

Abstract
Background
Methylphenidate (MPH), a psychostimulant drug for the treatment of attention-deficit hyperactivity disorder (ADHD), produces the effects of increasing alertness and improving attention, while its misuse has been associated with an increased risk of aggression and psychosis. In this study, we sought to determine the molecular mechanism underlying the complex actions of MPH.
Methods
Adolescent (4-week-old) rats were given one injection of MPH at different doses. The impact of MPH on glutamatergic signaling in pyramidal neurons of prefrontal cortex (PFC) was measured. MPH-induced behavioral changes were also examined in parallel.
Results
We found that administration of low-dose (0.5 mg/kg) MPH selectively potentiated NMDAR-mediated excitatory synaptic currents (EPSCs) via adrenergic receptor activation, while the high-dose (10 mg/kg) MPH suppressed both NMDAR- and AMPAR-EPSCs. The dual effects of MPH on EPSCs were associated with bi-directional changes in the surface level of glutamate receptor subunits. Behavioral tests also indicated that low-dose MPH facilitated the PFC-mediated temporal order recognition memory (TORM) and attention, while animals injected with high-dose MPH exhibited significantly elevated locomotive activity. Inhibiting the function of SNAP-25, a key SNARE proteins involved in NMDAR exocytosis, blocked the increase of NMDAR-EPSC by low-dose MPH. In animals exposed to repeated stress, administration of low-dose MPH effectively restored NMDAR function and TORM via a mechanism dependent on SNAP-25.
Conclusions
Our results have provided a potential mechanism underlying the cognitive enhancing effects of low-dose MPH, as well as the psychosis-inducing effects of high-dose MPH.
Keywords: Methylphenidate, prefrontal cortex, NMDA receptors, AMPA receptors, SNAP-25, stress
Introduction
Methylphenidate (MPH) is a psychostimulant widely used for the treatment of Attention deficit hyperactivity disorder (ADHD) in adolescents and adults (1). Therapeutic dose of MPH effectively improves the cognitive function and reduces the hyperactivity in ADHD (2) as well as normal human subjects and animals (3,4). However, overdose of MPH produce agitation, restlessness and hallucinations in human beings (5) as well as hyper-locomotion and impaired cognition in animals (6). Subchronic administration of methylphenidate in juvenile rodents has been found to induce long-lasting behavioral adaptations (7,8). To achieve therapeutic benefit and minimal side-effects, it is suggested that the doses of MPH should be titrated to an optimal level.
The biochemical action of MPH is well characterized. The dopamine transporter (DAT) and norepinephrine transporter (NET) are blocked by MPH, resulting in elevated concentration of dopamine (DA) and norepinephrine (NA) at synapses (3,9,10). However, it remains unclear about the mechanisms by which therapeutic dose of MPH acutely improves cognitive functions, while overdose of MPH induces psychosis.
Prefrontal cortex (PFC) is a key brain region mediating cognitive and executive functions, including working memory, sustained attention, inhibitory response control, and cognitive flexibility (11,12). A delayed maturation in PFC (13), dysfunction of the frontostriatal circuitry (14), and hypoactivation in the frontal cortex (15,16) have been implicated in ADHD patients. Also, PFC is identified as the primary target of MPH (17). The glutamatergic pyramidal neurons are one of the major cellular constituents in the PFC. Glutamatergic transmission that controls PFC activity is pivotal for cognitive function such as working memory (11,18). Disturbed glutamate receptors are implicated in the cognitive dysfunction associated with many mental disorders (19). Thus, we speculate that glutamate receptors are potential targets of MPH critically involved in PFC-mediated cognitive functions. In this study, we examined the impact of low- vs. high-dose MPH on glutamatergic transmission in PFC of adolescent rats and its relevance to behavioral outcomes.
Materials and Methods
Animals and Reagents
Male Sprague-Dawley rats were purchased from Harlan (Indianapolis, Indiana). Upon arrival, animals were allowed 4–5 days to acclimate before the experiments. Rats at the early adolescent period (p25–30) (20) were paired-housed on a 12 hour light-dark cycle and provided ad lib access to food and water. Rats from more than one litter were contributed to each treatment to avoid litter effects. All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee of the State University of New York at Buffalo. See Supplementary Methods for details of reagents.
Animal Surgery
The delivery of peptides to the PFC was conducted as we previously described (22). See Supplementary Methods for details.
Electrophysiological Recordings
Recordings of evoked synaptic currents in prefrontal cortical slices used standard whole-cell voltage-clamp technique as we previously described (23,24). The paired pulse ratio (PPR) of NMDAR-EPSCs was calculated as described previously (25). See Supplementary Methods for details.
Biochemical Measurement of Surface and Total Proteins
Surface and total AMPA and NMDA receptors were detected as we described previously (23,24). See Supplementary Methods for details.
Repeated stress paradigm
Repeated restraint stress was carried out as we previously described (24,26). In brief, SD rats were placed in air-accessible cylinders for 2 h daily (10:00am–12:00pm) for 5–7 days (starting at p21–23). The container size was similar to the animal size, which made the animal almost immobile in the container. Experiments were performed 24 hr after the last stressor exposure.
Behavioral Testing
Temporal order recognition memory (TORM), a cognitive behavior controlled by prefrontal cortex (27), locomotor activity and attentional set-shifting tasks were performed as previously described (24,26,28). See Supplementary Methods for details.
Statistics
Experiments with two groups were analyzed statistically using unpaired Student’s t-tests. Experiments with more than two groups were subjected to one- or two-way analysis of variance (ANOVA), followed by Bonferroni’s post-hoc tests.
Results
In vivo administration of a low-dose MPH enhances NMDAR-mediated synaptic currents, while a high-dose MPH reduces glutamatergic transmission in cortical neurons
To investigate the impact of MPH on glutamate signaling, we examined the NMDAR- and AMPAR-mediated excitatory postsynaptic currents (EPSCs) in the pyramidal neurons of prefrontal cortex (PFC) from adolescent male rats (4-week-old) subjected to a single administration of low-dose (0.5 mg/kg) or high-dose (10 mg/kg) MPH. As shown in Figure 1A and 1B, two-way ANOVA analysis revealed a significant main effect of MPH treatment on NMDAR- or AMPAR-EPSC (NMDA: F2, 150 = 49.5, p < 0.001; AMPA: F2, 205 = 18.7, p <0.001). Post-hoc analysis indicated that low-dose MPH significantly potentiated NMDAR-EPSC (38%–57% increase, n = 10–13 cells/4 rats per group, p < 0.05), but not AMPAR-EPSC (<10% change, n = 14–21 cells/4 rats per group, p > 0.05). In contrast, high-dose MPH markedly reduced both NMDAR- and AMPAR-EPSC (NMDA: 26%–48% decrease, n = 10 cells/4 rats per group, p < 0.05; AMPA: 36%–47% decrease, n = 10–21 cells/4 rats per group, p < 0.01). These results suggest that MPH exerts a dose-dependent effect on glutamatergic transmission in the prefrontal cortex.
Figure 1. Low-dose MPH selectively enhances NMDAR-EPSC, while high-dose MPH reduces both NMDAR- and AMPAR-EPSC.

(A, B) Input-output curves of NMDAR-EPSC (A) or AMPAR-EPSC (B) evoked by a series of stimulation intensities in PFC pyramidal neurons from rats with a single injection (i.p.) of saline, low-dose MPH (0.5 mg/kg) or high-dose MPH (10 mg/kg). *: p < 0.05, **: p < 0.01. Inset: representative EPSC traces. Scale bars: 50 pA, 100 ms (A); 50 pA, 20 ms (B). (C, D) Bar graph showing the paired-pulse ratio (PPR) of NMDAR-EPSC (interstimulus interval: 100ms) (C) or decay time constant of NMDAR-EPSC (D) in PFC pyramidal neurons taken from animals injected with saline, low-dose MPH or high-dose MPH. Inset: representative NMDAR-EPSC traces evoked by paired pulses. #: p < 0.001. Scale bar: 50 pA, 100 ms.
To test whether the effects of MPH on the NMDAR-EPSC result from a pre- or postsynaptic mechanism, we measured the paired pulse ratio (PPR), a readout that is affected by the presynaptic transmitter release (29). As shown in Figure 1C, PPR was unchanged by low-dose MPH, but was significantly elevated by high-dose MPH (saline: 1.42 ± 0.07, n = 12, low-dose MPH: 1.41 ± 0.06, n = 13, high-dose MPH: 1.85 ± 0.09, n = 12, F2, 36 = 11.24, p < 0.001, ANOVA). It suggests that low-dose MPH regulates glutamatergic transmission mainly via a postsynaptic mechanism, while high-dose MPH might affect presynaptic glutamate release and/or postsynaptic glutamate receptors. In addition, the decay time constant was not statistically changed in animals treated with MPH at low or high doses (saline: 202.0 ± 15.9, n = 11, low-dose MPH: 252.0 ± 18.8, n = 15, high-dose MPH: 197.4 ± 12.4, n = 11, F2, 47 = 0.93, p > 0.05, ANOVA), suggesting that the elevated NMDAR-EPSC is mediated by both NR2A and NR2B subunits.
In vivo administration of a low-dose MPH increases the surface level of NMDAR subunits, while a high-dose MPH decreases surface NMDAR and AMPAR subunits
As the surface expression of glutamate receptors could determine the strength of glutamatergic transmission, we performed biotinylation and western blotting to examine the surface level of NMDAR and AMPAR subunits in cortical slices from rats treated with saline or MPH. As shown in Figure 2A, low-dose MPH (0.5mg/kg) significantly enhanced the surface level of NMDAR subunits (NR1: 89.0% ± 15.3% increase, NR2A: 117.3% ± 18.4% increase, NR2B: 242.1% ± 47.0% increase, n = 4 pairs, p < 0.001, ANOVA), but only slightly (not significantly) increased the surface level of AMPAR subunits (GluR1: 39.0% ± 9.8% increase, GluR2: 36.1% ± 21.3% increase, n = 4 pairs, p > 0.05, ANOVA). Total protein levels of all these glutamate receptor subunits were unchanged by the low-dose MPH (n = 5 pairs, p > 0.05, ANOVA).
Figure 2. Low-dose MPH increases the surface level of NMDAR subunits, while high-dose MPH decreases the surface NMDAR and AMPAR expression.

(A) Immunoblots and quantification analysis of the surface and total NMDAR and AMPAR subunits in PFC slices from rats injected with saline or MPH (0.5 mg/kg, or 2.5 mg/kg, i.p.). *: p < 0.05, #: p < 0.001. (B) Immunoblots and quantification analysis of the surface and total NMDAR and AMPAR subunits from the rats treated with saline or high-dose MPH (10 mg/kg, i.p.). *: p < 0.05.
In animals injected with a medium-dose MPH (2.5 mg/kg) (30,31), only the surface NR1 level was modestly increased (Figure 2A, 36.2% ± 15.8% increase, n = 4 pairs, p < 0.05, ANOVA), while other subunits had no significant change in their surface expression. However, a single administration of high-dose MPH (10 mg/kg) induced a substantial reduction of the surface levels of both NMDAR and AMPAR subunits (Figure 2B, surface NR1: 45.0% ± 12.6% decrease, surface NR2A: 32.7% ± 7.8% decrease, surface NR2B: 21.9% ± 7.9% decrease, surface GluR1: 34.6% ± 6.3% decrease, surface GluR1: 37.5% ± 10.6% decrease, n = 7 pairs, p < 0.05, t-test), without changing the total levels of glutamate receptors (p>0.05, t-test). Taken together, these results indicate that MPH exerts a dose-dependent, bi-directional regulation of the surface expression of glutamate receptors, which may underlie the dual effects of MPH on NMDAR- and AMPAR-mediated synaptic currents.
In vivo administration of a low-dose MPH facilitates recognition memory and attention, while a high-dose MPH induces hyperlocomotion
Since cortical glutamatergic transmission mediates many behavioral tasks, we examined the behavioral impact of MPH at different dosage in adolescent rats. Temporal order recognition memory (TORM), a cognitive process controlled by medial PFC (24,27), was found to be significantly enhanced in animals with a single injection of the low-dose (0.5 mg/kg) MPH (Figure 3A, discrimination ratio (DR) in saline: 29.1% ± 3.8%, n = 6; DR in low-dose MPH: 51.1% ± 8.4%, n = 5, p < 0.05, t-test). In the test of perceptual attentional set shifting, an aspect of attention mediated by medial frontal cortex (28), rats injected with low-dose MPH exhibited selective improvement in the extradimensional (ED) shift, taking less trials to learn the new discrimination (Figure 3B, trials to criterion, saline: 13.8 ± 0.98, n = 6; low-dose MPH: 8.6 ± 0.4, n = 5, p < 0.01, t-test). Locomotor activity was unchanged by the low-dose MPH injection (Figure 3D, number of midline crossing, saline: 11.6 ± 1.7, n = 11; low-dose MPH: 12.1 ± 2.5, n = 7, p > 0.05, ANOVA).
Figure 3. Low-dose MPH enhances the temporal order recognition memory and attentional set shifting, while high-dose MPH elevates locomotor activity.

(A, C) Bar graph (mean ± SEM) showing the discrimination ratio (DR) of temporal order recognition memory (TORM) tasks in animals treated with saline vs. MPH (A, 0.5 mg/kg, i.p.; C, 10mg/kg, i.p.) *: p < 0.05. (B) Bar graph showing the number of trials to criterion (6 consecutive correct trials) for each discrimination stage of the attentional set-shifting task in animals treated with saline or a low-dose MPH (0.5 mg/kg, i.p.). **: p < 0.01. (D) Bar graph showing the number of midline crossing in locomotion apparatus for animals injected with saline vs. MPH (low or high dose). #: p < 0.001.
On the other hand, a single injection of the high-dose (10 mg/kg) MPH profoundly impaired the TORM (Figure 3C, DR in saline: 32.0% ± 6.4%, n = 4; DR in high-dose MPH: −7.7% ± 14.2%, n = 9, p < 0.05, t-test). A significant increase of locomotor activity was observed in rats injected with the high-dose MPH (Figure 3D, number of midline crossing, saline: 11.6 ± 1.7, n = 11, high-dose MPH: 34.0 ± 3.6, n = 7; F2, 22 = 24.5, p < 0.001, ANOVA). The hyperlocomotion made these animals fail to complete the attentional set-shifting task.
Our results are consistent with previous animal and human subject studies showing the behavior changes by MPH at different dosage (2,3,5,6). The potentiated NMDAR signaling by low-dose MPH may underlie the enhanced recognition memory (24,27), while the reduced glutamate signaling by high-dose MPH may underlie the increased locomotion since NMDAR antagonists profoundly stimulate locomotion in animals (32).
Norepinephrine neurotransmission mediates the potentiating effect of low-dose MPH on NMDARs
Given the positive effects of low-dose MPH on NMDARs and cognitive behaviors, we next examined the molecular mechanisms underlying low-dose MPH. It is known that MPH blocks the norepinephrine transporter (NET) and the dopamine transporter (DAT) in the presynaptic terminals, resulting in elevated synaptic levels of these neurotransmitters (3,9,10). To determine whether dopaminergic or adrenergic neurotransmission is involved, we examined NMDAR-EPSC in animals treated with specific NET or DAT inhibitors. As shown in Figure 4A, animals injected with Maprotiline (20 mg/kg, i.p), a highly selective NET inhibitor (33), exhibited enhanced NMDA-EPSC (47–57% increase, n = 11–12 cells/3 rats per group, F1, 84 = 42.6, p < 0.01, ANOVA), similar to what was found in animals injected with low-dose MPH. Furthermore, animals injected with a higher dose of Maprotiline (50 mg/kg) exhibited reduced NMDAR-EPSC (Figure S1). The dose-dependent effects of Maprotiline parallel well with those of MPH. In contrast, animals injected with GBR-12909 (5 mg/kg, i.p), a highly selective DAT inhibitor (34), showed unaltered NMDAR-EPSC (Figure 4B, n = 6–9 cells/3 rats per group, F1, 65 = 1.76, p > 0.05, ANOVA).
Figure 4. Low-dose MPH potentiates NMDAR-EPSC via norepinephrine reuptake inhibition and adrenergic receptor activation.

(A, B) Summarized input-output curves of NMDAR-EPSC in PFC pyramidal neurons from rats treated with saline, Maprotiline (A, 20 mg/kg, i.p.), or GBR-12909 (B, 5 mg/kg, i.p.). Inset: representative traces of NMDAR-EPSC. Scale bar: 50 pA, 200 ms. **: p < 0.01. (C) Summarized input-output curves of NMDAR-EPSC in saline- vs. MPH- (0.5 mg/kg, i.p.) injected rats pre-treated with prazosin and yohimbine (Prazosin, 1 mg/kg, Yohimbine, 5 mg/kg, i.p., injected 0.5 hr before MPH injection). Inset: representative NMDAR-EPSC traces. Scale bar: 50 pA, 100 ms. (D) Summarized input-output curves of NMDAR-EPSC in saline- vs. MPH-injected rats pre-treated with SCH23390 and sulpiride (SCH23390, 1 mg/kg, sulpiride, 50 mg/kg, i.p., injected 0.5 hr before MPH injection). Inset: representative traces. Scale bar: 50 pA, 100 ms. **: p < 0.01.
To further confirm that MPH regulates NMDAR responses by preferentially targeting adrenergic neurotransmission, we pretreated animals with Prazosin, an antagonist of 1 adrenergic receptor (35), and Yohimbine, an antagonist of α2 adrenergic receptor (36). As shown in Figure 4C, blocking adrenergic receptors with Prazosin and Yohimbine completely abolished the effect of low-dose MPH on NMDAR-EPSC (−12%–11% increase, n = 8–10 cells/3 rats per group, F1, 160 = 0.29, p > 0.05, ANOVA). On the contrary, when applying SCH23390, a D1-class receptor antagonist (37), and sulpiride, a D2-class receptor antagonist (38), the enhancement of NMDAR-EPSC by low-dose MPH remained the same (Figure 4D, 29%–60% increase, n = 8–9 cells/3 rats per group, F1, 98 = 76.5, p < 0.01, ANOVA). These results suggest that low-dose MPH potentiates NMDAR-EPSC primarily by inhibiting norepinephrine transporter and activating adrenergic receptors.
SNAP-25 mediates the enhancement of NMDARs and cognition by low-dose MPH
The potentiated NMDAR currents by low-dose MPH are accompanied by elevated surface expression of NMDARs, suggesting that the membrane delivery of NMDARs might be affected. It is known that SNARE proteins are the key protein family involved in the membrane fusion in eukaryotic cells (39). In particular, SNAP-25, a member of SNAREs, has been implicated in the incorporation of NMDARs to postsynaptic membrane (40,41). Thus, we examined the role of SNAP-25 in the potentiation of surface NMDARs by low-dose MPH. Since intravenous (i.v.) injection can reliably deliver TAT peptides into central nervous system neurons (22,42), we gave animals an i.v. injection of the SNAP-25 blocking peptide (0.6 pmol/g) 30 min before MPH administration. This peptide mimics the N-terminal domain of SNAP-25 and thus disrupts the interaction of SNAP-25 with N-ethylmaleimide-sensitive factor (NSF), which is critical for the assembly and disassembly cycle of SNARE complexes (21,44). As shown in Figure 5A, two-way ANOVA analysis revealed a significant main effect on treatments (F 3,157 = 25.7, p < 0.001). Post-hoc tests indicated that the enhancing effect of low-dose MPH on NMDAR-EPSC was blocked by the SNAP-25 blocking peptide (2%–9% increase, n = 8–10 cells/4 rats per group, p > 0.05), but not a scrambled peptide (43%–77% increase, n = 8–13 cells/4 rats per group, p < 0.05). Biotinylation assays also showed that the increasing effects of low-dose MPH on surface NMDAR subunits was abolished by SNAP-25 blocking peptide (Figure 5B and 5C, surface NR1: 9.8% ± 10.4% decrease, surface NR2A: 27.4% ± 9.7% decrease, surface NR2B: 13.7% ± 21.0% decrease; n = 4 pairs, p > 0.05, ANOVA), but not the scrambled peptide (surface NR1: 73.3% ± 10.6% increase, surface NR2A: 117.2% ± 43.8% increase, surface NR2B: 218% ± 47.9% increase; n = 4 pairs, p < 0.01, ANOVA). Taken together, these results suggest that SNAP-25 mediates the enhanced exocytosis of NMDARs by low-dose MPH.
Figure 5. SNAP-25 participates in the potentiation of NMDAR-EPSC and cognitive functions by low-dose MPH.

(A) Summarized input-output curves of NMDAR-EPSC in saline- vs. MPH- (0.5 mg/kg, i.p.) injected rats pre-treated with SNAP-25 blocking peptide (SNAP-25 pep, 0.6 pmol/g, i.v.) or a scrambled peptide (sc pep, 0.6 pmol/g, i.v.). Inset: representative EPSC traces. Scale bar: 50 pA, 200 ms. *: p < 0.05. (B, C) Immunoblots (B) and quantification analysis (C) of the surface and total NMDAR subunits in rat PFC slices from saline- vs. MPH-injected rats pre-treated with SNAP-25 blocking peptide or a scrambled peptide. **: p < 0.01. (D, E) Bar graphs showing the discrimination ratio (DR) of temporal order recognition memory (TORM) tasks (D) or number of trials to criterion at each discrimination stage of the attentional set-shifting task (E) in MPH (0.5 mg/kg, i.p.)-injected animals pre-treated with a scrambled peptide or SNAP-25 blocking peptide. *: p < 0.05.
Next, we examined the role of SNAP-25 in MPH regulation of cognitive functions. As shown in Figure 5D, in rats injected with SNAP-25 peptide, low-dose MPH failed to enhance temporal order recognition memory (DR, SNAP-25 pep+MPH: 29.4% ± 5.4%, n = 5, con pep+MPH: 48.3% ± 5.3%, n = 6, p < 0.05, t-test). Moreover, injection of SNAP-25 peptide blocked the beneficial effect of low-dose MPH in the attentional set-shifting task, resulting in more trials to achieve the criterion in ED shift (trials to criterion, con pep+MPH: 8.6 ± 0.7, n = 5; SNAP25+MPH: 12.0 ± 0.8, n = 5, p < 0.05, t-test).
To avoid potential non-specific effects with the systemic administration of SNAP-25 peptide, we performed the stereotaxic injection of peptides to PFC bilaterally, followed by MPH injection (i.p.). Electrophysiological recordings showed that PFC infusion of SNAP-25 peptide (3 pmol/side) blocked the increase of NMDAR-EPSC by low-dose MPH (Figure 6A, SNAP-25 pep: ~10% increase, con pep: ~55% increase, n = 16–30 cells/4 rats per group, F3,382 = 22.3, p < 0.001, ANOVA). Behavioral tests indicated that PFC infusion of SNAP-25 peptide blocked the increase of TORM by low-dose MPH (Figure 6B, DR, SNAP-25 pep: ~0 fold increase, con pep: ~0.8 fold increase, n = 5 pairs, F3.20 = 5.89, p < 0.01, ANOVA). These data suggest that SNAP-25 in PFC is critical for the potentiation of NMDARs and cognition by low-dose MPH.
Figure 6. PFC infusion of SNAP-25 blocking peptide abolished the low-dose MPH-induced enhancement of NMDAR-EPSC and temporal order recognition memory.
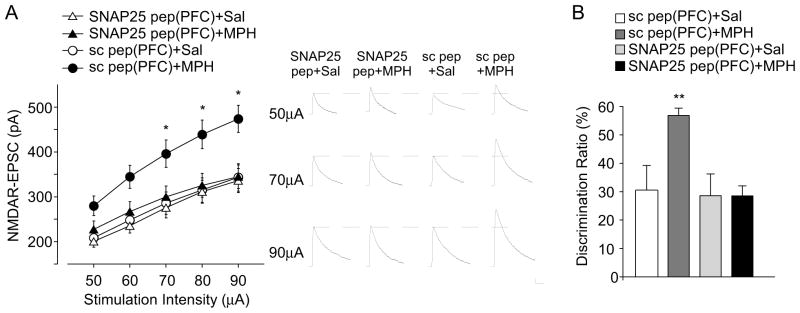
(A, B) Summarized input-output curves of NMDAR-EPSC (A) or bar graph (mean ± SEM) showing the discrimination ratio (DR) of temporal order recognition memory (TORM) tasks (B) in saline vs. MPH (0.5 mg/kg, i.p.)-injected animals with PFC infusion of a scrambled peptide or SNAP-25 blocking peptide (3 pmol/site). Inset: representative EPSC traces. Scale bar: 50 pA, 200 ms. *: p < 0.05, **: p<0.01.
Since protein kinase C (PKC) phosphorylation of SNAP-25 could affect the surface expression of NMDARs (40), we also examined the involvement of PKC in MPH effects. Low-dose MPH failed to enhance NMDAR-EPSC in the presence of a PKC inhibitor, chelerythrine (3 mg/kg, i.p.) (45) (Figure S2, ~7% increase, n = 10–12 cells/3 rats per group, F1, 100 = 0.59, p > 0.05, ANOVA). These results suggest that PKC, which may be activated by low-dose MPH, is important for facilitating SNAP-25-dependent NMDAR surface delivery.
Low-dose MPH rescues the impaired NMDAR and cognitive function in animals exposed to repeated stress
Since low-dose MPH enhances NMDAR function and memory processes in naïve animals, we next examined whether low-dose MPH restores the impaired NMDAR and cognitive function in animals exposed to repeated stress (24). A significant main effect was found in treatment groups (Figure 7A, F5, 277 = 159.8, p < 0.001, two-way ANOVA). Post-hoc tests indicated that NMDAR-EPSC was markedly decreased in PFC pyramidal neurons from young male rats exposed to repeated (7 days) restraint stress (76%–96% reduction, n = 13–17 cells/4 rats per group, p < 0.001), consistent with our previous results (29,30). A single injection of low-dose MPH (0.5 mg/kg, i.p.) after the repeated stress exposure restored NMDAR-EPSC to the control level (n = 13–17 cells/4 rats per group, p > 0.05). The recovery was blocked in animals pretreated with SNAP-25 blocking peptide (0.6 pmol/g, i.v., 37%–81% reduction, n = 8–18 cells/3 rats per group, p < 0.001).
Figure 7. Low-dose MPH restores the impaired NMDAR function and recognition memory in animals exposed to repeated stress.

A, summarized input-output curves of NMDAR-EPSC in control or repeatedly stressed (RS) rats treated with saline or MPH (0.5 mg/kg, i.p.) without or with SNAP-25 peptide (S26p, 0.6 pmol/g, i.v.) pretreatment. *: p < 0.05, #: p < 0.001. Inset: representative NMDAR-EPSC traces. Scale bar: 50 pA, 200 ms. B, C, Bar graphs (mean ± SEM) showing the DR (B) and total exploration time (C) of TORM tasks in repeatedly stressed animals injected with saline or MPH without or with SNAP-25 peptide pretreatment. **: p < 0.01.
Behavioral studies found that the repeatedly stressed rats had the impaired TORM, which was remarkably recovered by a single injection of low-dose MPH (Figure 7B, DR, stress+saline: 6.6% ± 7.0%, n = 7, stress+MPH: 56.3% ± 11.4%, n = 9, F2, 23 =5.7, p < 0.01, ANOVA). The recovering effect of low-dose MPH was abolished by pre-treatment with SNAP-25 blocking peptide (DR, stress+SNAP-25: 3.7% ± 10.9%, n = 4; stress+SNAP-25+MPH: 1.2% ± 5.8%, n = 6, p > 0.05). The total exploration time in the two sample phases and the subsequent test trial was unchanged by any of these treatments (Figure 7C, p > 0.05, ANOVA). These results suggest that low-dose MPH is capable of rescuing the impaired NMDAR function and cognitive deficits in stressed animals through a mechanism involving SNAP-25.
Discussion
Despite the widespread use of MPH as a cognitive enhancer, little is known about the causal mechanism underlying its behavioral actions. The dopamine and adrenergic system has been primarily studied for MPH, however considering that the glutamatergic system is critically involved in synaptic plasticity and cognitive processes (16,25), regulation of glutamate signaling might underlie the neuronal mechanism of MPH. Since MPH is commonly prescribed for the treatment of ADHD in children and adolescents, it is of importance to use adolescent rats to study the effect of MPH exposure in early life. In the present study, we found that a low-dose MPH, which yields clinically relevant plasma levels (3), remarkably potentiated NMDAR-mediated synaptic responses and the surface expression of NMDA receptors in adolescent rats. On the other hand, we also found that a high-dose MPH substantially decreased glutamatergic transmission, via a mechanism involving both decreasing presynaptic glutamate release probability and reducing postsynaptic glutamate receptor surface expression. In contrast, a previous study showed that one hour after a single injection of MPH (1 mg/kg, i.p.), NMDAR-mediated currents and NMDAR total protein levels were decreased in PFC of juvenile rats (p15–25) (46). We have not seen such reducing effects with MPH (1 mg/kg) injection.
In parallel with the dose-dependent bi-directional effects of MPH on PFC glutamatergic signaling, our behavioral studies found that the low-dose MPH enhanced temporal order recognition memory (TORM) and attentional set shifting, while the high-dose MPH impairs TORM and elevated locomotor activity. It is consistent with previous work in animals and human subjects showing that the therapeutic dose of MPH effectively improves cognitive functions (2,3), while overdose of MPH is associated with aggression and hyperactivity (4). Given that ADHD children exhibit prefrontal hypoactivity (15,16), the elevated NMDAR function by low-dose MPH might underlie its beneficial effects on memory, attention and other cognitive aspects. On the other hand, since NMDAR antagonists, such as phencyclidine or ketamine, can lead to the formation of psychotic symptoms including hyperlocomotion (32,47), the reduced glutamate signaling by high-dose MPH might underlie its psychosis-inducing effects.
MPH acts as a NET and DAT inhibitor, and our data indicate that low-dose MPH potentiates NMDAR functions mainly through norepinephrine system. Consistently, MPH is shown to have higher affinity for NET than DAT in vitro (48), preferentially affect norepinephrine at low doses in vivo (49), and significantly occupy NET at clinically relevant doses in humans (10). Norepinephrine system has been implicated in many PFC functions, including working memory, attention and emotional control (50,51). An in vitro study suggests that the enhancement of NMDAR-EPSC by bath application of MPH (50 M) in PFC slices is mediated by Sigma-1 receptors instead of adrenergic or dopamine receptors (31). The inconsistency may be due to different routes of drug administrations and MPH concentrations.
Since low-dose MPH increases NMDAR surface expression, we have examined the potential molecule downstream of adrenergic receptors that is involved in NMDAR exocytosis. SNARE (Soluble N-ethylmaleimide sensitive fusion proteins Attachment Protein REceptor) proteins, comprising SNAP-25/23, syntaxins and synaptobrevin/VAMP, form SNARE complexes in the late stage of synaptic vesicle exocytosis mediating vesicles docking and fusion (39). SNAP-25 (Synaptosomal-associated protein 25), a key component of SNARE complex expressed in excitatory neurons (52), participates in the delivery of NMDAR vesicles at postsynaptic sites (21,40,41). More importantly, dysfunction of SNAP-25 is linked to various human mental disorders, such as schizophrenia, ADHD and early-onset bipolar disorder (53–55). The mouse carrying a deletion of SNAP-25 gene has been used as an ADHD animal model (56). In the present study, we demonstrate that SNAP-25 mediates the increase of NMDAR exocytosis by low-dose MPH.
In addition to enhancing cognitive function, MPH is able to combat stress (57). Chronic or severe stress is a trigger for many mental illnesses (58). Previous studies have found that repeated stress suppresses PFC glutamatergic signaling, resulting in cognitive impairment (24,26,59,60). In this study, we found that low-dose MPH restored the impaired NMDAR function and object recognition memory in animals exposed to repeated stress through a mechanism dependent on SNAP-25-mediated exocytosis of NMDARs. It provides a molecular mechanism for MPH to be used as a potential therapeutic strategy for stress treatment.
A remaining question is the long-term effect of MPH on glutamatergic transmission and PFC-dependent cognitive function. Previous studies suggest that glutamatergic pathways are involved in the acute and chronic MPH regulation of locomotion in adult rats (61), and exposing rats to MPH during adolescence period results in increased stress reactivity (7). It awaits to be tested whether PFC network activity is altered after long-term exposure to different doses of MPH.
Conclusion
The current study reveals that administration of a low-dose MPH potentiates NMDAR trafficking and function, enhances PFC-mediated cognition, and counteracts the detrimental effects of repeated stress in adolescent rats, via a mechanism involving adrenergic receptors and SNAP-25. In contrast, administration of a high-dose MPH suppresses PFC glutamatergic transmission and induces hyperlocomotion. It provides a potential mechanism underlying the cognitive enhancing effects of low-dose MPH, and the psychosis-inducing effects of high-dose MPH.
Supplementary Material
Acknowledgments
This work was supported by NIH grants (MH85774, MH65434) to ZY and NSFC grants (81220108010, 81171197) to GJC.
We would like to thank Xiaoqing Chen for her excellent technical support. Dr. Yulei Deng provided kind help in some experiments.
Footnotes
All authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Biederman J. Attention-deficit /hyperactivity disorder: A selective overview. Biol Psychiatry. 2005;57:1215–1220. doi: 10.1016/j.biopsych.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 2.Elliott R, Sahakian BJ, Matthews K, Bannerjea A, Rimmer J, Robbins TW. Effects of methylphenidate on spatial working memory and planning in healthy young adults. Psychopharmacology (Berl) 1997;131:196–206. doi: 10.1007/s002130050284. [DOI] [PubMed] [Google Scholar]
- 3.Berridge CW, Devilbiss DM, Andrzejewski ME, Arnsten AF, Kelley AE, Schmeichel B, et al. Methylphenidate preferentially increases catecholamine neurotransmission within the prefrontal cortex at low doses that enhance cognitive function. Biol Psychiatry. 2006;60:1111–1120. doi: 10.1016/j.biopsych.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 4.Smith ME, Farah MJ. Are prescription stimulants “smart pills”? The epidemiology and cognitive neuroscience of prescription stimulant use by normal healthy individuals. Psychol Bull. 2011;137:717–41. doi: 10.1037/a0023825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein-Schwartz W. Abuse and toxicity of methylphenidate. Curr Opin Pediatr. 2002;14:219–23. doi: 10.1097/00008480-200204000-00013. [DOI] [PubMed] [Google Scholar]
- 6.Gerasimov MR, Franceschi M, Volkow ND, Gifford A, Gatley SJ, Marsteller D, et al. Comparison between intraperitoneal and oral methylphenidate administration: A microdialysis and locomotor activity study. J Pharmacol Exp Ther. 2000;295:51–7. [PubMed] [Google Scholar]
- 7.Wiley MD, Poveromo LB, Antapasis J, Herrera CM, Bolaños Guzmán CA. Kappa-opioid system regulates the long-lasting behavioral adaptations induced by early-life exposure to methylphenidate. Neuropsychopharmacology. 2009;34:1339–50. doi: 10.1038/npp.2008.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warren BL, Iñiguez SD, Alcantara LF, Wright KN, Parise EM, Weakley SK, Bolaños-Guzmán CA. Juvenile administration of concomitant methylphenidate and fluoxetine alters behavioral reactivity to reward- and mood-related stimuli and disrupts ventral tegmental area gene expression in adulthood. J Neurosci. 2011;31:10347–58. doi: 10.1523/JNEUROSCI.1470-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spencer TJ, Biederman J, Ciccone PE, Madras BK, Dougherty DD, Bonab AA, et al. PET study examining pharmacokinetics, detection and likeability, and dopamine transporter receptor occupancy of short- and long-acting oral methylphenidate. Am J Psychiatry. 2006;163:387–95. doi: 10.1176/appi.ajp.163.3.387. [DOI] [PubMed] [Google Scholar]
- 10.Hannestad J, Gallezot JD, Planeta-Wilson B, Lin SF, Williams WA, van Dyck CH, et al. Clinically relevant doses of methylphenidate significantly occupy norepinephrine transporters in humans in vivo. Biol Psychiatry. 2010;68:854–60. doi: 10.1016/j.biopsych.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–85. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 12.Dalley JW, Cardinal RN, Robbins TW. Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neurosci Biobehav Res. 2004;28:771–84. doi: 10.1016/j.neubiorev.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 13.Shaw P, Eckstrand K, Sharp W, Blumenthal J, Lerch JP, Greenstein D, et al. Attention-deficit/hyperactivity disorder is characterized by a delay in cortical maturation. Proc Natl Acad Sci U S A. 2007;104:19649–19654. doi: 10.1073/pnas.0707741104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnsten AF. Fundamentals of attention-deficit/hyperactivity disorder: circuits and pathways. J Clin Psychiatry. 2006;67:7–12. [PubMed] [Google Scholar]
- 15.Fernández A, Quintero J, Hornero R, Zuluaga P, Navas M, Gómez C, et al. Complexity analysis of spontaneous brain activity in attention-deficit/hyperactivity disorder: diagnostic implications. Biol Psychiatry. 2009;65:571–7. doi: 10.1016/j.biopsych.2008.10.046. [DOI] [PubMed] [Google Scholar]
- 16.Cortese S, Kelly C, Chabernaud C, Proal E, Di Martino A, Milham MP, Castellanos FX. Toward systems neuroscience of ADHD: a meta-analysis of 55 fMRI studies. Am J Psychiatry. 2012;169:1038–55. doi: 10.1176/appi.ajp.2012.11101521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnsten AF. Toward a new understanding of attention-deficit hyperactivity disorder pathophysiology: An important role for prefrontal cortex dysfunction. CNS Drugs. 2009;23:33– 41. doi: 10.2165/00023210-200923000-00005. [DOI] [PubMed] [Google Scholar]
- 18.Lisman JE, Fellous JM, Wang XJ. A role for NMDA-receptor channels in working memory. Nat Neurosci. 1998;1:273–275. doi: 10.1038/1086. [DOI] [PubMed] [Google Scholar]
- 19.Kantrowitz J, Javitt DC. Glutamatergic transmission in schizophrenia: from basic research to clinical practice. Curr Opin Psychiatry. 2012;25:96–102. doi: 10.1097/YCO.0b013e32835035b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24:417–63. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- 21.Lledo PM, Zhang X, Sudhof TC, Malenka RC, Nicoll RA. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279:399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- 22.Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS, Yan Z. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry. 2011;16:156–70. doi: 10.1038/mp.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuen EY, Liu W, Karatsoreos IN, Feng J, McEwen BS, Yan Z. Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc Natl Acad Sci U S A. 2009;106:14075–9. doi: 10.1073/pnas.0906791106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron. 2012;73:962–77. doi: 10.1016/j.neuron.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lou X, Fan F, Messa M, Raimondi A, Wu Y, Looger LL, et al. Reduced release probability prevents vesicle depletion and transmission failure at dynamin mutant synapses. Proc Natl Acad Sci U S A. 2012;109:515–23. doi: 10.1073/pnas.1121626109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei J, Yuen EY, Liu W, Li X, Zhong P, Karatsoreos IN, et al. Estrogen protects against the detrimental effects of repeated stress onglutamatergic transmission and cognition. Mol Psychiatry. 2013 doi: 10.1038/mp.2013.83. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 27.Barker GR, Bird F, Alexander V, Warburton EC. Recognition memory for objects, place, and temporal order: a disconnection analysis of the role of the medial prefrontal cortex and perirhinal cortex. J Neurosci. 2007;27:2948–2957. doi: 10.1523/JNEUROSCI.5289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birrell JM, Brown VJ. Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci. 2000;20:4320–4. doi: 10.1523/JNEUROSCI.20-11-04320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- 30.Yang Pamela B, Swann Alan C, Dafny Nachum. Acute and chronic methylphenidate dose–response assessment on three adolescent male rat strains. Brain Research Bulletin. 2006;71:301–310. doi: 10.1016/j.brainresbull.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang CL, Feng ZJ, Liu Y, Ji XH, Peng JY, Zhang XH, et al. Methylphenidate enhances NMDA-receptor response in medial prefrontal cortex viasig ma-1 receptor: a novel mechanism for methylphenidate action. PLoS One. 2012;7:51910. doi: 10.1371/journal.pone.0051910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hargreaves EL, Cain DP. MK801-induced hyperactivity: duration of effects in rats. Pharmacol Biochem Behav. 1995;51:13–9. doi: 10.1016/0091-3057(94)00321-9. [DOI] [PubMed] [Google Scholar]
- 33.Barbaccia ML, Ravizza L, Costa E. Maprotiline: an antidepressant with an unusual pharmacological profile. J Pharmacol Exp Ther. 1986;236:307–12. [PubMed] [Google Scholar]
- 34.Andersen PH. The dopamine inhibitor GBR 12909: selectivity and molecular mechanism of action. Eur J Pharmacol. 1989;166:493–504. doi: 10.1016/0014-2999(89)90363-4. [DOI] [PubMed] [Google Scholar]
- 35.Cambridge D, Davey MJ, Massingham R. Prazosin, a selective antagonist of post-synaptic alpha-adrenoceptors. Br J Pharmacol. 1977;59:514–515. [PMC free article] [PubMed] [Google Scholar]
- 36.Hamano N, Inada T, Iwata R, Asai T, Shingu K. The alpha2-adrenergic receptor antagonist yohimbine improves endotoxin-induced inhibition of gastrointestinal motility in mice. Br J Anaesth. 2007;98:484–90. doi: 10.1093/bja/aem011. [DOI] [PubMed] [Google Scholar]
- 37.Alburges ME, Hunt ME, McQuade RD, Wamsley JK. D1-receptor antagonists: comparison of [3H]SCH39166 to [3H]SCH23390. J Chem Neuroanat. 1992;5:357–66. doi: 10.1016/0891-0618(92)90051-q. [DOI] [PubMed] [Google Scholar]
- 38.Anderson SM, Schmidt HD, Pierce RC. Administration of the D2 dopamine receptor antagonist sulpiride into the shell, but not the core, of the nucleus accumbens attenuates cocaine priming-induced reinstatement of drug seeking. Neuropsychopharmacology. 2006;31:1452–61. doi: 10.1038/sj.npp.1300922. [DOI] [PubMed] [Google Scholar]
- 39.Jahn R, Scheller RH. SNAREs – engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 40.Lau CG, Takayasu Y, Rodenas-Ruano A, Paternain AV, Lerma J, Bennett MV, Zukin RS. SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking. J neurosci. 2010;30:242–254. doi: 10.1523/JNEUROSCI.4933-08.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng J, Liu W, Duffney LJ, Yan Z. SNARE proteins are essential in the potentiation of NMDA receptors by group II metabotropic glutamate receptors. J Physiol. 2013;591:3935–47. doi: 10.1113/jphysiol.2013.255075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, et al. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- 43.Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Mel. 2003;9:1180–6. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 44.Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–7. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lavaur J, Mineur YS, Picciotto MR. The membrane cytoskeletal protein adducin is phosphorylated by protein kinase C in D1 neurons of thenucleus accumbens and dorsal striatum following cocaine administration. J Neurochem. 2009;111:1129–37. doi: 10.1111/j.1471-4159.2009.06405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Urban KR1, Li YC, Gao WJ. Treatment with a clinically-relevant dose of methylphenidate alters NMDA receptor composition and synaptic plasticity in the juvenile rat prefrontal cortex. Neurobiol Learn Mem. 2013;101:65–74. doi: 10.1016/j.nlm.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steinpreis R. The behavioral and neurochemical effects of phencyclidine in humans and animals: some implications for modeling psychosis. Behav Brain Res. 1996;74:45–55. doi: 10.1016/0166-4328(95)00162-x. [DOI] [PubMed] [Google Scholar]
- 48.Eshleman AJ, Carmolli M, Cumbay M, Martens CR, Neve KA, Janowsky A. Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. J Pharmacol Exp Ther. 1999;289:877–885. [PubMed] [Google Scholar]
- 49.Kuczenski R, Segal DS. Exposure of adolescent rats to oral methylphenidate: Preferential effects on extracellular norepinephrine and absence of sensitization and cross-sensitization to methamphetamine. J Neurosci. 2002;22:7264–7271. doi: 10.1523/JNEUROSCI.22-16-07264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arnsten AF, Mathew R, Ubriani R, Taylor JR, Li BM. Alpha-1 noradrenergic receptor stimulation impairs prefrontal cortical cognitive function. Biol Psychiatry. 1999;45:26–31. doi: 10.1016/s0006-3223(98)00296-0. [DOI] [PubMed] [Google Scholar]
- 51.Berridge CW, Shumsky JS, Andrzejewski ME, McGaughy JA, Spencer RC, Devilbiss DM, Waterhouse BD. Differential sensitivity to psychostimulants across prefrontal cognitive tasks: differential involvement of noradrenergic α1 - and α2-receptors. Biol Psychiatry. 2012;71:467–73. doi: 10.1016/j.biopsych.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwab Y, Mouton J, Chasserot-Golaz S, Marty I, Maulet Y, Jover E. Calcium-dependent translocation of synaptotagmin to the plasma membrane in the dendrites of developing neurones. Mol Brain Res. 2001;96:1–13. doi: 10.1016/s0169-328x(01)00244-3. [DOI] [PubMed] [Google Scholar]
- 53.Thompson PM, Sower AC, Perrone-Bizzozero NI. Altered levels of the synaptosomal associated protein SNAP-25 in schizophrenia. Biol Psychiatry. 1998;43:239–43. doi: 10.1016/S0006-3223(97)00204-7. [DOI] [PubMed] [Google Scholar]
- 54.Barr CL, Feng Y, Wigg K, Bloom S, Roberts W, Malone M, et al. Identification of DNA variants in the SNAP-25 gene and linkage study of these polymorphisms and attention-deficit hyperactivity disorder. Mol Psychiatry. 2000;5:405–9. doi: 10.1038/sj.mp.4000733. [DOI] [PubMed] [Google Scholar]
- 55.Etain B, Dumaine A, Mathieu F, Chevalier F, Henry C, Kahn JP, et al. A SNAP25 promoter variant is associated with early-onset bipolar disorder and a high expression level in brain. Mol Psychiatry. 2010;15:748–55. doi: 10.1038/mp.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilson MC. Coloboma mouse mutant as an animal model of hyperkinesis and attention deficit hyperactivity disorder. Neurosci Biobehav Rev. 2000;24:51–7. doi: 10.1016/s0149-7634(99)00064-0. [DOI] [PubMed] [Google Scholar]
- 57.Zehle S, Bock J, Jezierski G, Gruss M, Braun K. Methylphenidate treatment recovers stress-induced elevated dendritic spine densities in the rodent dorsal anterior cingulate cortex. Dev Neurobiol. 2007;67:1891–900. doi: 10.1002/dneu.20543. [DOI] [PubMed] [Google Scholar]
- 58.De Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- 59.Cerqueira JJ, Mailliet F, Almeida OF, Jay TM, Sousa N. The prefrontal cortex as a key target of the maladaptive response to stress. J Neurosci. 2007;27:2781–2787. doi: 10.1523/JNEUROSCI.4372-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, Hof PR, et al. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. 2006;26:7870–7874. doi: 10.1523/JNEUROSCI.1184-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wanchoo SJ, Swann AC, Dafny N. Descending glutamatergic pathways of PFC are involved in acute and chronic action of methylphenidate. Brain Res. 2009;1301:68–79. doi: 10.1016/j.brainres.2009.08.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.