Such results support the potential efficacy of (partial) NMDA agonists, such as DCS, for reducing generalization of conditioned fear by strengthening the accuracy of the conditioning memory and thereby reducing the frequency of “generalization errors.” This possibility receives support from animal findings demonstrating that DCS dose dependently improves acquisition of conditioning and results in less generalization to stimuli resembling the CS+34. Such findings support the potential utility of DCS for reversing overgeneralization associated with clinical anxiety by increasing the strength and accuracy of the encoded conditioning memory – an effect apt for testing given the favorable neurotoxicity profile of DCS in human beings.
VII. Conclusion
Advances in the neurobiology of Pavlovian fear-conditioning are setting the stage for systems neuroscience approaches to conceptualizing and treating clinical anxiety. The scientific traction of fear-conditioning, in this context, is fueled by its conservation across species and demonstrated relevance to clinical anxiety. This convergence of attributes affords a wealth of neurobiological data from elegant, but invasive, animal research with which to hypothesize, test, and interpret candidate neural loci of conditioning aberrancies associated with anxiety disorders. One Pavlovian correlate of anxiety pathology with particular promise is the over-generalization of conditioned fear to stimuli resembling the conditioned danger cue. Animal findings together with emerging human data, implicate a set of interrelated neural processes that may determine levels of generalization. One such process involves hippocampally-mediated pattern separation of CS+ from CS− that may both activate brain areas associated with fear inhibition (vmPFC) and deactivate areas associated with fear excitation (amygdala, anterior insula) resulting in both successful discrimination of CS+ from CS− and little generalization. Additionally, generalization may be subserved in part by cholinergically mediated plasticity in neural representations of the CS+, shown to blur perceptual distinctions between the CS+ and CS−, that then bias the hippocampus toward pattern completion. These ideas represent the beginnings of a neural account of a central conditioning correlate of anxiety pathology that, together with other emerging findings, are giving form to a working, brain-based conceptualization of clinical anxiety with promise for promoting novel, neurally targeted therapeutics.
Acknowledgments
This work was supported by award #R00MH080130 from the National Institute of Mental Health.
References
- 1.Mineka S, Zinbarg R. Conditioning and ethological models of anxiety disorders: stress-in-dynamic-context anxiety models. In: Hope DA, editor. Nebraska symposium on motivation: Vol. 43. Perspectives on anxiety, panic, and fear. Lincoln: University of Nebraska Press; 1996. [PubMed] [Google Scholar]
- 2.Lissek S, Powers AS, McClure EB, Phelps EA, Woldehawariat G, Grillon C, Pine DS. Classical fear-conditioning in the anxiety disorders: A meta-analysis. Behaviour Research and Therapy. 2005;43:1391–1424. doi: 10.1016/j.brat.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Bouton ME, Mineka S, Barlow DH. A modern learning theory perspective on the etiology of panic disorder. Psychol Rev. 2001;108:4–32. doi: 10.1037/0033-295x.108.1.4. [DOI] [PubMed] [Google Scholar]
- 4.Foa EB, Kozak MJ. Emotional processing of fear: exposure to corrective information. Psychol Bull. 1986;99:20–35. [PubMed] [Google Scholar]
- 5.Johansen JP, Cain CK, Ostroff LE, LeDoux JE. Molecular Mechanisms of Fear Learning and Memory. Cell. 2011;147:509–524. doi: 10.1016/j.cell.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE. Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem. 2001;8:229–242. doi: 10.1101/lm.30901. [DOI] [PubMed] [Google Scholar]
- 7.Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004;44:75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 8.Quirk GJ, Garcia R, González-Lima F. Prefrontal mechanisms in extinction of conditioned fear. Biological Psychiatry. 2006;60:337–343. doi: 10.1016/j.biopsych.2006.03.010. (2006) [DOI] [PubMed] [Google Scholar]
- 9.Pape H, Pare D. Plastic Synaptic Networks of the Amygdala for the Acquisition, Expression, and Extinction of Conditioned Fear. Physiol Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis M, Walker DL, Miles L, Grillon C. Phasic vs sustained fear in rats and humans: Role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology Reviews. 2010;35:105–135. doi: 10.1038/npp.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lissek S, Rabin SJ, Heller RE, Luckenbaugh D, Geraci M, Pine DS, Grillon C. Overgeneralization of conditioned fear as a pathogenic marker of panic disorder. American Journal of Psychiatry. 2010;167:47–55. doi: 10.1176/appi.ajp.2009.09030410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lissek S, Grillon C. Learning models of PTSD. In: Beck JG, Sloan DM, editors. The Oxford handbook of traumatic disorders. Oxford University Press; in press. [Google Scholar]
- 13.Lissek S, Biggs AL, Rabin S, Cornwell BR, Alvarez RP, Pine DS, Grillon C. Generalization of conditioned fear-potentiated startle in humans: Experimental validation and clinical relevance. Behav Res Ther. 2008;46:678–87. doi: 10.1016/j.brat.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pavlov IP. Conditioned Reflexes: An Investigation of the Physiological Activity of the Cerebral Cortex. London: Oxford University Press; 1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grillon C, Ameli R, Woods SW, Merikangas K, Davis M. Fear-potentiated startle in humans: effects of anticipatory anxiety on the acoustic blink reflex. Psychophysiology. 1991;28:588–595. doi: 10.1111/j.1469-8986.1991.tb01999.x. [DOI] [PubMed] [Google Scholar]
- 16.Bouton ME, Mineka S, Barlow DH. A modern learning theory perspective on the etiology of panic disorder. Psychol Rev. 2001;108:4–32. doi: 10.1037/0033-295x.108.1.4. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein AJ, Chambless DL. A reanalysis of agoraphobia. Behav Ther. 1978;9:47–59. [Google Scholar]
- 18.Mineka S, Zinbarg R. A contemporary learning theory perspective on the etiology of anxiety disorders: it’s not what you thought it was. Am Psychol. 2006;61:10–26. doi: 10.1037/0003-066X.61.1.10. [DOI] [PubMed] [Google Scholar]
- 19.Dugas MJ, Marchand A, Ladouceur R. Further validation of a cognitive-behavioral model of generalized anxiety disorder: diagnostic and symptom specificity. Journal of Anxiety Disorders. 2005;19:329–343. doi: 10.1016/j.janxdis.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 20.Mineka S. The positive and negative consequences of worry in the aetiology of generalized anxiety disorder: A learning theory perspective. In: Yiend J, editor. Cognition, Emotion and Psychopathology Theoretical, Empirical and Clinical Directions. Cambridge University Press; 2004. pp. 29–48. [Google Scholar]
- 21.Lissek S. Generalization of Pavlovian cue-shock conditioning in the anxiety disorders. In: Pine DS chair, editor. Dimensions of Psychopathology: Implications for Treatment and Research in Anxiety Disorders; Presented as part of the ADAA Annual Research Symposium; New Orleans, LA. 2011. [Google Scholar]
- 22.Solomon PR, Moore JW. Latent inhibition and stimulus generalization of the classically conditioned nictitating membrane response in rabbits (Orcytolagus cuniculus) following dorsal hippocampal ablation. Journal of Comparative and Physiological Psychology. 1975;89:1192–1203. doi: 10.1037/h0077183. [DOI] [PubMed] [Google Scholar]
- 23.Wild JM, Blampied NM. Hippocampal lesions and stimulus generalization in rats. Physiology of Behavior. 1972;9:505–511. doi: 10.1016/0031-9384(72)90005-4. [DOI] [PubMed] [Google Scholar]
- 24.Jarrell T, Gentile C, Romanski L, McCabe P, Schneiderman N. Involvement of cortical and thalamic auditory regions in retention of differential bradycardiac conditioning to acoustic conditioned stimuli in rabbits. Brain Research. 1987;412:285–294. doi: 10.1016/0006-8993(87)91135-8. [DOI] [PubMed] [Google Scholar]
- 25.Teich AH, McCabe PM, Gentile CG, Jarrell TW, Winters RW, Liskowsky DR, et al. Role of auditory cortex in the acquisition of differential heart rate conditioning. Physiology and Behavior. 1988;44:405–412. doi: 10.1016/0031-9384(88)90044-3. (1988) [DOI] [PubMed] [Google Scholar]
- 26.Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S. NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci. 2004;5:361–372. doi: 10.1038/nrn1385. [DOI] [PubMed] [Google Scholar]
- 27.Kapp BS, Whalen PJ, Supple WF, Pascoe JP. Amygdaloid contributions to conditioned arousal and sensory information processing. In: Aggleton J, editor. The Amygdala: Neurobiological Aspects of Emotion, Memory, and Mental Dysfunction. New York: Wiley-Liss; 1992. pp. 229–254. [Google Scholar]
- 28.Weinberger NM. Physiological memory in primary auditory cortex: characteristics and mechanisms. Neurobiology Learning and Memory. 1998;70:226–251. doi: 10.1006/nlme.1998.3850. [DOI] [PubMed] [Google Scholar]
- 29.Weinberger NM. Associative representational plasticity in the auditory cortex: a synthesis of two disciplines. Learning and Memory. 2007;14:1–16. doi: 10.1101/lm.421807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohl FW, Scheich H. Learning-induced dynamic receptive field changes in primary auditory cortex of the unanaesthetized Mongolian gerbil. Journal of Comparative Physiology A: Neuroethology, Sensory, Neural, and Behavioral Physiology. 1997;181:685–696. doi: 10.1007/s003590050150. [DOI] [PubMed] [Google Scholar]
- 31.Bremner JD, Vermetten E, Schmahl C, Vaccarino V, Vythilingam M, Afzal N, et al. Positron emission tomographic imaging of neural correlates of a fear acquisition and extinction paradigm in women with childhood sexual-abuse-related post-traumatic stress disorder. Psychological Medicine. 2005;35:791–806. doi: 10.1017/s0033291704003290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monahan JB, Handelmann GE, Hood WF, Cordi AA. D-cycloserine, a positive modulator of the N-methyl-D-aspartate receptor, enhances performance of learning tasks in rats. Pharmacol Biochem Behav. 1989;34:649–653. doi: 10.1016/0091-3057(89)90571-6. [DOI] [PubMed] [Google Scholar]
- 33.Land C, Riccio DC. d-Cycloserine: Effects on long-term retention of a conditioned response and on memory for contextual attributes. Neurobiology of Learning and Memory. 1999;72:158–168. doi: 10.1006/nlme.1998.3897. [DOI] [PubMed] [Google Scholar]
- 34.Thompson LT, Disterhoft JF. Age- and dose-dependent facilitation of associative eyeblink conditioning by D-cycloserine in rabbits. Behavioral Neuroscience. 1997;111:1303–1312. doi: 10.1037//0735-7044.111.6.1303. [DOI] [PubMed] [Google Scholar]