

Abstract
Mucolipidosis type II (MLII), or I-Cell Disease, is a rare, but severe disorder affecting localization of enzymes to the lysosome, generally resulting in death before the 10th birthday. Although hematopoietic stem cell transplant (HSCT) has been used to successfully treat some lysosomal storage diseases, there have been only two case reports in the use of HSCT to treat MLII. For the first time, we describe the combined international experience in the use of HSCT for MLII in 22 patients. Although 95% of the patients engrafted, the overall survival was low with only 6 patients (27%) alive at last follow-up. The most common cause of death post-transplant was cardiovascular complications, most likely due to disease progression. Survivors were globally delayed in development, and often required complex medical support such as gastrostomy tubes for nutrition, and tracheostomy with mechanical ventilation. Although HSCT has demonstrated efficacy in treating some lysosomal storage disorders, the neurologic outcome and survival for patents with MLII were poor. Therefore new medical and cellular therapies should be sought for these patients.
Introduction
Mucolipidosis type II (MLII), or I-cell Disease, is a rare autosomal recessive disorder caused by mutation in the GNPTAB gene on chromosome 12. This gene encodes the α/β subunits of the enzyme N-acetylglucosamine-1-phosphotransferase (GNPT) which acts to couple phosphate groups to mannose residues (mannose-6-phosphate moieties, M6P) on enzymes destined to be targeted to the lysosome. Mutation in GNPTAB, with resultant abnormal GNPT function, can lead to inappropriate trafficking of lysosomal enzyme to the extracellular compartment. Pathologically, dense and dark granules, visible on phase-contrast microscopy, fill the cytoplasm of cultured fibroblasts, so-called inclusion cells, or I-cells. A complete absence of GNPT activity results in the severe storage disease, MLII, characteristically evident in infancy or even prenatally.
Clinical findings include coarse facial features, dysostosis multiplex, growth failure, global development delay, generalized hypotonia but stiff joints, and recurrent respiratory infections progressive. Death often occurs in the first decade of life from cardio-pulmonary disease1-3. For patients with lysosomal storage disorders, HSCT aims to provide donor-derived hematopoietic cells that produce lysosomal enzymes with M6P moiety allowing for intracellular uptake with appropriate trafficking to the lysosomal for substrate degradation. This “cross-correction” provided by transplanted cells has been used successfully for treatment of several enzyme-specific lysosomal storage disorders notably mucopolysaccharidosis type I, Gaucher disease, and alpha-mannodidosis4-11. However, in the case of I-cell Disease, the gene product is not a soluble enzyme capable of internalization through binding to M6P receptors on the cell surface. The use of HSCT to treat I-cell Disease has not been previously reported beyond two case reports 12,13. We now describe the largest collection of outcome data on 22 patients with I-cell Disease who underwent HSCT.
Methods
Data Collection
Patient-, disease- and transplant-related data were obtained from the Center for International Blood and Marrow Transplant Research (CIBMTR). The CIBMTR is a voluntary working group of more than 450 transplantation centers that contribute detailed data on consecutive allogeneic and autologous transplantations to a Statistical Center at the Medical College of Wisconsin in Milwaukee or the National Marrow Donor Program in Minneapolis. Participating centers are required to report all transplants consecutively and compliance is monitored by on-site audits. Patients are followed longitudinally until death or until they are lost to follow-up. All patients provided written informed consent for data submission and research participation. The study was approved by the Institutional Review Boards of the Medical College of Wisconsin and the National Marrow Donor Program.
Inclusion criteria
Patients were identified by query of the CIBMTR database for the diagnoses “mucolipidosis, type II” or “I-cell Disease.” Records were reviewed to verify the molecular or enzyme-based diagnosis. All patients undergoing first or second allogeneic HSCT for MLII were considered.
Outcomes
The primary endpoint was overall survival (OS). Other outcomes studied include time to neutrophil engraftment (defined as the first day achieving an absolute neutrophil count ≥0.5 × 109/L for three consecutive measurements), platelet engraftment (defined as the first day when platelets remained ≥ 20 × 109/L without transfusions for 7 days), incidence of acute graft-versus-host disease (GvHD), and chronic GvHD. Acute and chronic GVHD were defined using standard criteria 14,15.
Results
Patient and Graft Characteristics
We identified 22 patients reported to the CIBMTR who underwent allogeneic HSCT for MLII or I-cell disease (summarized in Table 1). Patients were transplanted at a median age of 9 months (range 2 – 23 months) and at a median of 3 months after diagnosis (range 2 – 20 months). There were 12 females and 10 males in this cohort with the majority having Lansky performance scores of 80 or higher at transplantation. Cell sources varied with 14 patients receiving unrelated umbilical cord blood (UCB) with the majority of these being 5/6 or 6/6 HLA-matched to the recipient. Of the remaining 8 patients, 3 patients received HLA-matched sibling bone marrow (carrier status was unknown), one patient received bone marrow from the mother, another received bone marrow from the father, and three patients received bone marrow from unrelated adult donors (n=1 HLA-matched; n=2 HLA-mismatched).
Table 1.
Patient and Transplant Characteristics. Performance scores are given as Lansky scores. Chemotherapy regimens consisted of anti-thymoglobulin (ATG), busulfan (Bu), cytoxan (Cy), melphalan (Mel), fludarabine (Flu) and the conditioning intent classified as either myeloablative (MAC) or reduced intensity conditioning (RIC). Graft versus host prophylaxis: cyclosporin (CSA), mycophenolate mofetil (MMF), methotrexate (MTX). Data which was not available is listed as n/a.
| Patient ID |
Year of Transpl ant |
Time from Dx to Transplant (months) |
Age at transplant (months) |
Sex | Performance score at transplant |
HLA-Match and Graft Source |
Conditioning regimen |
GVHD prophylaxis | aGVHD grade Gluksberg |
cGVHD | HSCT to last contat (months) |
Status | Cause of death | GNTPAB Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2005 | n/a | 9 | Female | 100 | 5/6 UCB | ATG, Bu, Cy (MAC) | Seroids + CSA | 2 | No | 8.5 | Dead | Organ Failure | Diagnosed by enzyme testing, no molecualr data. |
| 2 | 2006 | n/a | 4 | Male | 100 | 6/8 unrelated BM | ATG, Bu, Cy (MAC) | ATG, CSA, MTX | 3 | No | 54.1 | Alive | Diagnosed by enzyme testing, no molecualr data. | |
| 3 | 2007 | n/a | 13 | Male | 90 | 5/6 UCB | Mel, Clof, 200 cGyTBI (RIC) | CSA, MMF | 0 | No | 62 | Alive | Frameshift detected, second mutation not detected by standard sequencing. | |
| 4 | 2000 | n/a | 14 | Male | 90 | 4/8 unrelated BM | Bu, Cy (MAC) | TCD, ATG, Seroids, CSA | 0 | No | 27.6 | Dead | Organ Failure | Frameshift + missense mutations giving a diagnosis of either intermediate MLII or ML III. |
| 5 | 2002 | n/a | 4 | Female | 70 | 6/6 UCB | ATG, Bu, Cy (MAC) | Steroids, CSA | 2 | Yes | 5.5 | Dead | Interstitial pnueumonia | Diagnosed by enzyme testing, no molecualr data. |
| 6 | 2002 | n/a | 13 | Male | 60 | 6/6 unrelated BM | Campath, Flu (RIC) | Tacrolimus | 2 | Yes | 117.4 | Alive | Diagnosed by enzyme testing, no molecualr data. | |
| 7 | 2007 | 19 | Female | 100 | 6/6 UCB | ATG, Bu, Cy (MAC) | Tacrolimus, MMF | 0 | No | 62.8 | Alive | Compound heterozygous frameshift mutation + nonsense muation. | ||
| 8 | 1999 | 3 | 4 | Male | n/a | HLA match unknown UCB | ATG, Bu, Mel (MAC) | Steroids, CSA | n/a | 6.1 | Dead | Organ Failure | Diagnosed by enzyme testing, no molecualr data. | |
| 9 | 1997 | 20 | 20 | Female | 80 | Sibling BM | Bu, Cy, 750 cGy TBI (MAC) | TCD | 2 | No | 171.1 | Alive | Frameshift + a splice change givng a milder than expected phenotype, intermediate MLII/MLIII. | |
| 10 | 2000 | 2 | 4 | Male | >=80 | 5/6 related BM from Mother | ATG, Bu, Cy (MAC) | Steroids, CSA | 0 | No | 40.1 | Dead | Primary Disease | Compound heterozygous frameshift mutations. |
| 11 | 2004 | 3 | 23 | Female | n/a | 6/6 UCB | ATG, Bu, Cy (MAC) | Steroids, CSA | 0 | n/a | 98 | Dead | Unknown | Compound heterozygous frameshift mutation + nonsense muation. |
| 12 | 2005 | 3 | 6 | Female | >=80 | 5/6 UCB | ATG, Cy (RIC) | CSA, MMF | 2 | No | 57.2 | Dead | Infection | Compound heterozygous frameshift mutation + nonsense muation. |
| 13 | 2005 | 5 | 18 | Female | >=80 | 6/6 UCB | ATG, Cy (RIC) | CSA, MMF | 0 | No | 90 | Dead | Primary Disease | Homozygous frameshift mutation. |
| 14 | 1991 | n/a | n/a | Male | n/a | Other related BM | Cy, TBI (MAC) | CSA | n/a | n/a | 2.9 | Dead | Unknown | Diagnosed by enzyme testing, no molecualr data. |
| 15 | 1996 | 2 | 9 | Male | 90 | Silbling BM | ATG, Bu, Cy (MAC) | CSA, MTX | 1 | No | 31.7 | Dead | Interstitial Pnueumonia | Diagnosed by enzyme testing, no molecualr data. |
| 16 | 2008 | 3 | 15 | Male | 90 | 5/6 UCB | ATG, Bu, Cy (MAC) | CSA, MMF | 2 | Yes | 22.7 | Dead | Organ failure | Frameshift + a splice change giving a milder than expected phenotype, intermediate MLII/MLIII. |
| 17 | 2008 | 10 | 10 | Female | 100 | 5/6 UCB | Campath, Bu, Cy (MAC) | Tacrolimus, MTX | 0 | No | 0.6 | Dead | Interstitial pnueumonia | Diagnosed by enzyme testing, no molecualr data. |
| 18 | 2008 | 8 | 8 | Female | n/a | 5/6 UCB | ATG, Bu, Flu (MAC) | Steroids, CSA | 2 | No | 5.3 | Dead | Organ failure | Compound heterozygous frameshift mutation + nonsense muation. |
| 19 | 2010 | 3 | 3 | Female | 100 | 6/6 UCB | Campath, Mel, Clof, 200 cGy TBI (RIC) | CSA, MMF | 0 | No | 8.3 | Dead | Primary Disease | Homozygous nonsense mutation. |
| 20 | 2010 | 2 | 2 | Female | 100 | Sibling BM | ATG, Bu, Flu (MAC) | CSA | 0 | No | 5.4 | Dead | Organ Failure | Compound heterozygous frameshift mutation + nonsense muation. |
| 21 | 2010 | 10 | 10 | Male | 100 | 5/6 UCB | ATG, Bu, Cy (MAC) | Steroids, CSA | 2 | No | 89 | Alive | No repsonse from center. | |
| 22 | 2011 | 5 | 5 | Female | 90 | 4/6 UCB | ATG, Bu, Cy (MAC) | Steroids, CSA | 0 | No | 26 | Dead | Pulmonary Failure | Homozygous frameshift mutation. |
Preparative regimens varied. Most were busulfan–based and myeloablative in intent, though five patients received a reduced intensity regimen. GVHD prophylaxis consisted largely of cyclosporine (CSA) and steroids, though other regimens were also employed.
Thirteen of 22 patients had documented DNA mutation analysis confirming the diagnosis of MLII. Interestingly, three patients (patients 4, 9, and 17) were found to have a genotype consistent with the less severe Intermediate MLII/MLIII. It has been reported that these affected patients have mutations in the same GNPTAB gene as MLII, though retain some GNTP activity and have a milder phenotype16. However, one of these three died of organ failure more than 2 years following transplant, which suggests that mortality in this case resulted from underlying disease. On the other hand, one of these also died of pneumonitis shortly after transplant, a complication that was likely due to the transplant procedure itself.
Outcomes
Primary engraftment was obtained in 19/22 patients with a median time to neutrophil and platelet engraftment of 17 days and 37.5 days respectively. Engraftment status was not known in two patients; however both died within 6 months. One patient had early secondary graft failure defined as sustained loss of neutrophil recovery in the absence of infection. This patient (patient#6) received a second transplant within two months from the first transplant and a third transplant 15 months after the second transplant. The donor source was adult unrelated donor. This patient is alive, approximately 10 years from first transplantation. Another patient (patient#9) developed a post-transplant lymphoma (PTL) in the liver and was treated with surgical resection and two infusions (one month apart) of sibling donor lymphocytes at 10.5 and 11.5 months post-transplant with resolution of her PTL. Five of the six survivors listed in Table 1 had 100% donor chimerism at 1 – 2 years post HSCT (no chimerism data was available for patient#21).
Grade 1 - 2 acute GvHD was documented in nine patients. Grade 3 aGVHD was seen in only one patient. There were three patients with documented chronic GVHD, and both had been previously diagnosed with Grade 2 aGVHD. The probability of 5-year overall survival was 33% (95% CI 15 - 55) with a median follow-up 67 months (Figure 1); 27% of patients (n=6) were alive at last contact. The median time to death was 27.6 months. The most common cause of death given was “organ failure” (6 patients) followed by primary disease progression (3 patients) and interstitial pneumonia (3 patients).
Figure 1.
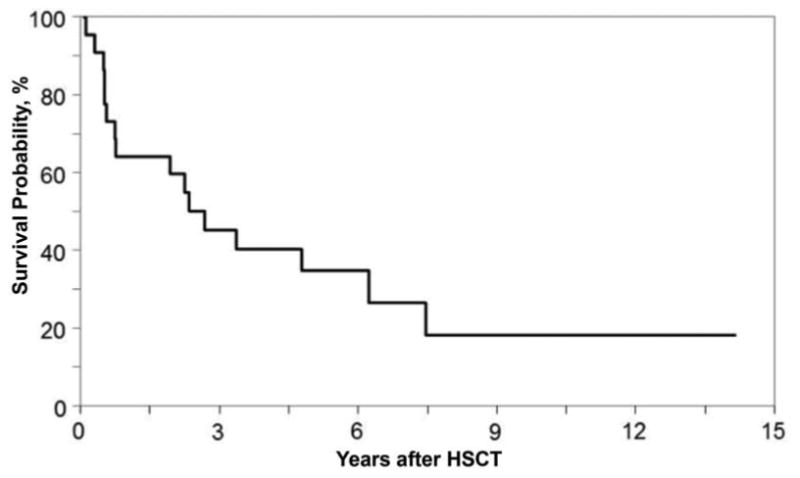
Kaplan-Meier estimate of overall survival in I-Cell patients after transplant.
While numerous reports have provided increasing amounts of information regarding survival and transplant related outcomes for lysosomal storage disorders, the prevalence and severity of developmental delays associated with these disorders has been more difficult to measure. Consistent neuropsychiatric measures that are used across multiple centers would be ideal in these assessments, but were not available in this cohort of patients. We attempted a basic functional evaluation by sending a simple questionnaire to transplant centers to inquire as to the status of surviving patients and received information on six patients, one of whom had genetic testing indicating an Intermediate form of MLII/MLIII (Table 2). There was significant morbidity in these post-transplant MLII patients that was likely related to their underlying disease, as they were described as requiring gastrostomy tubes for nutrition, ventilatory support per a tracheostomy, having an impaired ability to ambulate, and being very delayed in speech and learning. Though this data is far from comprehensive, it documents that a high degree of medical support was required for children with MLII post-transplant, and that much of this support appears to be related to the underlying disease rather than the transplant procedure.
Table 2.
Summary of results of questionnaire to assess neurologic status in MLII patients after transplantation. Patient 9 had a molecular diagnosis consistent with Intermediate MLII/III. Two patients died soon after the assessment was taken.
| Patient ID | Gastrostomy tube | Tracheostomy | Wheelchair use | Can they walk? | Speech? | Chronologic age | In School? | Pottytrainied | Status |
|---|---|---|---|---|---|---|---|---|---|
| 3 | yes | yes | yes | never | no | 7 years | functions at a 6-9 month old level | no | Alive |
| 7 | yes | yes | yes | 8 years | never in school, homebound teacher 1 day per week | Alive | |||
| 9 | no | no | sometimes | yes | some | 18 years | functions at a 4 year old level | yes | Alive |
| 13 | yes | yes | yes | never | no | 9 years | no | no | Died |
| 21 | yes | yes | no | no, and no wheelchair | 4 years | no | Alive | ||
| 22 | yes | no | custom wheelchair | never walked | no | 2.5 years | never achieved age for education | no | Dead |
Discussion
We have described the largest series of patients undergoing HSCT for Mucolipidosis type II, or I-cell Disease. HSCT has been used for the metabolic “cross-correction” of lysosomal storage diseases for over 30 years, with the majority of patients having severe mucopolysaccharidosis type I (MPSI-H). From these patients we have learned that HSCT can arrest the fatal neurodegeneration of MPSI-H allowing patients to live significantly longer with improved function and quality of life. Patients undergoing HSCT for MPSI-H earlier in life (ideally prior to age 2 years) have been shown to lose less developmental ground and perform better. However, while HSCT may help alleviate visceral and neurological disease associated with MPSI-H, its impact on orthopedic and cardiac valve pathology may be limited 8,17-19.
The overall survival at last follow-up in our cohort is strikingly low at 27%, with the median time to death of 27.6 months. This suggests that these fatalities were not due to peri-transplant related toxicity, but due to disease related complications that were not alleviated with transplantation. Specifically, although several reported causes of death were listed as “organ failure,” as these occurred later after transplant, we presume that some of these were due to progressive disease. Because MLII is rare, its natural history is documented mainly as case reports or small case series. These reports suggest that most patients do not survive past the first decade and die from cardio-respiratory complications 3,20. Although our data show that there are two patients post-transplant over the age of 10 (patients 6 and 9), patient 9 (who has the longest follow-up) was later determined to have an intermediate form of MLII/MLIII on review of her mutation. Interestingly, patient 9 was one of the first case reports of HSCT for I-Cell Disease12.
The correlation of phenotype to genotype has only recently been reported by Cathey et al. who described, for the first time, 61 patients with mutations in GNPTAB and found that some mutations fell into clinical separations of MLII (I-Cell), MLIII, and an intermediate form of MLII/MLIII. On this basis, we cannot conclude that HSCT favorably changed the clinical outcome for this patient, as her survival and neurologic status are not clearly different than that of an Intermediate MLII/III patient. In comparison to classic MLII, Intermediate MLII/III patients tend to be taller, develop complications at a later age, have less pronounced skeletal issues, less severe neurocognitive deficits, and a greater life span16. Furthermore, upon evaluation of the available molecular mutations for patients in this cohort, three patients had mutations consistent with a milder phenotype than the classical MLII (Table 1).
Our limited neurologic follow-up showed that after transplant, many of the children are still severely affected by their disease, requiring significant medical interventions. It is unclear if they displayed any “true” improvement in neurological development. HSCT has been attempted for several lysosomal storage diseases, though not all diseases are improved after HSCT. Also, HSCT outcomes are generally poor for patients who are in an advanced neurologic decline prior to transplant. We can speculate on several reasons for poor outcome after HSCT. It is believed that delivery of lysosomal enzymes by donor-derived hematopoietic cells requires the 6-mannose phosphate moiety to allow targeting to the lysosome. In the case of MLII, disruption of the pathway by which the M6P signal is associated with the enzyme affects not just one enzyme, but numerous enzymes. This results in inappropriate secretion and insufficient delivery of multiple enzymes to the lysosome. The result is the accumulation of macromolecular substrates that create inclusions in cells (giving rise to the namesake I-Cells), ultimately, making MLII a very severe form of lysosomal storage disease due to insufficient activity of most of the lysosomal enzymes. Evaluation of lysosomal enzyme activity in MPSI patients can show correction toward normal after HSCT, but in MLII patients, the inappropriate plasma secretion of several enzymes (ß-galactosidase, ß-hexosaminidase A, α-mannosidase, ß-mannosidase, ß-glucoronidase, α-glucosaminidase, α-L-fucosidase) remains significantly elevated months to years after successful HSCT (personal observation). One could speculate that this represents an incomplete lysosomal correction of disease by HSCT insufficient to prevent continued pathologic consequences.
In conclusion, we have little to no evidence that HSCT leads to improved clinical/neurological outcomes in MLII. Overall patient survival is poor, as is quality of life among those who do survive. New modifications to HSCT or complementary therapies such as gene therapy, enzyme replacement, or substrate reduction are needed to improve outcomes in this population of children.
Acknowledgments
The CIBMTR is supported by Public Health Service Grant/Cooperative Agreement U24-CA076518 from the National Cancer Institute (NCI), the National Heart, Lung and Blood Institute (NHLBI) and the National Institute of Allergy and Infectious Diseases (NIAID); a Grant/Cooperative Agreement 5U10HL069294 from NHLBI and NCI; a contract HHSH250201200016C with Health Resources and Services Administration (HRSA/DHHS); two Grants N00014-12-1-0142 and N00014-13-1-0039 from the Office of Naval Research; and grants from *Actinium Pharmaceuticals; Allos Therapeutics, Inc.; *Amgen, Inc.; Anonymous donation to the Medical College of Wisconsin; Ariad; Be the Match Foundation; *Blue Cross and Blue Shield Association; *Celgene Corporation; Chimerix, Inc.; Fred Hutchinson Cancer Research Center; Fresenius-Biotech North America, Inc.; *Gamida Cell Teva Joint Venture Ltd.; Genentech, Inc.;*Gentium SpA; Genzyme Corporation; GlaxoSmithKline; Health Research, Inc. Roswell Park Cancer Institute; HistoGenetics, Inc.; Incyte Corporation; Jeff Gordon Children's Foundation; Kiadis Pharma; The Leukemia & Lymphoma Society; Medac GmbH; The Medical College of Wisconsin; Merck & Co, Inc.; Millennium: The Takeda Oncology Co.; *Milliman USA, Inc.; *Miltenyi Biotec, Inc.; National Marrow Donor Program; Onyx Pharmaceuticals; Optum Healthcare Solutions, Inc.; Osiris Therapeutics, Inc.; Otsuka America Pharmaceutical, Inc.; Perkin Elmer, Inc.; *Remedy Informatics; *Sanofi US; Seattle Genetics; Sigma-Tau Pharmaceuticals; Soligenix, Inc.; St. Baldrick's Foundation; StemCyte, A Global Cord Blood Therapeutics Co.; Stemsoft Software, Inc.; Swedish Orphan Biovitrum; *Tarix Pharmaceuticals; *TerumoBCT; *Teva Neuroscience, Inc.; *THERAKOS, Inc.; University of Minnesota; University of Utah; and *Wellpoint, Inc. The views expressed in this article do not reflect the official policy or position of the National Institute of Health, the Department of the Navy, the Department of Defense, Health Resources and Services Administration (HRSA) or any other agency of the U.S. Government.
Footnotes
Corporate Members
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cathey SS, Kudo M, Tiede S, et al. Molecular order in mucolipidosis II and III nomenclature. Am J Med Genet A. 2008;146A(4):512–513. doi: 10.1002/ajmg.a.32193. [DOI] [PubMed] [Google Scholar]
- 2.Wenger DA, Sattler M, Clark C, Wharton C. I-cell disease: activities of lysosomal enzymes toward natural and synthetic substrates. Life Sci. 1976;19(3):413–420. doi: 10.1016/0024-3205(76)90047-3. [DOI] [PubMed] [Google Scholar]
- 3.Leroy JG, Spranger JW, Feingold M, Opitz JM, Crocker AC. I-cell disease: a clinical picture. J Pediatr. 1971;79(3):360–365. doi: 10.1016/s0022-3476(71)80142-7. [DOI] [PubMed] [Google Scholar]
- 4.Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science. 1968;162(3853):570–572. doi: 10.1126/science.162.3853.570. [DOI] [PubMed] [Google Scholar]
- 5.Prasad VK, Kurtzberg J. Transplant outcomes in mucopolysaccharidoses. Semin Hematol. 2010;47(1):59–69. doi: 10.1053/j.seminhematol.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 6.Beck M. Therapy for lysosomal storage disorders. IUBMB Life. 2010;62(1):33–40. doi: 10.1002/iub.284. [DOI] [PubMed] [Google Scholar]
- 7.Hoogerbrugge PM, Brouwer OF, Bordigoni P, et al. Allogeneic bone marrow transplantation for lysosomal storage diseases. The European Group for Bone Marrow Transplantation. Lancet. 1995;345(8962):1398–1402. doi: 10.1016/s0140-6736(95)92597-x. [DOI] [PubMed] [Google Scholar]
- 8.Lund TC. Hematopoietic stem cell transplant for lysosomal storage diseases. Pediatr Endocrinol Rev. 2013;11(Suppl 1):91–98. [PubMed] [Google Scholar]
- 9.Martin PL, Carter SL, Kernan NA, et al. Results of the cord blood transplantation study (COBLT): outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with lysosomal and peroxisomal storage diseases. Biol Blood Marrow Transplant. 2006;12(2):184–194. doi: 10.1016/j.bbmt.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 10.Krivit W. Allogeneic stem cell transplantation for the treatment of lysosomal and peroxisomal metabolic diseases. Springer seminars in immunopathology. 2004;26(1-2):119–132. doi: 10.1007/s00281-004-0166-2. [DOI] [PubMed] [Google Scholar]
- 11.Mynarek M, Tolar J, Albert MH, et al. Allogeneic hematopoietic SCT for alpha-mannosidosis: an analysis of 17 patients. Bone Marrow Transplant. 2012;47(3):352–359. doi: 10.1038/bmt.2011.99. [DOI] [PubMed] [Google Scholar]
- 12.Grewal S, Shapiro E, Braunlin E, et al. Continued neurocognitive development and prevention of cardiopulmonary complications after successful BMT for I-cell disease: a long-term follow-up report. Bone marrow transplantation. 2003;32(9):957–960. doi: 10.1038/sj.bmt.1704249. [DOI] [PubMed] [Google Scholar]
- 13.Tang X, Hinohara T, Kato S, Watanabe K, Tsutsumi Y. I-cell disease: report of an autopsy case. Tokai J Exp Clin Med. 1995;20(2):109–120. [PubMed] [Google Scholar]
- 14.Przepiorka D, Weisdorf DJ, Martin P, et al. 1994 Concensus conference on acute GVHD grading. Bone Marrow Transplant. 1995;15(6):825–828. [PubMed] [Google Scholar]
- 15.Flowers ME, Kansu E, Sullivan KM. Pathophysiology and treatment of graft-versus-host disease. Hematol Oncol Clin North Am. 1999;13(5):1091–1112. doi: 10.1016/s0889-8588(05)70111-8. [DOI] [PubMed] [Google Scholar]
- 16.Cathey SS, Leroy JG, Wood T, et al. Phenotype and genotype in mucolipidoses II and III alpha/beta: a study of 61 probands. Journal of medical genetics. 2010;47(1):38–48. doi: 10.1136/jmg.2009.067736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boelens JJ, Aldenhoven M, Purtill D, et al. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler's syndrome after myeloablative conditioning. Blood. 2013;121(19):3981–3987. doi: 10.1182/blood-2012-09-455238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boelens JJ, Wynn RF, O'Meara A, et al. Outcomes of hematopoietic stem cell transplantation for Hurler's syndrome in Europe: a risk factor analysis for graft failure. Bone Marrow Transplant. 2007;40(3):225–233. doi: 10.1038/sj.bmt.1705718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braunlin EA, Berry JM, Whitley CB. Cardiac findings after enzyme replacement therapy for mucopolysaccharidosis type I. Am J Cardiol. 2006;98(3):416–418. doi: 10.1016/j.amjcard.2006.02.047. [DOI] [PubMed] [Google Scholar]
- 20.Okada S, Owada M, Sakiyama T, Yutaka T, Ogawa M. I-cell disease: clinical studies of 21 Japanese cases. Clin Genet. 1985;28(3):207–215. doi: 10.1111/j.1399-0004.1985.tb00388.x. [DOI] [PubMed] [Google Scholar]