

Abstract
Rationale
Synthetic hallucinogenic tryptamines, especially those originally described by Alexander Shulgin, continue to be abused in the United States. The range of subjective experiences produced by different tryptamines suggests that multiple neurochemical mechanisms are involved in their actions, in addition to the established role of agonist activity at serotonin-2A (5-HT2A) receptors.
Objectives
This study evaluated the interaction of a series of synthetic tryptamines with biogenic amine neurotransmitter transporters and with serotonin (5-HT) receptor subtypes implicated in psychedelic effects.
Methods
Neurotransmitter transporter activity was determined in rat brain synaptosomes. Receptor activity was determined using calcium mobilization and DiscoveRx PathHunter® assays in HEK293, Gα16-CHO, and CHOk1 cells transfected with human receptors.
Results
Twenty-one tryptamines were analyzed in transporter uptake and release assays, and 5-HT2A, serotonin 1A (5-HT1A), and 5-HT2A β-arrestin functional assays. Eight of the compounds were found to have 5-HT-releasing activity. Thirteen compounds were found to be 5-HT uptake inhibitors or were inactive. All tryptamines were 5-HT2A agonists with a range of potencies and efficacies, but only a few compounds were 5-HT1A agonists. Most tryptamines recruited β-arrestin through 5-HT2A activation.
Conclusions
All psychoactive tryptamines are 5-HT2A agonists, but 5-HT transporter (SERT) activity may contribute significantly to the pharmacology of certain compounds. The in vitro transporter data confirm structure-activity trends for releasers and uptake inhibitors whereby releasers tend to be structurally smaller compounds. Interestingly, two tertiary amines were found to be selective substrates at SERT, which dispels the notion that 5-HT-releasing activity is limited only to primary or secondary amines.
Keywords: Tryptamines, Shulgin, psychedelic, serotonin, serotonin transporter, serotonin releaser, serotonin 2A receptor, serotonin 1A receptor, β-arrestin recruitment, psilocybin
Introduction
Synthetic hallucinogenic tryptamines such as N,N-diisopropyl-5-methoxytryptamine (Foxy, 1) and N-isopropyl-N-methyl-5-methoxytryptamine (Moxy, 2) shown in Figure 1 continue to be abused in the United States, despite the recent emergence of synthetic “bath salt” cathinone compounds. Many of these synthetic tryptamines were originally described by Alexander Shulgin and synthesized as potential “entactogens” (Nichols 1986), intended originally as candidates for augmentation of psychotherapeutic sessions, similar to early efforts by Sandoz with lysergic acid diethylamide (3, LSD) (Busch and Johnson 1950) and more recent efforts with 3,4-methylenedioxymethamphetamine (4, MDMA or “ecstasy”) (Oehen et al. 2013; Parrott 2007). The therapeutic strategy for psychedelic-assisted therapy is to find compounds that improve the outcome of psychiatric sessions by facilitating “the production of memories, fantasies and insights and to enhance the therapeutic alliance” (Grinspoon and Bakalar 1981; 1986) without inducing strong, possibly aversive hallucinations. Recently, interest in the possible therapeutic effects of hallucinogens has been rekindled. Psilocybin (5) is being explored in human laboratory experiments to explore the “mystical” properties of these types of compounds (Griffiths et al. 2008; Griffiths et al. 2011), and has shown efficacy as a treatment for cluster headaches (Johnson et al. 2012), for psychotherapy in general (MacLean et al. 2011), for smoking cessation (Johnson 2013), and for alcohol abuse (Bogenschutz 2013).
Figure 1.

Psychedelic and Non-Psychedelic Compounds
The psychoactive properties of various tryptamines make them attractive to recreational drug users. As with the “bath salt” cathinone compounds, non-medical misuse of tryptamines appears to be driven by Internet availability and by increasing interest in synthetic pharmaceutical and “designer” drugs in general (Carroll et al. 2012). Synthetic tryptamines are based on natural product psychedelics such as psilocybin (5), dimethyltryptamine (6, DMT), and N,N-dimethyl-5-methoxytryptamine (7, 5-MeO-DMT). These compounds all contain an indole ring, a structural theme shared by psychedelics including LSD (3) and ibogaine (8). The unique psychoactive properties of psychedelics have generally been attributed to agonist activity at 5-HT2A receptors. However, the wide range of subjective experiences produced by these compounds suggests that multiple neurochemical pathways are involved.
Most psychedelics have a high affinity for 5-HT2A receptors, but not all 5-HT2A agonists are psychedelics, as highlighted by the existence of 5-HT2A agonists such as lisuride (9). Lisuride is not psychoactive in humans or animals (Callahan and Appel 1990; Halberstadt and Geyer 2013; White and Appel 1982), indicating that some type of functional selectivity at the 5-HT2A receptor may be responsible for eliciting psychedelic effects. One recent study described a hallucinogen-specific signaling pathway mediated by the 5-HT2A receptor in cortical neurons whereby both hallucinogenic and non-hallucinogenic compounds induced c-fos expression and activated Gαq/11 proteins, while only hallucinogenic compounds induced egr-2 expression and activated Gαi/o proteins (Gonzalez-Maeso et al. 2007). Additional reports have implicated a 5-HT2A-metabotropic glutamate 2 receptor (5-HT2A-mGluR2) heterodimeric complex as being responsible for a unique hallucinogen-specific downstream signaling pattern, but more studies are warranted in order to fully understand the biological role of this complex (Delille et al. 2012; Fribourg et al. 2011; Gonzalez-Maeso et al. 2008; Moreno et al. 2011). Other studies have examined 5-HT2A receptor function and shown that hallucinogens such as 2,5-dimethoxy-4-iodoamphetamine (DOI) and 5-MeO-DMT (7) activate downstream effectors independently of β-arrestin-2, while the non-hallucinogenic endogenous agonist 5-HT requires β-arrestin-2 for activation of the same downstream effectors (Schmid and Bohn 2010; Schmid et al. 2008). Collectively, these reports suggest that functional selectivity at the 5-HT2A receptor is important in mediating the psychoactive behavioral effects of hallucinogenic compounds.
Although 5-HT2A receptor activity plays a major role in the pharmacology of psychedelic compounds, additional signaling pathways have been shown to be significant as well. As recently outlined in an excellent review on the pharmacology of hallucinogens (Nichols 2004), serotonin 2C receptor (5-HT2C) agonism, 5-HT1A agonism, and SERT uptake inhibition have all been implicated in the activity of hallucinogens. Dopamine (DA) receptors (Marona-Lewicka et al. 2009; Marona-Lewicka et al. 2005; Seeman et al. 2005), the trace amine receptor (Bunzow et al. 2001) and the sigma-1 receptor (Fontanilla et al. 2009; Su et al. 2009) have also been suggested to modulate the effects of hallucinogenic compounds. The hallucinogen salvinorin A (10), a natural product derived from Salvia divinorum or “magic mint” was unexpectedly found to be a highly selective kappa opioid receptor agonist (Roth et al. 2002), providing yet another possible neurochemical pathway for psychoactivity. More recent evidence suggests cannabinoid receptor involvement in the behavioral effects of salvinorin A (Braida et al. 2008; Walentiny et al. 2010).
Synthetic psychoactive tryptamines are close analogs of the neurotransmitter 5-HT. Accordingly, tryptamines may block 5-HT uptake by the SERT or may be SERT substrates which induce 5-HT release via reversal of normal transporter flux. Indeed, several tryptamines have already been shown to interact with SERT (Cozzi et al. 2009; Nagai et al. 2007). It is well known that MDMA (4) is a SERT-mediated releaser (Callaway et al. 1990), as is trifluoromethylphenylpiperazine (11), which has been used in conjunction with benzylpiperazine to mimic MDMA as so-called “Legal X” (Baumann et al. 2004). The precise role of SERT-mediated release in the psychotropic actions of most of these compounds is not known, but likely includes indirect activation of 5-HT receptor subtypes by released neurotransmitter. It is certainly intriguing that the two compounds most commonly investigated for use in psychotherapy are LSD and MDMA, compounds with different primary mechanisms of action (5-HT2A agonist activity vs SERT substrate activity). An understanding of the pharmacology of LSD, MDMA and related compounds is needed to develop novel therapeutics, for psychotherapy as well as other clinical applications. Psychoactive tryptamines represent a good starting point for the study of hallucinogenic and psychedelic mechanisms because they are simple chemical structures that are relatively easy to synthesize, when compared to more complicated substances such as LSD and ibogaine. To date, a comprehensive investigation of the interactions of tryptamines with biogenic amine transporters has not been reported. Here, we describe the transporter and 5-HT receptor activities of a group of synthetic tryptamines, many of which are known to have psychedelic properties.
Methods
Dopamine Transporter (DAT), Norepinephrine Transporter (NET), and SERT Assays
Rats were euthanized by CO2 narcosis, and brains were processed to yield synaptosomes as previously described (Baumann et al., 2013b; Rothman et al., 2003). Synaptosomes were prepared from rat striatum for the DAT assays, whereas synaptosomes were prepared from whole brain minus striatum and cerebellum for the NET and SERT assays. For uptake inhibition assays, 5 nM [3H]DA, 10 nM [3H]norepinephrine (NE) and 5 nM [3H]5-HT were used to assess transport activity at DAT, NET, and SERT, respectively. The selectivity of uptake assays was optimized for a single transporter by including unlabeled blockers to prevent uptake of [3H]transmitter by competing transporters. Uptake inhibition assays were initiated by adding 100 µl of tissue suspension to 900 µl Krebs-phosphate buffer (126 mM NaCl, 2.4 mM KCl, 0.83 mM CaCl2, 0.8 mM MgCl2, 0.5 mM KH2PO4, 0.5 mM Na2SO4, 11.1 mM glucose, 0.05 mM pargyline, 1mg/mL bovine serum albumin, and 1 mg/mL ascorbic acid, pH 7.4) containing test drug and [3H]transmitter. Uptake inhibition assays were terminated by rapid vacuum filtration through Whatman GF/B filters, and retained radioactivity was quantified by liquid scintillation counting. For release assays, 9 nM [3H]1-methyl-4-phenylpyridinium ([3H]MPP+) was used as the radiolabeled substrate for DAT and NET, while 5 nM [3H]5-HT was used as a substrate for SERT. All buffers used in the release assay methods contained 1 µM reserpine to block vesicular uptake of substrates. The selectivity of release assays was optimized for a single transporter by including unlabeled blockers to prevent the uptake of [3H]MPP+ or [3H]5-HT by competing transporters. Synaptosomes were preloaded with radiolabeled substrate in Krebs-phosphate buffer for 1 h (steady state). Release assays were initiated by adding 850 µl of preloaded synaptosomes to 150 µl of test drug. Release was terminated by vacuum filtration and retained radioactivity was quantified as described for uptake inhibition.
Calcium Mobilization Assays
Cells stably expressing the desired human receptor were plated into 96-well black-walled assay plates in growth medium. 5HT2A HEK293 cells were plated at 35,000 cells/well (plates precoated with PEI) in DMEM-HG supplemented with 10% fetal bovine serum, 100 units of penicillin and streptomycin, 15mM HEPES, and 100 µg/mL normocin™. 5-HT1A Gα16-CHO cells were plated at 25,000 cells/well in Ham’s F12 supplemented with 10% fetal bovine serum, 100 units of penicillin and streptomycin, and 100 µg/mL normocin™. After incubating at 37°C, 5% CO2 overnight, the growth medium was removed and the cells were gently washed with 100 µL of pre-warmed (37°C) assay buffer (1X HBSS, 20 mM HEPES, 2.5 mM probenecid, pH 7.4 at 37°C). The cells were incubated for 45 minutes at 37°C, 5% CO2 in 200 µL of a calcium-sensitive fluorescent dye (½ the manufacturer’s recommended concentration, calcium 5 assay kit, Molecular Devices). During the incubation period, 8-point concentration curves of the test compounds (10X) were prepared in assay buffer/1% DMSO and aliquoted into 96-well polypropylene plates. After 45 minutes, 25 µL of pretreatment (assay buffer/10% DMSO) was added to the wells and, following a 15 minute incubation period at 37°C, the plate was read in a FlexStation II (Molecular Devices). Calcium-mediated changes in fluorescence were monitored every 1.52 seconds over a 60 second time period, with the FlexStation II adding 25 µL of test compound at the 19 second time point (excitation at 485 nm, detection at 525 nm). Peak kinetic reduction (SoftMax, Molecular Devices) relative fluorescent units (RFU) were plotted against compound concentration. Data were fit to a three-parameter logistic curve to generate EC50 values (Prism, version 6.0, GraphPad Software, Inc., San Diego, CA). EC50 and % Emax values are reported as means ± SEM and are the result of three independent experiments performed in duplicate unless otherwise noted.
β-Arrestin Recruitment Assay (PathHunter Detection Kit, DiscoveRx)
CHOk1 cells stably expressing the 5HT2A receptor fused to the Prolink gene were plated into 96-well white-walled assay plates at a density of 15,000 cells/well in Cell Plating Reagent 21 (DiscoveRx) and incubated at 37°C, 5% CO2 overnight. The next day, test compounds were prepared at 10X concentration in DPBS/1% DMSO and 10 µL was added to the cells. Following a 3 hour incubation at 37°C, detection reagent (prepared according to the manufacturer’s specifications) was added to each well. Luminescence was measured at 1hr post detection reagent addition using a FlexStation III (Molecular Devices, 1000ms integration time). Relative luminescence units (RLU) were plotted against compound concentration. Data were fit to a three-parameter logistic curve to generate EC50 values (Prism, version 6.0, GraphPad Software, Inc., San Diego, CA). EC50 and % Emax values are reported as means ± SEM and are the result of three independent experiments performed in duplicate unless otherwise noted.
Compounds
Compounds 1, 2, 6, 7, 19, 20, 21, 22, 23, 25, 26, and 28 were synthesized as described by Shulgin (Shulgin and Shulgin 1997). Compounds 15, 17, 18, and 27 were synthesized using similar methods. Compounds 12 and 13 were purchased commercially. Compounds 14, 16, and 24 were procured from the National Institute of Drug Abuse Drug Supply program.
Results
A set of twenty-one tryptamines was studied in biogenic amine uptake inhibition and release assays, as well as in 5-HT2A and 5-HT1A calcium mobilization and 5-HT2A β-arrestin recruitment assays (Tables 1 and 2). The compounds were procured commercially, obtained from the National Institute of Drug Abuse Drug Supply program, or synthesized either as reported (Shulgin and Shulgin 1997) or following a similar synthetic route as reported. These compounds differed by N-substitution and indole ring substituents. Ten of the compounds were unsubstituted, nine contained methoxy groups in the 5-position, one had a hydroxyl in the 5-position and one had a hydroxyl in the 4-position. In the unsubstituted and 5-methoxylated series, compounds were synthesized such that their N-alkyl groups increased in size and complexity from unsubstituted to the N,N-diisopropyl groups found on Foxy. As shown by Shulgin (Shulgin and Shulgin 1997; Shulgin and Carter 1980), these simple structural changes induce a variety of psychoactive effects, involving both auditory and visual systems, making them ideal for a pharmacological mechanistic study. Some highlights of their reported psychoactivity are listed in Tables 1 and 2 as described in TiHKAL (Shulgin and Shulgin 1997). In order to assess their activity at the biogenic amine transporters, the compounds were characterized as substrates/releasers or uptake inhibitors as previously described (Rothman et al. 2001; Rothman et al. 2002). Any release activity was confirmed by substrate reversal. In order to assess the activity of compounds at 5-HT G protein-coupled receptors (GPCRs), an in vitro calcium mobilization assay was used to measure 5-HT2A receptor activation in HEK293 cells and 5-HT1A receptor activation in CHO-Gα16 cells in stably transfected cell lines using the human receptors. 5-HT2A receptor mediated β-arrestin recruitment was measured using CHO-β-arrestin-2 cells stably expressing the human 5-HT2A receptor fused to the small enzyme fragment ProLink (DiscoveRx PathHunter® technology).
Table 1.
DAT, SERT, and NET-mediated Releasing Properties and 5-HT2A, 5-HT2A β-arrestin, and 5-HT1A Activity of Psychedelic Tryptamine Analogs
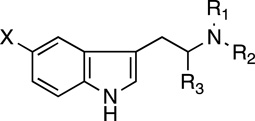 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Release EC50, (nM)a | 5-HT2Ab | 5-HT1Ab | 5-HT2A β-arrestinc | ||||||||||||
| # | Name | X | R1 | R2 | R3 | DAT | SERT | NET | EC50, (nM) |
Emax, (%) |
EC50, (nM) |
Emax, (%) |
EC50, (nM) | Emax, (%) |
Psychoactive Effectse |
| 12 | T | H | H | H | H | 164 ± 16 | 32.6 ± 2.6 | 716 ± 46 | 7.36 ± 0.56 | 104 ± 4 | IA | 3485 ± 234 | 108 ± 16 | Psychoactive, short acting due to metabolism, increased blood pressure, similar to LSD | |
| 13 | AMT | H | H | H | Me | 78.6 ± 4.0 | 21.7 ± 1.0 | 112 ± 6 | 23.1 ± 2.4 | 103 ± 3 | IA | 4855 ± 1416 | 95 ± 17 | Psychedelic but varied, fast/slow onset, good/bad experiences, long lasting, no visuals, speed-like, tachycardia, jaw clenching | |
| 14 | NMT | H | Me | H | H | 321 ± 23 | 22.4 ± 1.4 | 733 ± 94 | 50.7 ± 7.1 | 96 ± 2 | IA | IA | NR | ||
| 15 | NETPg | H | Et | H | H | 6660 ± 551 | 18.6 ± 1.6 | 3862 ± 635d | 38.3 ± 2.7 | 99 ± 2 | IA | 4582f | 64f | NR | |
| 6 | DMT | H | Me | Me | H | >10,000 | 114 ± 15 | 4166 ± 317 | 38.3 ± 0.81 | 83 ± 0.4 | IA | IA | Heavy intoxicant and hallucinogen, visuals, intense colors | ||
| 16 | Dimethyl - serotonin | OH | Me | Me | H | >10,000 | 30.5 ± 8.6 | >10,000 | 3.49 ± 0.08 | 100 ± 2 | 366 ± 67 | 95 ± 1 | ND | Some visuals (lines and dots), tightness in chest, nausea, breathing issues, mind feels crowded | |
| 17 | 5MeO-NMT | OMe | Me | H | H | >10,000 | 1114 ± 700 | >10,000 | 3.78 ± 0.73 | 84 ± 20 | 220 ± 12 | 72 ± 4 | 611 ± 149 | 66 ± 9 | NR |
| 18 | 5MeO-NET | OMe | Et | H | H | >10,000 | 284 ± 41.6 | >10,000 | 1.92 ± 0.29 | 112 ± 3 | 262 ± 27 | 70 ± 7 | 568 ± 174 | 103 ± 12 | NR |
IA = Inactive at 10 µM in agonist screen.
NR = Not reported in TiHKAL.
ND = Not determined.
Release EC50 values are reported as means ± SD and are the result of N= 3 performed in triplicate.
Calcium mobilization EC50 and Emax values are reported as means ± SEM and are the result of three independent experiments performed in duplicate.
β-arrestin recruitment EC50 and Emax values are reported as means ± SEM and are the result of three independent experiments performed in duplicate.
IC50 for uptake inhibition.
Tabulated from anecdotal reports in TiHKAL.
Single determination.
This compound is referred to as “NET” in TiHKAL but was altered to avoid confusion with the norepinephrine transporter.
Table 2.
DA, 5HT, and NE Uptake Inhibition Properties and 5-HT2A, 5-HT2A β-arrestin, and 5-HT1A Activity of Psychedelic Tryptamines
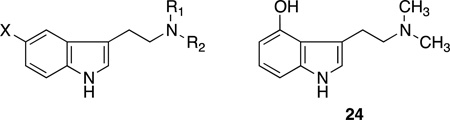 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # | Name | X | R1 | R2 | IC50, (nM)a | 5-HT2Ab | 5-HT1Ab | 5-HT2A β-arrestinc | Psychoactive Effectsd |
||||||
| DAT | SERT | NET | EC50, (nM) |
Emax, (%) |
EC50, (nM) |
Emax, (%) |
EC50, (nM) |
Emax, (%) |
|||||||
| 19 | DET | H | Et | Et | >10,000 | 258 ± 13 | >10,000 | 67.8 ± 4.4 | 90 ± 6 | IA | IA | Psychoactive, visuals, vertigo | |||
| 20 | NIPT | H | iPr | H | >10,000 | 1487 ± 93 | >10,000 | 116 ± 8.4 | 96 ± 3 | IA | >10,000 | Light-headed intoxicant, pleasant buzz | |||
| 21 | DPT | H | Pr | Pr | 2218 ± 168 | 157 ± 16 | 3202 ± 330 | 26.1 ± 1.9 | 97 ± 3 | IA | 1692 ± 78.9 | 63 ± 8 | Intensely visual, long lasting, things appear fast, religious experiences | ||
| 22 | DIPT | H | iPr | iPr | 4788 ± 416 | 288 ± 2.3 | >10,000 | 33.5 ± 1.5 | 110 ± 2 | IA | >10,000 | Auditory, not visual, heard voices, sounds had “golden spikes”, religious overtones | |||
| 23 | MIPT | H | iPr | Me | >10,000 | >10,000 | >10,000 | 44.9 ± 1.7 | 74 ± 5 | IA | IA | Mild hallucinations, stimulant, fast onset, psychedelic | |||
| 24 | Psilocin | 4-OH | Me | Me | >10,000 | 662 ± 41 | >10,000 | 45.2 ± 4.1e | 76 ± 7e | ND | ND | Very visual, lots of colors, better at night | |||
| 25 | 5MeO-T | OMe | H | H | >10,000 | 4000 | >10,000 | 0.503 ± 0.09 | 119 ± 2 | 183 ± 30 | 66 ± 4 | 535 ± 123 | 135 ± 20 | Not particularly active, possibly due to metabolism similar to 12 | |
| 7 | 5MeO-DMT | OMe | Me | Me | >10,000 | 2184 ± 222 | >10,000 | 3.87 ± 0.35 | 101 ± 4 | 791 ± 133 | 68 ± 3 | 385 ± 112 | 56 ± 6 | Time distortions, stoned feeling, hallucinogenic at high doses | |
| 26 | 5MeO-DET | OMe | Et | Et | >10,000 | 2410 ± 222 | >10,000 | 8.11 ± 0.36 | 101 ± 0.8 | 911 ± 186 | 67 ± 2 | 1065 ± 607 | 64 ± 8 | Auditory changes, negative experiences, anxiety | |
| 27 | 5MeO-NIPT | OMe | iPr | H | >10,000 | 5442 ± 377 | >10,000 | 9.24 ± 1.3 | 101 ± 2 | >10,000 | 1075 ± 234 | 99 ± 6 | NR | ||
| 28 | 5MeO-DPT | OMe | Pr | Pr | 16998 ± 2192 | 1031 ± 140 | >10,00 | 6.00 ± 0.43 | 101 ± 3 | 476 ± 102 | 43 ± 5 | 993 ± 421 | 68 ± 2 | Comfortable at low doses, oscillating good and bad sounds, negative side dominated as buzz continues | |
| 1 | 5MeO-DIPT | OMe | iPr | iPr | >10,000 | 646 ± 48 | >10,000 | 6.22 ± 1.1 | 109 ± 5 | >10,000 | 946 ± 141 | 124 ± 12 | Erotic, senses distorted, short-lived, could not make intuitive leaps | ||
| 2 | 5MeO-MIPT | OMe | iPr | Me | >10,000 | >10,000 | >10,000 | 7.82 ± 1.5 | 101 ± 3 | >10,000 | 566 ± 110 | 82 ± 13 | Fast onset, stimulates conceptual thought, depersonalization | ||
IA = Inactive at 10 µM in agonist screen.
NR = Not reported in TiHKAL.
ND = Not determined.
Uptake inhibition IC50 values are reported as means ± SD and are the result of N= 3 performed in triplicate.
Calcium mobilization EC50 and Emax values are reported as means ± SEM and are the result of three independent experiments performed in duplicate.
β-arrestin recruitment EC50 and Emax values are reported as means ± SEM and are the result of three independent experiments performed in duplicate.
Tabulated from anecdotal reports in TiHKAL.
Values are means ± SD and are the result of two independent experiments performed in duplicate.
The tryptamines binned into two groups, depending on their SERT activity. Eight of the compounds were found to have 5-HT releasing activity (Table 1) and the remaining thirteen compounds were found to be either 5-HT uptake inhibitors (Table 2), or were inactive (2 and 23). As expected, the smaller, less sterically encumbered compounds such as the primary amines (12 and 13), the N-methyl derivatives (15 and 17), and N-ethyl derivatives (15 and 18) were found to be releasers (Table 1). Most of the tryptamines in Table 2 were found to be 5-HT uptake inhibitors, with very little activity at either DAT or NET. These compounds have much larger N-alkyl groups and were uptake inhibitors, presumably because the compounds were not transportable.
Discussion
Our findings with the tryptamine compounds fit with the general hypothesis that psychedelic compounds are serotonergic in nature. Specifically, all of the compounds were active as 5-HT2A agonists and most were either SERT uptake inhibitors or transporter substrate releasers. SERT-mediated release potencies varied widely between the 5-methoxy and unsubstituted compounds. The unsubstituted indole compounds were more active as 5-HT releasers with EC50 values in the 18.6 to 32.6 nM range. Tryptamine (12),-methyltryptamine (13), and N-methyltryptamine (14) also displayed releasing activity at DA transporters and NE transporters, although with much lower potency when compared to activity at the SERT. The most potent and selective 5-HT releaser was N-ethyltryptamine (15, NETP), which had an EC50 for SERT-mediated release of 18.6 nM and was relatively inactive at the other two transporters. The 5-methoxylated version of DMT (5-MeO-DMT, 7) was a weak 5-HT uptake inhibitor (IC50 value = 2184 nM). This was somewhat surprising since the 5-hydroxy analog, 16, was a potent SERT-mediated releaser with an EC50 value of 30.5 nM. 5-OH-DMT (16), also known as bufotenin, is a compound reportedly found in schizophrenics and proposed as a possible disease biomarker (Emanuele et al. 2010; Faurbye and Pind 1968). 5-MeO-DMT (7) and DMT (6), are also found naturally in psychoactive plants such as Psychotria viridis and Virola calophylla (Freedland and Mansbach 1999). The N,N-dimethyl-4-hydroxy analog (24, psilocin), is the active metabolite of psilocybin (5), the O-phosphoryl analog of psilocin and the hallucinogenic component of the psychoactive Psilocybe genus of mushrooms which is currently being studied clinically. Psilocin was found to have reasonable uptake inhibitory properties at the SERT, with an IC50 value of 662 nM. The transposition of the hydroxyl group from the 5-position (16) to the 4-position (24) changed the activity from a SERT-mediated releaser to a SERT uptake inhibitor, meaning the 5-substituted compound was a substrate, but the 4-substituted analog was not.
The uptake inhibition data for 5-MeO-DIPT (1), 5-MeO-MIPT (2) and 5-MeO-DMT (7) in Table 2 correlates well with data reported by Nagai et al. who used a similar assay protocol with rat brain synaptosomes (Nagai et al. 2007). None of the 5-methoxy compounds had appreciable DA or NE uptake inhibitory properties. In all three cases, the IC50 values for 5-HT uptake inhibition were slightly lower than reported previously, 646 nM, >10,000 nM, and 2184 nM, respectively (see Table 2), compared to 2200 nM, 6400 nM and 4100 nM (Nagai et al. 2007), though the rank order of potency was the same. Nagai et al. also studied AMT (18) and found that it was a fairly potent releaser at transporters for DA, NE and 5-HT, with EC50 values of 180 nM, 68 nM, and 79 nM respectively. Our findings with AMT were similar to those of Nagai et al., as shown in Table 1. Sogawa et al. have also reported that 5-MeO-DIPT is a potent and selective 5-HT uptake inhibitor using rat brain synaptosomes with an IC50 value of 1800 nM (Sogawa et al. 2007). Their value is 3-fold higher than our observation of 646 nM.
As with the releasers, the 5-methoxy analogs were much less potent at the SERT when compared to their unsubstituted counterparts. For example, 5-MeO-DET (26) was found to have an IC50 of 2184 nM, whereas DET was 10-fold more potent (258 nM). In general, increasing the overall size of a chemical structure was associated with lower potency at SERT uptake inhibition. However, the diisopropyl compounds, DIPT (22) and 5-MeO-DIPT (1), inhibited 5-HT uptake much better than their mono-substituted analogs. NIPT (20) and 5-MeO-NIPT (27) were found to be weak 5-HT uptake inhibitors with IC50 values of 1487 nM and 5442 nM respectively. The addition of a second isopropyl group (DIPT and 5-MeO-DIPT) improved SERT activity by almost an order of magnitude, to 288 nM and 646 nM respectively. Even more surprising was the finding that substitution of one of the isopropyls with a methyl group to form MIPT (23) and 5-MeO-MIPT (2, Moxy) rendered the compounds totally inactive as 5-HT uptake inhibitors.
Our findings also fit with the general hypothesis that releasers are transporter substrates, similar to 5-HT, while uptake inhibitors are not. This hypothesis posits that structurally small compounds tend to be releasers because they are able to be transported and hence induce transporter-mediated release. On the other hand, larger compounds cannot be substrates because steric interactions with the transporter prevent translocation. These compounds either bind at the surface of the transporter, thereby blocking reuptake of neurotransmitter, or do nothing. As noted above, N-ethyltryptamine (15) was a SERT-mediated releaser, but the addition of a second N-ethyl group caused the compound to become a modestly potent 5-HT uptake inhibitor (19, DET) with an IC50 for inhibition of 258 nM. This finding reinforces the notion that bulky substituents render compounds that are too large to be transported. Presumably, both NETP and DET bind to the site of translocation, but the second ethyl group of DET prevents the transporter from making the required conformational changes to induce translocation.
One of the most interesting structural observations with the transporter data involve the N,N-dimethyl analogs. Both N,N-dimethyltryptamine (6, DMT) and N,N-dimethyl-5-hydroxytryptamine (16) were found to be SERT-selective releasers, with EC50 values of 114 nM and 30.5 nM, respectively. This observation was somewhat surprising because our previous experience has demonstrated that the vast majority of transporter substrates are primary or secondary amines. Cozzi, et al. also observed substrate activity for DMT, as well as for DPT (21), DIPT (22), and MIPT (23) which are also N,N-dialkyl analogs and much larger than DMT (Cozzi et al. 2009). Our assay protocol uses rat synaptosomes and compares activity in SERT release and uptake inhibition. Using this protocol the latter 3 compounds were found to be SERT uptake inhibitors (Table 2). Mechanistically, substrate-type releasers are active in both release and uptake inhibition assays, while uptake inhibitors are only active in the uptake inhibition assay. The protocol used by Cozzi et al. to identify SERT substrates was an indirect method of determining mechanism by comparing binding affinity to potency in uptake inhibition. Compounds with high binding-to-uptake ratios were considered SERT substrate releasers. The binding-to-uptake ratio protocol was originally developed by the Rothman laboratory (Rothman et al. 1999), but this method was found to be inconsistent and replaced with a protocol similar to the release assays used here (Rothman et al. 2002)
All of the neurotransmitter releasing tryptamines (Table 1) were potent and efficacious 5-HT2A agonists as measured in a calcium mobilization assay. The most potent and efficacious compound of this group is 5-methoxy-N-ethyltryptamine (18), which had an EC50 value of 1.9 nM with 112% efficacy. Compounds that exhibited Gαq/11 mediated functional responses also promoted the recruitment of β-arrestin, except for NMT (14) and DMT (6), which were inactive at 10 µM. The two methoxy derivatives (17 and 18) had similar potencies in the β-arrestin recruitment assay, but 17 was 67% efficacious as a partial agonist while 18 was fully efficacious. This difference in efficacy was also present in the 5-HT2A calcium assay, where 17 was less efficacious than 18. Interestingly, compound 17 and 18 displayed similar 70% efficacy in the 5-HT1A receptor assay. These results indicate that adding a methoxy group to the 5-position on the phenyl ring leads to small changes in 5-HT2A potency but large changes in efficacy. One additional compound (16) was active at 5-HT1A with an EC50 value of 366 nM and was 95% efficacious as opposed to 17 and 18.
All of the compounds in Table 2 were also potent and efficacious 5-HT2A agonists in the calcium mobilization assay, with compounds 23 and 24 being the least efficacious (74 and 76%) and compound 25 being the most potent (EC50 of 0.5 nM) and efficacious (119%). Interestingly, in contrast to their weak 5-HT uptake inhibiting properties, the compounds containing a methoxy group on the phenyl ring (25, 7, 26, 27, 28, 1, 2) displayed low nanomolar potency at the 5-HT2A receptor, as opposed to the unsubstituted compounds which were less active. The methoxy compounds were also among the most potent in the β-arrestin recruitment assay, a trend that was also observed for two of the 5-HT releasers as well (17, 18). These compounds have a range of efficacies starting at 56% and increasing to 124%, which does not seem to follow any trend related to N-substitution patterns. Four of the compounds (25, 7, 26, 28) were potent at 5-HT1A, but only partially efficacious (43–66%).
Functional selectivity is becoming an important aspect in GPCR ligand development because of the possibility that an agonist might be able to activate one downstream signaling pathway over another leading to differences in pharmacological effects (Schmid and Bohn 2010; Schmid et al. 2008). All of the compounds in Tables 1 and 2 were efficacious as 5-HT2A agonists in the calcium mobilization assay, but interestingly the unsubstituted compounds were weak agonists or inactive in the β-arrestin recruitment assay. The methoxy-substituted compounds were all active in the β-arrestin recruitment assay, although several (7, 17, 26, 28) exhibited lower efficacies (56% to 68%). The implications of these findings are unknown since additional experimentation would be required to validate functional selectivity, although such differences in β-arrestin recruitment could alter behavioral effects. It would be interesting to extend these studies to other downstream signaling pathways such as activation of Akt and/or Erk phosphorylation.
A description of reported psychoactive effects in humans has been included in Tables 1 and 2 tabulated from Shulgin reports (Shulgin and Shulgin 1997) in order to provide the reader with a sense of the wide range of psychological effects produced by tryptamine agents. There did not seem to be a correlation between in vitro activity and psychoactive effects for SERT-mediated activity, 5-HT2A receptor activity or 5-HT1A receptor activity. The psychoactive effects of five of the compounds have not been reported. Further pharmacological characterization of these compounds would need to be conducted in order to further understand their behavioral outcomes, in particular related to metabolism by monoamine oxidases (Halberstadt et al. 2008; Reimann and Schneider 1993) and their pharmacokinetic profiles (Shen et al. 2011), which could be critical. As noted above, psychoactivity can also be influenced by a number of other receptors such as DA receptors, trace amine-associated receptors, cannabinoid receptors, the sigma-1 receptor, and the kappa opioid receptor. Future studies should employ more comprehensive pharmacological screening to explore the role of additional biological targets in the mechanism of action for psychoactive tryptamines.
In conclusion, we have synthesized and studied a set of tryptamines, most of which have been reported to have psychoactivity in humans. Determining the pharmacology of hallucinogens like the tryptamines will be necessary to fully understand the recent clinical results with compounds like psilocybin noted above, and for the future design of new analogs. Some of the psychoactive tryptamines were 5-HT releasers while others blocked 5-HT uptake. All of the tryptamines were 5-HT2A agonists, but there was a mix of activity in the β-arrestin recruitment and 5-HT1A assays. The in vitro transporter data confirm general structure activity trends for releasers and uptake inhibitors. Releasers tend to be sterically small compounds, likely because they undergo transporter-mediated translocation. Larger compounds, which cannot be translocated and therefore cannot induce release, often block neurotransmitter uptake, possibly because they fit in the site of translocation but are too large to undergo translocation. Finally, we found no obvious relationships between the receptor/transporter activity and the reported in vivo effects of the compounds examined.
ACKNOWLEDGMENTS
We would like to thank NIDA for their financial support (DA12970). Portions of this work were supported by the Intramural Research Program, National Institute on Drug Abuse, NIH, DHHS.
Footnotes
CONFLICTS OF INTEREST: None.
References
- Baumann MH, Clark RD, Budzynski AG, Partilla JS, Blough BE, Rothman RB. Effects of "legal x" piperazine analogs on dopamine and serotonin release in rat brain. Annals of the New York Academy of Sciences. 2004;1025:189–197. doi: 10.1196/annals.1316.024. [DOI] [PubMed] [Google Scholar]
- Bogenschutz MP. Studying the effects of classic hallucinogens in the treatment of alcoholism: rationale, methodology, and current research with psilocybin. Curr Drug Abuse Rev. 2013;6:17–29. doi: 10.2174/15733998113099990002. [DOI] [PubMed] [Google Scholar]
- Braida D, Limonta V, Capurro V, Fadda P, Rubino T, Mascia P, Zani A, Gori E, Fratta W, Parolaro D, Sala M. Involvement of kappa-opioid and endocannabinoid system on Salvinorin A-induced reward. Biol Psychiatry. 2008;63:286–292. doi: 10.1016/j.biopsych.2007.07.020. [DOI] [PubMed] [Google Scholar]
- Bunzow JR, Sonders MS, Arttamangkul S, Harrison LM, Zhang G, Quigley DI, Darland T, Suchland KL, Pasumamula S, Kennedy JL, Olson SB, Magenis RE, Amara SG, Grandy DK. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Molecular pharmacology. 2001;60:1181–1188. doi: 10.1124/mol.60.6.1181. [DOI] [PubMed] [Google Scholar]
- Busch AK, Johnson WC. L.S.D. 25 as an aid in psychotherapy; preliminary report of a new drug. Dis Nerv Syst. 1950;11:241–243. [PubMed] [Google Scholar]
- Callahan PM, Appel JB. Differentiation between the stimulus effects of (+)-lysergic acid diethylamide and lisuride using a three-choice, drug discrimination procedure. Psychopharmacology. 1990;100:13–18. doi: 10.1007/BF02245782. [DOI] [PubMed] [Google Scholar]
- Callaway CW, Wing LL, Geyer MA. Serotonin release contributes to the locomotor stimulant effects of 3,4-methylenedioxymethamphetamine in rats. J Pharmacol Exp Ther. 1990;254:456–464. [PubMed] [Google Scholar]
- Carroll FI, Lewin AH, Mascarella SW, Seltzman HH, Reddy PA. Designer drugs: a medicinal chemistry perspective. Annals of the New York Academy of Sciences. 2012;1248:18–38. doi: 10.1111/j.1749-6632.2011.06199.x. [DOI] [PubMed] [Google Scholar]
- Cozzi NV, Gopalakrishnan A, Anderson LL, Feih JT, Shulgin AT, Daley PF, Ruoho AE. Dimethyltryptamine and other hallucinogenic tryptamines exhibit substrate behavior at the serotonin uptake transporter and the vesicle monoamine transporter. J Neural Transm. 2009;116:1591–1599. doi: 10.1007/s00702-009-0308-8. [DOI] [PubMed] [Google Scholar]
- Delille HK, Becker JM, Burkhardt S, Bleher B, Terstappen GC, Schmidt M, Meyer AH, Unger L, Marek GJ, Mezler M. Heterocomplex formation of 5-HT2A-mGlu2 and its relevance for cellular signaling cascades. Neuropharmacology. 2012;62:2184–2191. doi: 10.1016/j.neuropharm.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Emanuele E, Colombo R, Martinelli V, Brondino N, Marini M, Boso M, Barale F, Politi P. Elevated urine levels of bufotenine in patients with autistic spectrum disorders and schizophrenia. Neuro Endocrinol Lett. 2010;31:117–121. [PubMed] [Google Scholar]
- Faurbye A, Pind K. Occurrence of bufotenin in the urine of schizophrenic patients and normal persons. Nature. 1968;220:489. doi: 10.1038/220489a0. [DOI] [PubMed] [Google Scholar]
- Fontanilla D, Johannessen M, Hajipour AR, Cozzi NV, Jackson MB, Ruoho AE. The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science (New York, NY. 2009;323:934–937. doi: 10.1126/science.1166127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedland CS, Mansbach RS. Behavioral profile of constituents in ayahuasca, an Amazonian psychoactive plant mixture. Drug and alcohol dependence. 1999;54:183–194. doi: 10.1016/s0376-8716(98)00154-9. [DOI] [PubMed] [Google Scholar]
- Fribourg M, Moreno JL, Holloway T, Provasi D, Baki L, Mahajan R, Park G, Adney SK, Hatcher C, Eltit JM, Ruta JD, Albizu L, Li Z, Umali A, Shim J, Fabiato A, MacKerell AD, Jr, Brezina V, Sealfon SC, Filizola M, Gonzalez-Maeso J, Logothetis DE. Decoding the signaling of a GPCR heteromeric complex reveals a unifying mechanism of action of antipsychotic drugs. Cell. 2011;147:1011–23. doi: 10.1016/j.cell.2011.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Maeso J, Ang RL, Yuen T, Chan P, Weisstaub NV, Lopez-Gimenez JF, Zhou M, Okawa Y, Callado LF, Milligan G, Gingrich JA, Filizola M, Meana JJ, Sealfon SC. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature. 2008;452:93–97. doi: 10.1038/nature06612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Maeso J, Weisstaub NV, Zhou M, Chan P, Ivic L, Ang R, Lira A, Bradley-Moore M, Ge Y, Zhou Q, Sealfon SC, Gingrich JA. Hallucinogens recruit specific cortical 5-HT(2A) receptor-mediated signaling pathways to affect behavior. Neuron. 2007;53:439–452. doi: 10.1016/j.neuron.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Griffiths R, Richards W, Johnson M, McCann U, Jesse R. Mystical-type experiences occasioned by psilocybin mediate the attribution of personal meaning and spiritual significance 14 months later. Journal of psychopharmacology (Oxford, England) 2008;22:621–632. doi: 10.1177/0269881108094300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths RR, Johnson MW, Richards WA, Richards BD, McCann U, Jesse R. Psilocybin occasioned mystical-type experiences: immediate and persisting dose-related effects. Psychopharmacology. 2011;218:649–665. doi: 10.1007/s00213-011-2358-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinspoon L, Bakalar JB. The psychedelic drug therapies. Curr Psychiatr Ther. 1981;20:275–283. [PubMed] [Google Scholar]
- Grinspoon L, Bakalar JB. Can drugs be used to enhance the psychotherapeutic process? Am J Psychother. 1986;40:393–404. doi: 10.1176/appi.psychotherapy.1986.40.3.393. [DOI] [PubMed] [Google Scholar]
- Halberstadt AL, Buell MR, Masten VL, Risbrough VB, Geyer MA. Modification of the effects of 5-methoxy-N,N-dimethyltryptamine on exploratory behavior in rats by monoamine oxidase inhibitors. Psychopharmacology. 2008;201:55–66. doi: 10.1007/s00213-008-1247-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberstadt AL, Geyer MA. Characterization of the head-twitch response induced by hallucinogens in mice: detection of the behavior based on the dynamics of head movement. Psychopharmacology. 2013;227:727–739. doi: 10.1007/s00213-013-3006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MW. Facilitation of cognitive behavioral therapy for smoking cessation using the 5-HT2A agonist psilocybin College on Problems of Drug Dependence, San Diego. 2013 [Google Scholar]
- Johnson MW, Sewell RA, Griffiths RR. Psilocybin dose-dependently causes delayed, transient headaches in healthy volunteers. Drug and alcohol dependence. 2012;123:132–140. doi: 10.1016/j.drugalcdep.2011.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean KA, Johnson MW, Griffiths RR. Mystical experiences occasioned by the hallucinogen psilocybin lead to increases in the personality domain of openness. Journal of psychopharmacology (Oxford, England) 2011;25:1453–1461. doi: 10.1177/0269881111420188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marona-Lewicka D, Chemel BR, Nichols DE. Dopamine D4 receptor involvement in the discriminative stimulus effects in rats of LSD, but not the phenethylamine hallucinogen DOI. Psychopharmacology. 2009;203:265–277. doi: 10.1007/s00213-008-1238-0. [DOI] [PubMed] [Google Scholar]
- Marona-Lewicka D, Thisted RA, Nichols DE. Distinct temporal phases in the behavioral pharmacology of LSD: dopamine D2 receptor-mediated effects in the rat and implications for psychosis. Psychopharmacology. 2005;180:427–435. doi: 10.1007/s00213-005-2183-9. [DOI] [PubMed] [Google Scholar]
- Moreno JL, Holloway T, Albizu L, Sealfon SC, Gonzalez-Maeso J. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493:76–79. doi: 10.1016/j.neulet.2011.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai F, Nonaka R, Satoh Hisashi Kamimura K. The effects of non-medically used psychoactive drugs on monoamine neurotransmission in rat brain. European journal of pharmacology. 2007;559:132–137. doi: 10.1016/j.ejphar.2006.11.075. [DOI] [PubMed] [Google Scholar]
- Nichols DE. Differences between the mechanism of action of MDMA, MBDB, and the classic hallucinogens. Identification of a new therapeutic class: entactogens. Journal of psychoactive drugs. 1986;18:305–313. doi: 10.1080/02791072.1986.10472362. [DOI] [PubMed] [Google Scholar]
- Nichols DE. Hallucinogens. Pharmacology & therapeutics. 2004;101:131–181. doi: 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Oehen P, Traber R, Widmer V, Schnyder U. A randomized, controlled pilot study of MDMA (+/− 3,4-Methylenedioxymethamphetamine)-assisted psychotherapy for treatment of resistant, chronic Post-Traumatic Stress Disorder (PTSD) Journal of psychopharmacology (Oxford, England) 2013;27:40–52. doi: 10.1177/0269881112464827. [DOI] [PubMed] [Google Scholar]
- Parrott AC. The psychotherapeutic potential of MDMA (3,4-methylenedioxymethamphetamine): an evidence-based review. Psychopharmacology. 2007;191:181–193. doi: 10.1007/s00213-007-0703-5. [DOI] [PubMed] [Google Scholar]
- Reimann W, Schneider F. The serotonin receptor agonist 5-methoxy-N,Ndimethyltryptamine facilitates noradrenaline release from rat spinal cord slices and inhibits monoamine oxidase activity. Gen Pharmacol. 1993;24:449–453. doi: 10.1016/0306-3623(93)90331-q. [DOI] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Ayestas MA, Dersch CM, Baumann MH. Aminorex, fenfluramine, and chlorphentermine are serotonin transporter substrates. Implications for primary pulmonary hypertension. Circulation. 1999;100:869–875. doi: 10.1161/01.cir.100.8.869. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39:32–41. doi: 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Katsnelson M, Vu N, Partilla JS, Dersch CM, Blough BE, Baumann MH. Interaction of the anorectic medication, phendimetrazine, and its metabolites with monoamine transporters in rat brain. European journal of pharmacology. 2002;447:51–57. doi: 10.1016/s0014-2999(02)01830-7. [DOI] [PubMed] [Google Scholar]
- Schmid CL, Bohn LM. Serotonin, but not N-methyltryptamines, activates the serotonin 2A receptor via a ss-arrestin2/Src/Akt signaling complex in vivo. J Neurosci. 2010;30:13513–13524. doi: 10.1523/JNEUROSCI.1665-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, Raehal KM, Bohn LM. Agonist-directed signaling of the serotonin 2A receptor depends on beta-arrestin-2 interactions in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:1079–1084. doi: 10.1073/pnas.0708862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P, Ko F, Tallerico T. Dopamine receptor contribution to the action of PCP, LSD and ketamine psychotomimetics. Mol Psychiatry. 2005;10:877–883. doi: 10.1038/sj.mp.4001682. [DOI] [PubMed] [Google Scholar]
- Shen HW, Jiang XL, Yu AM. Nonlinear pharmacokinetics of 5-methoxy-N,N-dimethyltryptamine in mice. Drug metabolism and disposition: the biological fate of chemicals. 2011;39:1227–1234. doi: 10.1124/dmd.111.039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulgin A, Shulgin A. TiHKAL The Continuation. Transform Press, Berkeley. 1997 [Google Scholar]
- Shulgin AT, Carter MF. N, N-Diisopropyltryptamine (DIPT) and 5-methoxy-N,N-diisopropyltryptamine (5-MeO-DIPT). Two orally active tryptamine analogs with CNS activity. Communications in psychopharmacology. 1980;4:363–369. [PubMed] [Google Scholar]
- Sogawa C, Sogawa N, Tagawa J, Fujino A, Ohyama K, Asanuma M, Funada M, Kitayama S. 5-Methoxy-N,N-diisopropyltryptamine (Foxy), a selective and high affinity inhibitor of serotonin transporter. Toxicology letters. 2007;170:75–82. doi: 10.1016/j.toxlet.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Su TP, Hayashi T, Vaupel DB. When the endogenous hallucinogenic trace amine N,N-dimethyltryptamine meets the sigma-1 receptor. Sci Signal. 2009;2:pe12. doi: 10.1126/scisignal.261pe12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walentiny DM, Vann RE, Warner JA, King LS, Seltzman HH, Navarro HA, Twine CE, Jr, Thomas BF, Gilliam AF, Gilmour BP, Carroll FI, Wiley JL. Kappa opioid mediation of cannabinoid effects of the potent hallucinogen, salvinorin A, in rodents. Psychopharmacology. 2010;210:275–284. doi: 10.1007/s00213-010-1827-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FJ, Appel JB. Lysergic acid diethylamide (LSD) and lisuride: differentiation of their neuropharmacological actions. Science (New York, NY. 1982;216:535–537. doi: 10.1126/science.7071600. [DOI] [PubMed] [Google Scholar]