

Abstract
Recent studies have shown that CXCL1 upregulation in spinal astrocytes is involved in the maintenance of neuropathic pain. However, whether and how CXCL1 regulates inflammatory pain remains unknown. Here we show that intraplantar injection of CFA increased mRNA and protein expressions of CXCL1 and its major receptor CXCR2 in the spinal cord at 6 hours and 3 days after the injection. Immunofluorescence double staining showed that CXCL1 and CXCR2 were expressed in spinal astrocytes and neurons, respectively. Intrathecal injection of CXCL1 neutralizing antibody or CXCR2 antagonist SB225002 attenuated CFA-induced mechanical and heat hypersensitivity on post-CFA day 3. Patch-clamp recordings showed that CXCL1 potentiated NMDA-induced currents in lamina II neurons via CXCR2, and this potentiation was further increased in CFA-treated mice. Furthermore, intrathecal injection of CXCL1 increased COX-2 expression in dorsal horn neurons, which was blocked by pretreatment with SB225002 or MEK (ERK kinase) inhibitor PD98059. Finally, pretreatment with SB225002 or PD98059 decreased CFA-induced heat hyperalgesia and COX-2 mRNA/protein expression and ERK activation in the spinal cord. Taken together, our data suggest that CXCL1, upregulated and released by spinal astrocytes after inflammation, acts on CXCR2-expressing spinal neurons to increase ERK activation, synaptic transmission and COX-2 expression in dorsal horn neurons and contributes to the pathogenesis of inflammatory pain.
Keywords: CXCL1, CXCR2, ERK, COX-2, chemokines, astrocytes, astroglial-neuronal interaction, complete Freund's adjuvant, inflammatory pain
Introduction
Chronic pain resulted from inflammation, infection, nerve injury, or cancer is a major public health problem worldwide. Neuroinflammation, which is mediated by a variety of inflammatory mediators, including cytokines and chemokines, has been recently recognized to play an important role in the pathogenesis of chronic pain (Mennicken et al., 1999; Miller et al., 2008; Scholz and Woolf, 2007; White et al., 2007). Chemokines are a family of small (8–12 kDa) proteins involved in the modulation of numerous biological functions, including leukocyte migration and activation, cell adhesion, and T cell activation via G-protein-coupled receptors (GPCR). There are 4 families of chemokines: C family, CC family, CXC family and CX3C family (Murdoch and Finn, 2000). Recent studies implicated that several chemokines (e.g. CCL2, CX3CL1) are increased in the spinal cord after peripheral nerve injury and involved in the enhancement of neuropathic pain (Gao et al., 2009; Imai et al., 2013; Lindia et al., 2005)
Spinal astrocytes have been demonstrated to be a major source of inflammatory mediators under chronic pain conditions (Gao and Ji, 2010). Our previous study showed that incubation of primary culture of astrocytes with tumor necrosis factor α (TNF-α) induced a marked increase in the levels of several chemokines, including CCL2 and CXCL1 (Gao et al., 2009). It has been shown that CCL2 upregulation in spinal astrocytes is involved in regulating hypersensitivity in spinal nociceptive neurons via its receptor CCR2 and contributes to central sensitization and chronic pain (Gao et al., 2009). CCL2 can also be released from primary afferents to activate spinal microglia (Thacker et al., 2009; Zhang and De Koninck, 2006; Zhang et al., 2007). Compared to well-investigated role of CCL2 in pain regulation (Gao et al., 2009; Gosselin et al., 2005; Guo et al., 2012; Jung et al., 2009; Jung et al., 2008; Zhang and De Koninck, 2006; Zhang et al., 2007), little is known about the involvement of CXCL1 in persistent pain.
CXCL1 is a member of CXC family and also known as keratinocyte-derived chemokines (KC) or growth-related oncogene (GRO). CXCR2, the primary receptor of CXCL1 (Savarin-Vuaillat and Ransohoff, 2007) has been detected on neurons, microglia, and oligodendrocyte progenitors in the brain (Horuk et al., 1997; Nguyen and Stangel, 2001; Popivanova et al., 2003; Valles et al., 2006). Interestingly, CXCR2 is predominantly expressed in neurons of the spinal cord and increased after spinal nerve ligation (SNL) and paw incision (Sun et al., 2013; Zhang et al., 2013). Our recent study showed that intrathecal injection of CXCL1 induced rapid CXCR2-dependent activation of ERK (Zhang et al., 2013), a marker for central sensitization (Gao and Ji, 2009; Ji et al., 1999) in spinal neurons. These data suggest CXCL1 and CXCR2 are involved in astroglial-neuronal interaction in the spinal cord under chronic pain conditions. However, whether CXCL1 can regulate inflammatory pain, synaptic transmission, and gene expression in the spinal cord remains unclear.
In the present study, we investigated whether CXCL1 has a role in inflammatory pain using the well-established complete Freunds's adjuvant (CFA) model. We also examined the mRNA and protein expression for CXCL1 and CXCR2 and their cellular localization in the spinal cord. We further investigated the direct role of CXCL1/CXCR2 in regulating excitatory synaptic transmission and cyclooxygenase-2 (COX-2) expression in the spinal cord.
Materials and Methods
Animals and surgery
Adult ICR mice were (male, 7–8 weeks) purchased from the Experimental Animal Center of Nantong University. CD1 mice (male, 4–6 weeks) were purchased from Charles River Laboratory for the electrophysiology study. The animals were maintained at room temperature of 22 ± 1°C, humidity of 55 ± 10%, and a 12:12 light–dark cycle with free access to food and water. All animal procedures in this study were performed according to the guidelines of the International Association for the Study of Pain and were approved by the Animal Care and Use Committee of Nantong University or the Animal Care Committee of Duke University. Peripheral inflammation was induced by intraplantar injection of CFA (20 μl, Sigma, St Louis, MO) in the left hind paws under brief anesthesia with isofluorane. The sham-treated animals were injected same volume of normal saline.
Drugs and administration
SB225002, a potent and selective antagonist of CXCR2, was purchased from Tocris (Bristol, UK). MEK inhibitor PD98059 was purchased from Merck KGaA (Darmstadt, Germany). The CXCL1 neutralizing antibody was purchased from Boster (Wuhan, China). For intrathecal injection, spinal cord puncture was made with a 30 G needle between the L5 and L6 level to deliver the reagents to the cerebral spinal fluid (Hylden and Wilcox, 1980).
Real-time PCR (RT-PCR)
Total RNA was extracted from L4-5 spinal cord with the Trizol reagent (Invitrogen, Carlsbad, CA). One microgram of total RNA was converted into cDNA using PrimeScript RT reagent kit (Takara, Otsu, Shiga, Japan). The cDNA was amplified using the following primers: CXCL1 forward, 5'-GCT TGA AGG TGT TGC CCT CAG -3'; CXCL1 reverse, 5'-AGA AGC CAG CGT TCA CCA GAC-3'; CXCR2 forward, 5'-TCT GCT CAC AAA CAG CGT CGT A-3'; CXCR2 reverse, 5'-GAG TGG CAT GGG ACA GCA TC-3'; COX-2 forward, 5'-CTC CTG GAA CAT GGA CTC AC TCA-3'; COX-2 reverse, 5'-AGG CCT TTG CCA CTG CTT GTA-3'. GAPDH forward, 5'-AAA TGG TGA AGG TCG GTG TGA AC-3'; GAPDH reverse, 5'-CAA CAA TCT CCA CTT TGC CAC TG-3'. The SYBR Premix Ex Taq II kit (Takara) was used for all PCR reactions. The PCR reactions were run on a Rotor-Gene 3000 RT-PCR machine (Corbett Research, Mortlake, Australia). The PCR amplifications were performed at 95°C for 30 seconds, followed by 45 cycles at 95°C for 5 seconds, 56°C for 30 seconds, and 72°C for 30 seconds. The melting curves were performed to validate the utility and specificity of each PCR product. The data were analyzed using Rotor-Gene 6000 series software, and evaluated using the Comparative CT Method (2−ΔΔCT).
ELISA
Mouse CXCL1 ELISA kit was purchased from R&D systems (Minneapolis, MN). Animals were transcardially perfused with PBS at 6 h and 3 d after CFA or saline injection. The lumbar spinal cord segments were dissected. The tissues were homogenized in a lysis buffer containing protease and phosphatase inhibitors. Protein concentrations were determined by BCA Protein Assay (Thermal Fisher, Rockford, IL). For each reaction in a 96-well plate, 100 μg of proteins were used, and ELISA was performed according to manufacturer's protocol. The standard curve was included in each experiment.
Immunohistochemistry
After appropriate survival times, animals were deeply anesthetized with isoflurane and perfused through the ascending aorta with PBS followed by 4% paraformaldehyde with 1.5% picric acid in 0.16 M PB. After the perfusion, the L4-L5 spinal cord segments were removed and postfixed in the same fixative overnight. Spinal cord sections (30 μm, free-floating) were cut in a cryostat and processed for immunofluorescence as we described previously (Gao et al., 2009). The sections were first blocked with 2% donkey serum for 1 h at room temperature. The sections were then incubated overnight at 4°C with the following primary antibodies: CXCL1 antibody (rabbit, 1:100; Boster, Wuhan, China), CXCR2 antibody (rabbit, 1:100; Boster), GFAP antibody (mouse, 1:6000; Millipore, Billerica, MA), CD11b antibody (mouse, 1:200, AbD serotec, Raleigh, NC), neuronal-specific nuclear protein (NeuN) antibody (mouse, 1:800, Millipore), COX-2 (rabbit, 1:500, Boster, China). The sections were then incubated for 1 h at room temperature with Cy3- or FITC-conjugated secondary antibodies (1:1000, Jackson ImmunoResearch, Westgrove, PA). For double immunofluorescence, sections were incubated with a mixture of mouse and rabbit primary antibodies followed by a mixture of Cy3- and FITC-conjugated secondary antibodies. The stained sections were examined with a Leica fluorescence microscope, and images were captured with a Charge-coupled Device Spot camera.
Western blot
Protein samples were prepared in the same way as for ELISA analysis, and 30 μg of proteins were loaded for each lane and separated on SDS-PAGE gel (10%). After the transfer, the blots were incubated overnight at 4°C with antibody against CXCR2 (1:100, rabbit, Boster), COX-2 (1:500, rabbit, Cell Signaling), pERK (1:1000, rabbit, Cell Signaling). For loading control, the blots were incubated with GAPDH antibody (1:20000, mouse, Sigma). These blots were further incubated with HRP-conjugated secondary antibody, developed in ECL solution (Thermo Scientific), and exposed onto film (Millipore) for 1–5 min. Specific bands were evaluated by apparent molecular size. The intensity of the selected bands was analyzed using Image J software (NIH, Bethesda, MD).
Spinal slice preparation and patch-clamp recordings
Young mice (4–6 weeks old) were anesthetized with urethane (50 mg/kg, i.p.), and a portion of the lumbar spinal cord (L4 –L5) was removed and kept in pre-oxygenated ice-cold Krebs' solution. Transverse spinal cord slices (350–450 μm thick) were cut on a vibrating microslicer. The slices were stored in an incubation solution at room temperature (in mM: NaCl, 95; KCl, 1.8; KH2PO4, 1.2; CaCl2, 0.5; MgSO4, 7; NaHCO3, 26; glucose, 15; sucrose, 50; oxygenated with 95% O2, 5% CO2; pH 7.4). A slice was then transferred into a recording chamber and perfused with oxygenated recording solution at 3 ml/min at room temperature. The recording solution (Mg2+ free) was identical to the incubation solution except for (in mM): NaCl 127, CaCl2 2.4, MgSO4 0 and sucrose 0.
The whole-cell patch-clamp recordings were made from lamina IIo neurons in voltage-clamp mode as we previously reported (Park et al., 2011). Under a dissecting microscope with transmitted illumination, the substantia gelatinosa (lamina II) is clearly visible as a relatively translucent band across the dorsal horn. Standard whole-cell patch clamp recordings were performed with glass pipettes having a resistance of 5–10 MΩ in lamina IIo of spinal dorsal horn. The pipette solution consisted of (in mM): K-gluconate, 135; KCl, 5; CaCl2, 0.5; MgCl2, 2; EGTA, 5; HEPES, 5 and Mg-ATP, 5, pH 7.4 with KOH, measured osmolarity 300 mOsm. To measure NMDA-induced currents, the membrane potential was held at −40 mV and NMDA (50 μM) was puffed into the solution. CXCL1 (100 ng/ml) and SB225002 (0.1 μM) were given in the same way. The electrophysiological properties of the recorded neurons were investigated in voltage-clamp modes using an Axon 700B amplifier and pClamp10.0 data acquisition software. Signals were low-pass filtered at 5 kHz, sampled at 10 kHz and analyzed offline (Baba et al., 2003; Kawasaki et al., 2008).
Behavioral analysis
Animals were habituated to the testing environment daily for at least two days before baseline testing. The room temperature and humidity remained stable for all experiments. For testing mechanical sensitivity, animals were put in boxes on an elevated metal mesh floor and allowed 30 min for habituation before examination. The plantar surface of each hindpaw was stimulated with a series of von Frey hairs with logarithmically incrementing stiffness (0.02–2.56 grams, Stoelting, Wood Dale, IL), presented perpendicular to the plantar surface (2–3 seconds for each hair, 3 min interval between the tests). The 50% paw withdrawal threshold was determined using Dixon's up-down method (Chaplan et al., 1994). For testing heat sensitivity, animals were put in plastic boxes and allowed 30 min for habituation before examination. Heat sensitivity was tested by radiant heat using Hargreaves apparatus (IITC Life Science Inc., Woodland Hills, CA) and expressed as paw withdrawal latency (PWL). The test was repeated at least three times/animal allowing at least 5 min in between each test. The radiant heat intensity was adjusted so that basal PWL is between 10–14 seconds, with a cut-off of 18 seconds to prevent tissue damage.
Quantification and statistics
All data were expressed as mean ± SEM. The behavioral data was analyzed by two-way analysis of variance (ANOVA) followed by Bonferroni test as the multiple comparison analysis. The qPCR and ELISA data was analyzed by one-way ANOVA followed by Newman-Keuls post hoc test. For western blot, the density of specific bands was measured with Image J (NIH). COX-2 and pERK levels were normalized to loading control (GAPDH). Differences between two groups were compared using Student's t-test. The criterion for statistical significance was P < 0.05.
Results
CFA induces CXCL1 expression in spinal cord astrocytes
Unilateral injection of 20 μl CFA into a hindpaw of mice produces rapid and persistent inflammatory pain (Gao et al., 2010). To check the expression of CXCL1, we collected the spinal cord (L4-5) at 6 h and 3 d after CFA or saline injection. As shown in Fig.1A, CXCL1 mRNA was increased at both 6 h (4.0 ± 0.8 fold compared to sham, P < 0.05) and 3 d (2.7 ± 0.7 fold, P < 0.01) after CFA injection. CXCL1 mRNA was not changed after saline injection (Fig. 1A). Elisa results showed low level CXCL1 expression in the spinal cords of naïve animals (6.8 ± 0.4 pg/ml). CFA increased CXCL1 at 6 h (11.1 ± 0.5 pg/ml, P < 0.01) and 3 d (10.3 ± 0.6 pg/ml, P < 0.01, Fig. 1B). Saline injection had no effects on CXCL1 protein expression at the time points examined (Fig. 1B).
Figure 1.

CFA induces CXCL1 mRNA and protein expression in the spinal cord. (A) Real-time PCR results show the increase of CXCL1 mRNA expression in the spinal cord at 6 h and 3 d after CFA injection. * P < 0.05, compared to naïve. # P < 0.05, compared to saline. n = 4 mice/group. (B) Elisa results show that CXCL1 protein expression was also increased in the spinal cord after CFA. ** P < 0.01, compared to naïve. ## P < 0.01, compared to saline. n = 5 mice/group. (C–F) Immunostaining shows the CXCL1 expression in the spinal cord in naïve (C) and CFA (D) animals. CFA increased CXCL1-IR in the spinal cord at 3 days after injection (D). (E–G) Double staining shows CXCL1 was colocalized with astrocytic marker, GFAP (E), but not with neuronal marker NeuN (F) or microglial marker CD11b (G).
We then checked the distribution of CXCL1 in the spinal cord by immunofluorescence. CXCL1 was expressed in naïve animals (Fig.1C). CFA induced a marked increase of CXCL1-immunoreactive (IR) in the ipsilateral side of the lumbar spinal cord on post-CFA day 3 (Fig. 1D). CXCL1-IR cells were mainly found in the superficial layers (laminae I–III) of the dorsal horn. To define the cellular distribution of CXCL1, we performed double staining of CXCL1 with different cell markers. CXCL1-IR was colocalized with the astrocytic marker GFAP (Fig. 1E), but not with neuronal marker NeuN (Fig. 1F) or microglial marker CD11b (Fig. 1G), suggesting the localization of CXCL1 in spinal astrocytes.
CFA induces CXCR2 upregulation in spinal cord neurons after CFA injection
Since CXCR2 is the major receptor of CXCL1 (Savarin-Vuaillat and Ransohoff, 2007), we next examined CXCR2 expression in the spinal cord after CFA injection. CXCR2 mRNA was increased at 6 h (1.9 ± 0.2 fold compared to sham) and 3 d (2.9 ± 0.3 fold, P < 0.01, Fig. 2A). Western blot also showed a marked increase of CXCR2 (1.3 ± 0.03 fold) at 3 days after CFA injection compared to naïve (Fig. 2B). We further did immunostaining on spinal cord sections. In naïve animals, there was only low level expression of CXCR2 in the dorsal horn (Fig. 2C). At 3 days after CFA, CXCR2-IR was significantly increased in both the superficial and deep dorsal horn (Fig. 2D). To further test the cellular distribution of CXCR2, we did immunofluorescence double staining of CXCR2 and NeuN. The result showed that many CXCR2-IR cells were also NeuN-positive in the superficial and deep dorsal horn (Fig. 2E–G), suggesting that CXCR2 is expressed by spinal neurons.
Figure 2.

CFA induces CXCR2 mRNA and protein expression in the spinal cord. (A) Real-time PCR results show the increase of CXCR2 mRNA expression in the spinal cord at 6 h and 3 d after CFA injection. ** P < 0.01, compared to naïve. ## P < 0.01, compared to saline. n = 4 mice/group. (B) Western blot show CXCR2 protein expression in the spinal cord. CXCR2 increased 1.3-fold after CFA injection compared to naïve (P < 0.05). n = 3 mice/group. (C, D) Immunostaining shows the CXCR2 expression in the spinal cord in naïve (C) and CFA (D) animals. CXCR2-IR was enhanced at 3 days after CFA (D). (E–G) Double staining shows CXCR2 was colocalized with neuronal marker NeuN.
Intrathecal injection of CXCL1 neutralizing antibody or CXCR2 antagonist 3 days after inflammation attenuates inflammatory pain
We then checked whether inhibition of CXCL1 or CXCR2 could reverse CFA-induced inflammatory pain. Firstly, we intrathecally injected a CXCL1 neutralizing antibody at 3 days after CFA. CXCL1 neutralizing antibody, at the dose of 4 μg reduced CFA-induced mechanical allodynia at 3 h and 6 h (Fig. 3A) and CFA-induced heat hyperalgesia from 1 h to 24 h (Fig. 3B), whereas a higher dose CXCL1 neutralizing antibody (8 μg) attenuated both mechanical allodynia and heat hyperalgesia from 1 h to 24 h. These data suggest a dose-dependent inhibition of inflammatory pain by the CXCL1 neutralization.
Figure 3.
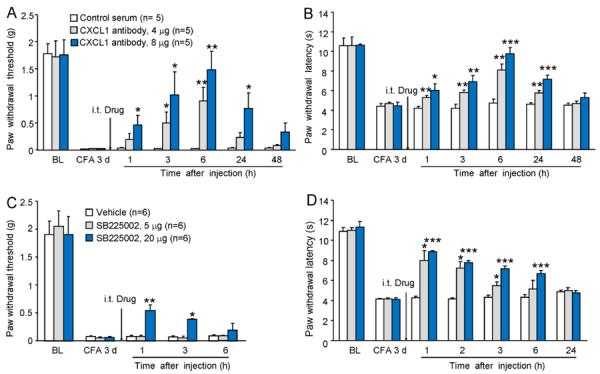
Intrathecal injection of CXCL1 neutralizing antibody or SB225002, a potent and selective CXCR2 antagonist, reduces CFA-induced mechanical allodynia and heat hyperalgesia at 3 days after CFA. (A, B) CXCL1 neutralizing antibody at a lower dose (4 μg) had mild effect on CFA-induced pain hypersensitivity, whereas at a higher dose (8 μg) the neutralizing antibody reversed CFA-induced mechanical allodynia (A) and heat hyperalgesia (B) for more than 24 hours. * P < 0.05, ** P < 0.01,*** P < 0.001 compared to control serum. BL, baseline. n = 6 mice/group. (C, D) SB2205002 dose-dependently attenuated CFA-induced mechanical allodynia (C) and heat hyperalgesia (D) at 3 days, with longer effect on the heat hyperalgesia. * P < 0.05; ** P < 0.01; *** P < 0.001 compared to vehicle. n = 6 mice/group.
We then checked the effect of CXCR2 antagonist on CFA-induced inflammatory pain at 3 days. SB225002 (5 μg and 20 μg), a selective and potent CXCR2 antagonist was intrathecally injected at 3 days after CFA. At a lower dose (5 μg) SB225002 had no effect on CFA-induced mechanical allodynia (P > 0.05, Fig. 3C), but attenuated CFA-induced heat hyperalgesia from 1 h (P < 0.05) to 3 h (P < 0.05) after intrathecal injection (Fig. 3D). At a higher dose (20 μg), intrathecal SB225002 significantly attenuated CFA-induced mechanical allodynia at 1 h (P < 0.01) and 3 h (P < 0.05) and heat hyperalgesia from 1 h to 6 h (P < 0.001, Fig. 3C, D).
CXCL1 increases excitatory synaptic transmission in spinal neurons
As CXCR2 was expressed in spinal neurons, we reasoned that CXCL1 would directly regulate spinal cord synaptic transmission via CXCR2. We prepared spinal cord slices from naïve and CFA-injected mice and performed patch-clamp recordings in lamina IIo neurons in which many nociceptive neurons are localized (Baba et al., 2003; Gao et al., 2009). Because NMDA receptors play an important role in mediating excitatory synaptic transmission and central sensitization, we examined the effect of the CXCL1 on NMDA-induced inward currents when holding the voltage at - 40 mV. In spinal cord slices from naïve animals, CXCL1 (100 ng/ml) significantly enhanced the inward currents elicited by NMDA (50 μM for 30 s, 1.27 ± 0.08 fold increase compared to NMDA alone; P < 0.05), which was blocked by SB225002 (0.1 μM) (Fig. 4A, B). In slices from CFA (3 d)-inflamed mice, NMDA induced greater inward currents than in naïve animals (−110 ± 2.4 pA for naïve vs. −212 ± 3.2 pA for CFA-3 d, P < 0.05, Fig. 4C, D). In addition, the enhancement by CXCL1 is stronger in CFA-treated animals than in naïve animals (1.27 ± 0.08 fold for naïve vs 2.63 ± 0.7 fold for CFA 3 d, P < 0.05, Fig. 4C, E). Together, these data indicate that CXCL1 can directly regulate excitatory synaptic transmission via CXCR2.
Figure 4.

Bath application of CXCL1 increases NMDA-induced currents in lamina IIo neurons in spinal cord slices. (A) Patch-clamp recording demonstrates increase in NMDA-induced currents after CXCL1 treatment (100 ng/ml, 2 min), which is blocked by SB225002 (0.1 μM). (B) Histogram shows the amplitude of NMDA-induced currents. * P < 0.05 compared to NMDA alone. # P < 0.05, compared to NMDA + CXCL1 treatment. n = 4–7 neurons/group. (C) Patch-clamp recording shows CXCL1 increases NMDA-induced current in naïve and CFA-treated animals. (D) Histogram shows the NMDA-induced inward current. * P < 0.05 compared to naïve. (E) Histogram shows the increased magnitude of NMDA-induced currents. * P < 0.05 compared to pretreatment baseline. # P < 0.05, CFA 3 days compared to naïve. n = 5–7 neurons/group.
Intrathecal injection of CXCL1 induces CXCR2- and ERK-dependent COX-2 upregulation
Our previous studies have shown that intrathecal injection of CXCL1 induced heat hyperalgesia and CXCR2-dependent ERK activation in spinal neurons (Zhang et al., 2013). To further examine the down-stream mechanisms of ERK activation, we examined COX-2 expression after CXCL1 treatment. As shown in Fig.5, intrathecal injection of CXCL1 (100 ng/ml) dramatically increased COX-2 expression in the dorsal horn of the spinal cord (Fig. 5A–C). Double staining showed that most COX-2-IR was colocalized with neuronal marker NeuN (Fig. 5D). RT-PCR further showed that COX-2 mRNA was significantly increased by CXCL1 (3.6 ± 0.5 fold compared to naive, P < 0.05). Pretreatment with SB225002, or ERK kinase (MEK) inhibitor PD98059 decreased CXCL1-induced COX-2 mRNA upregulation (SB225002, 2 ± 0.1 fold, P < 0.05; PD98059, 1.6 ± 0.2 fold, P < 0.05; Fig. 5E). These data suggest CXCL1 could induce CXCR2- and ERK-dependent COX-2 expression in spinal neurons.
Figure 5.

CXCL1 increases the expression of COX-2 in spinal cord neurons. (A–C) Intrathecal injection of CXCL1 (100 ng) increases COX-2 expression at 2 h after injection. (D) Majority of COX-2 immunoreactive cells in spinal cord express the neuronal marker NeuN. (E) CXCL1 also increased COX-2 mRNA expression in the spinal cord, which is blocked by pretreatment with SB225002 (20 μg) or PD98059. * P < 0.05, ** P < 0.01 compared to PBS. # P < 0.05 compared to CXCL1, one-way ANOVA followed by Newman-Keuls post hoc test. n = 4 mice/group.
Pretreatment with SB2205002 or PD98059 prevents CFA-induced heat hyperalgesia and COX-2 upregulation in the spinal cord
To further check whether the CXCR2/ERK pathway is involved in CFA-induced COX-2 expression in the spinal cord, we intrathecally injected SB225002 (20 μg) or PD98059 (10 μg) at 1 h before CFA injection. Behavioral results showed that both SB225002 and PD98059 partly prevented CFA-induced heat hyperalgesia (P < 0.001, Fig. 6A) at 6 h. CFA dramatically increased COX-2 mRNA expression (5.7 ± 0.5 fold compared to naive, P < 0.001). Pretreatment of SB225002 or PD98059 decreased CFA-induced COX-2 mRNA expression in the spinal cord (3.5 ± 0.7 fold for SB225002 treatment, P < 0.05; 3.4 ± 0.5 fold for PD98059 treatment, P < 0.05, Fig. 6B). Western blot further showed that pretreatment with SB225002 or PD98059 decreased CFA-induced COX-2 upregulation and ERK1/ERK2 activation in the spinal cord (Fig. 6C–E). These data suggest the CXCR2/ERK pathway is involved in COX-2 upregulation and the development of CFA-induced pain hypersensitivity.
Figure 6.
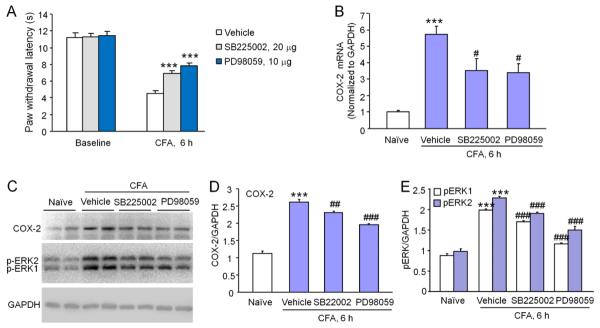
CFA induces CXCR2/ERK dependent COX-2 increase in the spinal cord. (A) Intrathecal pretreatment with SB225002 or PD98059 partly prevented CFA-induced heat hyperalgesia. *** P < 0.001 compared to vehicle. Student's t-test. n=6 mice/group. (B) Intrathecal pretreatment with SB225002 or PD98059 decreased CFA-induced COX-2 mRNA increase in the spinal cord. *** P < 0.001 compared to naive. # P < 0.01, compared to vehicle. n=6 mice/group. (C) Intrathecal pretreatment with SB225002 or PD98059 reduced CFA-induced COX-2 protein expression and pERK1/2 activation in the spinal cord. (D, E) Density of COX-2 bands (D) and pERK1, pERK2 bands (E), which is normalized to GAPDH loading control. *** P < 0.001 compared to naive. ## P < 0.01, ### P < 0.001 compared to vehicle. One-way ANOVA followed by Newman-Keuls post hoc test. n = 4 mice/group.
Discussion
Our recent study has shown (1) spinal nerve ligation-induced neuropathic pain is associated with a slow (3 d) but persistent (> 21 d) CXCL1 increase, which is secondary to TNF-α increase in the spinal cord and (2) CXCL1 is involved in the maintenance of neuropathic pain (Zhang et al., 2013). Different from these results, here we show CFA induced a rapid increase of CXCL1 and its major receptor CXCR2 in the spinal cord. Behavioral tests further showed that intrathecal injection of either CXCL1 neutralizing antibody or CXCR2-specific antagonist SB225002 attenuated CFA-induced pain hypersensitivity at 3 days. In addition, we for the first time examined the direct effect of CXCL1 on excitatory synaptic transmission and found that CXCL1 increased NMDA-induced currents via CXCR2 in the lamina IIo spinal cord neurons, and the increase was further potentiated after inflammation. We also explored the downstream mechanisms by which CXCR2 modulates inflammatory pain. Our results showed that intrathecal injection of CXCL1 induced COX-2 upregulation in spinal cord neurons, via the activation of the CXCR2/ERK pathway. Intrathecal injection of CXCR2 antagonist or MEK inhibitor partly prevented CFA-induced pain hypersensitivity, COX-2 expression and ERK activation in the spinal cord.
Rapid upregulation of CXCL1 in spinal astrocytes and the involvement of CXCL1 in the pathogenesis of inflammatory pain
It has been reported that CXCL1 expression is regulated in pathological pain conditions. CXCL1 expression is increased in the DRG at 3 days but not at 7 days after spinal nerve ligation and localized inflammation (Li et al., 2007; Xie et al., 2006). McTigue et al. reported that contusion injury of spinal cord induced 30-fold increase of CXCL1 mRNA in the spinal cord at 6 h post injury, which decayed rapidly thereafter (McTigue et al., 1998). Here we show that CFA induced mechanical alldodynia and heat hyperalgesia, which was associated with increased CXCL1 mRNA and protein expression, indicating a possible role of CXCL1 in CFA-induced inflammatory pain.
Although CXCL1 was shown to be expressed in brain neurons after epilepsy in rats (Johnson et al., 2011), our recent double-staining study of immunofluorescence or in situ hybridization and immunofluorescence showed that CXCL1 protein and mRNA were predominantly expressed in spinal astrocytes (Zhang et al., 2013). The immunostaining in this study further confirmed the expression of CXCL1 in spinal astrocytes, which was also consistent with some other studies. Pineau et al. reported that CXCL1 mRNA is upregulated in spinal astrocytes after spinal cord injury in mice (Pineau et al., 2010). CXCL1 is also induced in brain astrocytes by neuronal injury and intracerebroventricular administration of endothelin-1 (Katayama et al., 2009; Koyama et al., 2007). In humans, CXCL1 is selectively expressed in hypertropic astrocytes after active multiple sclerosis lesions (Omari et al., 2006; Omari et al., 2005). These data suggest CXCL1 may act as an astroglial mediator to modulate neuronal functions and neuroinflammation in the spinal cord and brain.
In parallel to the upregulation of CXCL1 mRNA and protein in spinal cord after inflammation, intrathecal injection of the CXCL1 neutralizing antibody 3 days after CFA injection attenuated CFA-induced mechanical allodynia and heat hyperalgesia for more than 24 h. In contrast, our previous data showed that CXCL1 neutralizing antibody only slightly attenuated SNL-induced mechanical allodynia and heat hyperalgesia for 1 h (Zhang et al., 2013), suggesting different roles of CXCL1 in inflammatory pain and neuropathic pain.
CXCL1/CXCR2 signaling and central sensitization
Chemokines act through a family of seven transmembrane G protein-coupled receptors to exert their biological effects (Cartier et al., 2005; Savarin-Vuaillat and Ransohoff, 2007). CXCR2 is the major receptor of CXCL1 (Savarin-Vuaillat and Ransohoff, 2007). Of interest, a recent study has shown that paw incision increased spinal mRNA level of CXCL1 and CXCR2, as a result of histone acetylation (Sun et al., 2013). Here we demonstrated that CFA also increased the expression of CXCL1 and CXCR2 mRNA and protein in the spinal cord dorsal horn. In addition, intrathecal injection of CXCR2 antagonist SB225002 dose-dependently alleviated CFA-induced mechanical allodynia and heat hyperalgesia. Repeated intraperitoneal injections of SB225002 displayed long-lasing antinociceptive effect following CFA injection or partial spinal nerve ligation (Manjavachi et al., 2010). Consistently, intrathecal injection of SB225002 attenuates SNL-induced pain hypersensitivity (Zhang et al., 2013), incision-induced mechanical allodynia (Sun et al., 2013), and carrageenan-induced mechanical hypernociception (Manjavachi et al., 2010), suggesting an important role of CXCR2 in both inflammatory and neuropathic pain. However, low dose of SB225002 attenuated CFA-induced heat hyperalgesia, but not mechanical allodynia, indicating that heat hyperalgesia and mechanical allodynia are mediated by different mechanisms (Gao et al., 2010). Since central sensitization plays a more important role in generating mechanical allodynia (Ji et al., 2003), our result also suggests CXCL1/CXCR2 drives inflammatory pain via central sensitization.
In the DRG, CXCR2 is expressed in neurons. In addition, CXCL1 increases the sodium currents, potassium currents in small diameter rat sensory neurons (Dong et al., 2012; Wang et al., 2008; Yang et al., 2009). Intrathecal injection of CXCL1 induced CXCR2-dependent heat hyperalgesia (Zhang et al., 2013). The neuronal expression of CXCR2 in the dorsal horn suggests that CXCL1 may directly regulate neuronal activity in the dorsal horn. This was supported by our electrophysiological results showing that spinal application of CXCL1 increased NMDA-induced currents via CXCR2. Moreover, the enhancement of NMDA-induced currents by CXCL1 was greater in CFA-treated animals than in naïve animals. Hyperactivity of NMDA receptors is the best known mechanism for the induction and maintenance of central sensitization (Ji et al., 2003; Woolf and Salter, 2000). These data suggest that CXCL1/CXCR2 may regulate central sensitization and inflammatory pain through astroglial-neuronal interactions in the spinal cord.
CXCL1 induces CXCR2/ERK dependent COX-2 expression in spinal neurons
Our recent studies showed that intrathecal injection of CXCL1 induced rapid ERK and CREB activation and c-Fos expression mainly in spinal cord neurons (Zhang et al., 2013). The ERK activation plays an important role in neuronal plasticity and central sensitization (Ji et al., 1999; Karim et al., 2001). Translocation of pERK to the nucleus activates the transcription factor CREB to initiate gene transcription and maintain central sensitization and chronic pain (Ji and Strichartz, 2004). CREB binding sites (CREs) are shown in the promoter regions of the genes encoding c-Fos, COX-2, NK-1, prodynorphin, and TrkB, which are important for the genesis of chronic pain (Ji and Strichartz, 2004; Simonetti et al., 2013). It was well-known that COX-2 was upregulated in spinal neurons after CFA injection and contributes to inflammatory pain (Lee et al., 2004; Samad et al., 2001; Seybold et al., 2003). In this study, we further demonstrated that CXCL1 is sufficient to induce COX-2 expression via the activation of the CXCR2/ERK pathway in the spinal cord. Consistently, CFA-induced COX-2 was reduced by pretreatment with CXCR2 antagonist and MEK inhibitor. These data suggest CXCL1 may be involved in the development of central sensitization and inflammatory pain in part through induction of pronociceptive genes such as Cox2.
In summary, we have demonstrated a distinct role of CXCL1/CXCR2 signaling in CFA-induced inflammatory pain. Different from its role in the maintenance of neuropathic pain, CXCL1/CXCR2 mainly contributes to the development of inflammatory pain. Moreover, CXCL1 may mediate astroglial-neuronal interaction in the spinal cord and drive central sensitization via enhancing the activity of NMDA receptors and inducing ERK-dependent COX-2 expression. Targeting this signaling pathway may help to alleviate inflammatory pain.
Highlights.
CFA induces upregulations of CXCL1 and CXCR2 in the spinal cord.
CXCL1 and CXCR2 are respectively expressed in spinal astrocytes and neurons.
Inhibition of CXCL1 or CXCR2 alleviates CFA-induced inflammatory pain.
CXCL1 increases NMDA-induced currents of lamina II neurons and induces CXCR2/ERK dependent COX-2 expression in spinal neurons.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (NSFC) 31171062, 31371121, Natural Science Research Program of Jiangsu Province 13KJB180016, the Priority Academic Program Development of Jiangsu Higher Education Institutions, and NIH grants DE17794 and DE22743.
Abbreviations
- CFA
complete Freund's adjuvant
- COX-2
cyclooxygenase-2
- ERK
extracellular signal-regulated kinase
- GFAP
glial fibrillary acidic protein
- NeuN
neuron-specific nuclear protein
- NMDA
N-methyl-d-aspartate
- RT-PCR
real-time quantitative polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baba H, Ji RR, Kohno T, Moore KA, Ataka T, Wakai A, Okamoto M, Woolf CJ. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Molecular and cellular neurosciences. 2003;24:818–830. doi: 10.1016/s1044-7431(03)00236-7. [DOI] [PubMed] [Google Scholar]
- Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. Journal of neuroscience methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Dong F, Du YR, Xie W, Strong JA, He XJ, Zhang JM. Increased function of the TRPV1 channel in small sensory neurons after local inflammation or in vitro exposure to the pro-inflammatory cytokine GRO/KC. Neurosci Bull. 2012;28:155–164. doi: 10.1007/s12264-012-1208-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. c-Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? Open Pain J. 2009;2:11–17. doi: 10.2174/1876386300902010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Targeting astrocyte signaling for chronic pain. Neurotherapeutics. 2010;7:482–493. doi: 10.1016/j.nurt.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Xu ZZ, Liu YC, Wen YR, Decosterd I, Ji RR. The c-Jun N-terminal kinase 1 (JNK1) in spinal astrocytes is required for the maintenance of bilateral mechanical allodynia under a persistent inflammatory pain condition. Pain. 2010;148:309–319. doi: 10.1016/j.pain.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29:4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin RD, Varela C, Banisadr G, Mechighel P, Rostene W, Kitabgi P, Melik-Parsadaniantz S. Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurones. Journal of neurochemistry. 2005;95:1023–1034. doi: 10.1111/j.1471-4159.2005.03431.x. [DOI] [PubMed] [Google Scholar]
- Guo W, Wang H, Zou S, Dubner R, Ren K. Chemokine signaling involving chemokine (C-C motif) ligand 2 plays a role in descending pain facilitation. Neurosci Bull. 2012;28:193–207. doi: 10.1007/s12264-012-1218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horuk R, Martin AW, Wang Z, Schweitzer L, Gerassimides A, Guo H, Lu Z, Hesselgesser J, Perez HD, Kim J, Parker J, Hadley TJ, Peiper SC. Expression of chemokine receptors by subsets of neurons in the central nervous system. J Immunol. 1997;158:2882–2890. [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. European journal of pharmacology. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Imai S, Ikegami D, Yamashita A, Shimizu T, Narita M, Niikura K, Furuya M, Kobayashi Y, Miyashita K, Okutsu D, Kato A, Nakamura A, Araki A, Omi K, Nakamura M, James Okano H, Okano H, Ando T, Takeshima H, Ushijima T, Kuzumaki N, Suzuki T. Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long-lasting neuropathic pain. Brain. 2013;136:828–843. doi: 10.1093/brain/aws330. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nature neuroscience. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends in neurosciences. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004;2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Johnson EA, Dao TL, Guignet MA, Geddes CE, Koemeter-Cox AI, Kan RK. Increased expression of the chemokines CXCL1 and MIP-1alpha by resident brain cells precedes neutrophil infiltration in the brain following prolonged soman-induced status epilepticus in rats. Journal of neuroinflammation. 2011;8:41. doi: 10.1186/1742-2094-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Bhangoo S, Banisadr G, Freitag C, Ren D, White FA, Miller RJ. Visualization of chemokine receptor activation in transgenic mice reveals peripheral activation of CCR2 receptors in states of neuropathic pain. J Neurosci. 2009;29:8051–8062. doi: 10.1523/JNEUROSCI.0485-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Toth PT, White FA, Miller RJ. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. Journal of neurochemistry. 2008;104:254–263. doi: 10.1111/j.1471-4159.2007.04969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau R.W.t. Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama T, Tanaka H, Yoshida T, Uehara T, Minami M. Neuronal injury induces cytokine-induced neutrophil chemoattractant-1 (CINC-1) production in astrocytes. J Pharmacol Sci. 2009;109:88–93. doi: 10.1254/jphs.08298fp. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama Y, Baba A, Matsuda T. Production of monocyte chemoattractant protein-1 and cytokine-induced neutrophil chemoattractant-1 in rat brain is stimulated by intracerebroventricular administration of an endothelin ETB receptor agonist. Neuroreport. 2007;18:1275–1279. doi: 10.1097/WNR.0b013e32825a67f1. [DOI] [PubMed] [Google Scholar]
- Lee KM, Kang BS, Lee HL, Son SJ, Hwang SH, Kim DS, Park JS, Cho HJ. Spinal NF-kB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity. Eur J Neurosci. 2004;19:3375–3381. doi: 10.1111/j.0953-816X.2004.03441.x. [DOI] [PubMed] [Google Scholar]
- Li H, Xie W, Strong JA, Zhang JM. Systemic antiinflammatory corticosteroid reduces mechanical pain behavior, sympathetic sprouting, and elevation of proinflammatory cytokines in a rat model of neuropathic pain. Anesthesiology. 2007;107:469–477. doi: 10.1097/01.anes.0000278907.37774.8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindia JA, McGowan E, Jochnowitz N, Abbadie C. Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J Pain. 2005;6:434–438. doi: 10.1016/j.jpain.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Manjavachi MN, Quintao NL, Campos MM, Deschamps IK, Yunes RA, Nunes RJ, Leal PC, Calixto JB. The effects of the selective and non-peptide CXCR2 receptor antagonist SB225002 on acute and long-lasting models of nociception in mice. European journal of pain (London, England) 2010;14:23–31. doi: 10.1016/j.ejpain.2009.01.007. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Tani M, Krivacic K, Chernosky A, Kelner GS, Maciejewski D, Maki R, Ransohoff RM, Stokes BT. Selective chemokine mRNA accumulation in the rat spinal cord after contusion injury. J Neurosci Res. 1998;53:368–376. doi: 10.1002/(SICI)1097-4547(19980801)53:3<368::AID-JNR11>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Mennicken F, Maki R, de Souza EB, Quirion R. Chemokines and chemokine receptors in the CNS: a possible role in neuroinflammation and patterning. Trends in pharmacological sciences. 1999;20:73–78. doi: 10.1016/s0165-6147(99)01308-5. [DOI] [PubMed] [Google Scholar]
- Miller RJ, Rostene W, Apartis E, Banisadr G, Biber K, Milligan ED, White FA, Zhang J. Chemokine action in the nervous system. J Neurosci. 2008;28:11792–11795. doi: 10.1523/JNEUROSCI.3588-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch C, Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood. 2000;95:3032–3043. [PubMed] [Google Scholar]
- Nguyen D, Stangel M. Expression of the chemokine receptors CXCR1 and CXCR2 in rat oligodendroglial cells. Brain Res Dev Brain Res. 2001;128:77–81. doi: 10.1016/s0165-3806(01)00128-6. [DOI] [PubMed] [Google Scholar]
- Omari KM, John G, Lango R, Raine CS. Role for CXCR2 and CXCL1 on glia in multiple sclerosis. Glia. 2006;53:24–31. doi: 10.1002/glia.20246. [DOI] [PubMed] [Google Scholar]
- Omari KM, John GR, Sealfon SC, Raine CS. CXC chemokine receptors on human oligodendrocytes: implications for multiple sclerosis. Brain. 2005;128:1003–1015. doi: 10.1093/brain/awh479. [DOI] [PubMed] [Google Scholar]
- Park CK, Lu N, Xu ZZ, Liu T, Serhan CN, Ji RR. Resolving TRPV1- and TNF-alpha-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J Neurosci. 2011;31:15072–15085. doi: 10.1523/JNEUROSCI.2443-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineau I, Sun L, Bastien D, Lacroix S. Astrocytes initiate inflammation in the injured mouse spinal cord by promoting the entry of neutrophils and inflammatory monocytes in an IL-1 receptor/MyD88-dependent fashion. Brain, behavior, and immunity. 2010;24:540–553. doi: 10.1016/j.bbi.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Popivanova BK, Koike K, Tonchev AB, Ishida Y, Kondo T, Ogawa S, Mukaida N, Inoue M, Yamashima T. Accumulation of microglial cells expressing ELR motif-positive CXC chemokines and their receptor CXCR2 in monkey hippocampus after ischemia-reperfusion. Brain Res. 2003;970:195–204. doi: 10.1016/s0006-8993(03)02343-6. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Savarin-Vuaillat C, Ransohoff RM. Chemokines and chemokine receptors in neurological disease: raise, retain, or reduce? Neurotherapeutics. 2007;4:590–601. doi: 10.1016/j.nurt.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nature neuroscience. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Seybold VS, Jia YP, Abrahams LG. Cyclo-oxygenase-2 contributes to central sensitization in rats with peripheral inflammation. Pain. 2003;105:47–55. doi: 10.1016/s0304-3959(03)00254-9. [DOI] [PubMed] [Google Scholar]
- Simonetti M, Hagenston AM, Vardeh D, Freitag HE, Mauceri D, Lu J, Satagopam VP, Schneider R, Costigan M, Bading H, Kuner R. Nuclear calcium signaling in spinal neurons drives a genomic program required for persistent inflammatory pain. Neuron. 2013;77:43–57. doi: 10.1016/j.neuron.2012.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Sahbaie P, Liang DY, Li WW, Li XQ, Shi XY, Clark JD. Epigenetic Regulation of Spinal CXCR2 Signaling in Incisional Hypersensitivity in Mice. Anesthesiology. 2013 doi: 10.1097/ALN.0b013e31829ce340. [DOI] [PubMed] [Google Scholar]
- Thacker MA, Clark AK, Bishop T, Grist J, Yip PK, Moon LD, Thompson SW, Marchand F, McMahon SB. CCL2 is a key mediator of microglia activation in neuropathic pain states. European journal of pain (London, England) 2009;13:263–272. doi: 10.1016/j.ejpain.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Valles A, Grijpink-Ongering L, de Bree FM, Tuinstra T, Ronken E. Differential regulation of the CXCR2 chemokine network in rat brain trauma: implications for neuroimmune interactions and neuronal survival. Neurobiol Dis. 2006;22:312–322. doi: 10.1016/j.nbd.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Wang JG, Strong JA, Xie W, Yang RH, Coyle DE, Wick DM, Dorsey ED, Zhang JM. The chemokine CXCL1/growth related oncogene increases sodium currents and neuronal excitability in small diameter sensory neurons. Mol Pain. 2008;4:38. doi: 10.1186/1744-8069-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Jung H, Miller RJ. Chemokines and the pathophysiology of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:20151–20158. doi: 10.1073/pnas.0709250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science (New York, N.Y. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Xie WR, Deng H, Li H, Bowen TL, Strong JA, Zhang JM. Robust increase of cutaneous sensitivity, cytokine production and sympathetic sprouting in rats with localized inflammatory irritation of the spinal ganglia. Neuroscience. 2006;142:809–822. doi: 10.1016/j.neuroscience.2006.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang RH, Strong JA, Zhang JM. NF-kappaB mediated enhancement of potassium currents by the chemokine CXCL1/growth related oncogene in small diameter rat sensory neurons. Mol Pain. 2009;5:26. doi: 10.1186/1744-8069-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. Journal of neurochemistry. 2006;97:772–783. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZJ, Cao DL, Zhang X, Ji RR, Gao YJ. Chemokine contribution to neuropathic pain: Respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain. 2013;154:2185–2197. doi: 10.1016/j.pain.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]