

Abstract
Vascular endothelial growth factor-A (VEGF-A) is best known as a key regulator of the formation of new blood vessels. Neutralization of VEGF-A with anti-VEGF therapy e.g. bevacizumab, can be painful, and this is hypothesized to result from a loss of VEGF-A-mediated neuroprotection. The multiple vegf-a gene products consist of two alternatively spliced families, typified by VEGF-A165a and VEGF-A165b (both contain 165 amino acids), both of which are neuroprotective. Under pathological conditions, such as in inflammation and cancer, the pro-angiogenic VEGF-A165a is upregulated and predominates over the VEGF-A165b isoform.
We show here that in rats and mice VEGF-A165a and VEGF-A165b have opposing effects on pain, and that blocking the proximal splicing event – leading to the preferential expression of VEGF-A165b over VEGF165a – prevents pain in vivo. VEGF-A165a sensitizes peripheral nociceptive neurons through actions on VEGFR2 and a TRPV1-dependent mechanism, thus enhancing nociceptive signaling. VEGF-A165b blocks the effect of VEGF-A165a.
After nerve injury, the endogenous balance of VEGF-A isoforms switches to greater expression of VEGF-Axxxa compared to VEGF-Axxxb, through an SRPK1-dependent pre-mRNA splicing mechanism. Pharmacological inhibition of SRPK1 after traumatic nerve injury selectively reduced VEGF-Axxxa expression and reversed associated neuropathic pain. Exogenous VEGF-A165b also ameliorated neuropathic pain.
We conclude that the relative levels of alternatively spliced VEGF-A isoforms are critical for pain modulation under both normal conditions and in sensory neuropathy. Altering VEGF-Axxxa/VEGF-Axxxb balance by targeting alternative RNA splicing may be a new analgesic strategy.
Abbreviations: VEGF-A, vascular endothelial growth factor-A; SRPK1, serine arginine protein kinase 1; SRSF1, serine arginine splice factor 1; VEGFR2, vascular endothelial growth factor receptor 2; IB4, isolectin B4; TRPV1, transient receptor potential vanilloid 1; CV, conduction velocity; PSNI, partial saphenous nerve ligation injury; DRG, dorsal root ganglia
Keywords: Vascular endothelial growth factor A, Alternative mRNA splicing, Neuropathy, Nociceptors
Graphical abstract

Highlights
-
•
The different vegf-a splice variants, VEGF-A165a and VEGF-A165b have pro- and anti-nociceptive actions respectively.
-
•
Pro-nociceptive actions of VEGF-A165a are dependent on TRPV1.
-
•
Alternative pre-mRNA splicing underpins peripheral sensitization by VEGF-A isoforms in normal and neuropathic animals.
Introduction
Neutralization of VEGF-A with anti-VEGF-A therapies, such as bevacizumab or VEGF-A receptor inhibitors (e.g., vandetanib) can result in pain, when given alone (Burger et al., 2007, Cohen and Hochster, 2007) or in combination with chemotherapies (Cohen et al., 2007, Garcia et al., 2008, Langenberg et al., 2011, Miller et al., 2007). The clinical findings that VEGF-A contributes to pain are supported by observations that inhibition of VEGF receptor 2 (VEGFR2) exacerbates peripheral neuronal damage, which is often associated with pain (Beazley-Long et al., 2013, Verheyen et al., 2012), and enhances pain behaviors in normal, nerve-injured and diabetic animals (Hulse et al., 2010a, Verheyen et al., 2012).
The vegf-a gene encodes two families of isoforms typified by VEGF-A165 a and VEGF-A165b (Harper and Bates, 2008). Both families have sister isoforms of the same length so they are referred collectively as VEGF-Axxxa and VEGF-Axxxb where xxx represents the number of amino acids. The isoform families differ only in their six C terminal amino acids (Harper and Bates, 2008), and they are both capable of binding to VEGFR2 with similar affinities, but the functional results of receptor activation are multivariate (Table 1) (Ballmer-Hofer et al., 2011). Control of relative isoform expression occurs by alternative pre-mRNA splicing of either proximal or distal splice sites in exon 8 (Fig. 1).
Table 1.
Overview of the C-terminal sequences, binding domains and interactions with VEGFR2 of the different VEGF-A splice variant isoforms.
| C terminal sequence | Binding domains present | Consequences of receptor binding | |
|---|---|---|---|
| VEGF-A165a | CDKPRR | VEGFR1, VEGFR2, NP-1 | Full agonist. Binds and stabilizes VEGFR + NP-1 interaction. Complete phosphorylation at Y1175. PIP2 hydrolysis, PKC activation. |
| VEGF-A165b | SLTRKD | VEGFR1, VEGFR2 | Partial VEGFR2 agonist/competitive inhibitor of VEGF-A165a binding. Very weak NP-1 interaction. Weak/incomplete phosphorylation at Y1175 No PIP2 hydrolysis, or PKC activation. Receptor internalization and degradation. (Ballmer-Hofer et al., 2011, Kisko et al., 2011) |
| VEGF-A159 | – | VEGFR1, VEGFR2 | Binds VEGFR, no activation. Very weak NP-1 interaction. |
| VEGF-A121a | CDKPRR | VEGFR1, VEGFR2 | Binds VEGFR. Very weak NP-1 interaction. Complete phosphorylation at Y1175 |
Fig. 1.
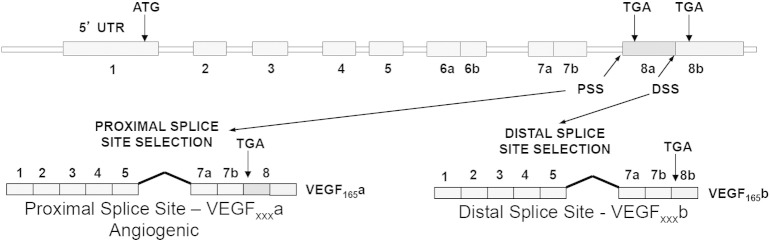
VEGF-A gene splice variant isoforms. VEGF-A pre-mRNA is alternatively spliced to form two families of mRNAs: VEGF-Axxxa and VEGF-Axxxb. The archetypal forms VEGF-A165a and VEGF-A165b are shown for illustration. VEGF-Axxxa proteins are translated from mRNAs that use the proximal splice site (PSS) and include all of exon 8, VEGF-Axxxb proteins from mRNAs that use the distal splice site (DSS) and contain only the b part of exon 8. The neuropilin-1 (NP-1) co-receptor binding site is located at the distal end of exon 7 and proximal exon 8a.
VEGF-Axxxa is the principal target of anti-VEGF and VEGFR therapies as these isoforms are upregulated and predominate in many pathologies. However, VEGF-Axxxb forms a significant proportion of total (pan-)VEGF-A protein in many normal tissues (Harper and Bates, 2008) so the therapeutic effects of VEGF-A sequestration with many current antibody therapies, or VEGFR2 inhibition are a net result of simultaneous blockade of the actions of both families. The impact of the neutralization of the VEGF-Axxxb family on treatment outcomes has only recently been exemplified, in terms of its ability to predict colorectal cancer patients that do not respond to bevacizumab (Bates et al., 2012).
rhVEGF-A165a exacerbated spinal cord contusion-associated pain and damage (Benton and Whittemore, 2003, Herrera et al., 2009, Nesic et al., 2010, Sundberg et al., 2011), and referred mechanical abdominal pain (Malykhina et al., 2012), but local VEGF-A delivery (presumed VEGF-Axxxa) partially reversed diabetic neuropathic mechanical hyperalgesia (Verheyen et al., 2013). Neutralization of all endogenous VEGF-A isoforms or VEGF receptor 2 inhibition increased pain sensitivity in chemotherapy-induced neuropathy (Verheyen et al., 2012), but conversely reversed neuropathic (Lin et al., 2010), and acute inflammatory hyperalgesia (Grosios et al., 2004).
These conflicting observations might be explained by different actions of the distinct isoforms, which have not been studied independently, and their differing actions on VEGFR2 (Ballmer-Hofer et al., 2011). We therefore tested the hypothesis that the alternatively spliced VEGF-A isoform families have different effects on pain. We investigated: a) the effects of specific VEGF-A isoforms on pain/nociception; b) the neuronal mechanisms through which effects on pain might occur; c) whether using control of alternative RNA splicing of VEGF-A could modulate nociception/pain, and d) whether either VEGF-A proteins or alternative splicing control may be potential novel analgesic targets.
Materials and methods
All procedures using animals were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986 and with University of Bristol and King's College London Ethical Review Groups approval. Human embryonic and adult tissues were obtained under ethical approval by University of Leiden and adult human DRG under ethical approval by Southmead Hospital Local Research Ethics Committee.
Antibody and pharmacological inhibitors
The following pharmacological interventions were used: pan-VEGF-A neutralization with mouse anti-VEGF-A antibody (Liang et al., 2006), specific VEGF-A165b neutralization using systemic treatment with anti-VEGF-A165b antibody (clone 56/1, (Woolard et al., 2004)) systemic and local VEGF receptor inhibition with selective (PTK787; (Wood et al., 2000)) and/or specific (ZM323881; (Whittles et al., 2002)). VEGFR2 tyrosine kinase inhibitors; systemic or local administration of VEGF-A165a and/or VEGF-A165b; systemic antagonism of TRPV1 with SB366791 (Varga et al., 2005); inhibition of serine-rich protein kinases with SRPIN340 (Fukuhara et al., 2006), and appropriate vehicles.
Measurement of mechanical and thermal nociceptive behaviors
A total of 64 adult male mice (C57Bl6, 25–30 g), 6 TRPV1 congenic knockouts and 6 wild-type strain-matched controls and 24 adult male Wistar rats were used to assess nociceptive behavior. TRPV1 homozygous knockout mice breeding pairs were generated and bred as described at King's College London, (Caterina et al., 2000, Fernandes et al., 2011, Fernandes et al., 2013) where breeding colonies were regularly backcrossed according to Jackson Laboratory guidelines to avoid sub-strain selection (Lambert, n.d.).
All animals were habituated to testing environments and handling prior to testing, and were allowed to habituate to the environment for at least 15 min at each test session. Nociceptive testing, as previously described (Hulse et al., 2008), consisted of measurement of mechanical allodynia by determination of von Frey hair mechanical withdrawal threshold and thermal hyperalgesia using the Hargreaves test (Hargreaves et al., 1988). Behavioral testing groups were randomized, and all operators were blinded to the drug and surgical treatment (nerve injury/sham) in each animal in all experiments.
Von Frey hair mechanical thresholds — mechanical allodynia
Animals were habituated to chambers with mesh floors. The plantar surface of each foot was stimulated with von Frey hairs (Linton, UK) of increasing gram force breaking points, over a range of 0.07–2 g (mice), or 1–100 g (rats) (Hulse et al., 2008). Each von Frey hair tested was applied a total of 5 times to each hind paw and the number of times an animal removed the paw from each stimulus was counted. The proportion of times that the animal withdrew from each stimulus was plotted against the breaking force, and the withdrawal threshold determined from the resultant stimulus response curve (the gram force at which paw removal occurred at 50% of the stimulations).
Hargreaves test for thermal hyperalgesia
Thermal hyperalgesia was measured using a radiant heat source directed against the plantar surface of the hind paws, through the Perspex floor of the testing chamber (Hargreaves et al., 1988), and the latency to withdrawal was measured. The stimulus intensity was determined at the beginning of each experimental series, to give a control withdrawal latency of ~ 10 s, and this intensity was subsequently used for each subsequent testing session for that experimental group. A maximum latency duration of 30 s was used to prevent tissue damage/sensitization to intense sustained stimulation. The mean withdrawal latency was determined from three repeated stimulations at an inter-stimulus interval of at least 5 min.
Model of neuropathic pain — partial saphenous nerve injury
24 mice and 18 rats underwent surgical partial saphenous nerve injury (PSNI) as previously described (Hulse et al., 2008, Walczak et al., 2005) under isofluorane anesthesia (2–3% in O2). A ~ 1 cm incision was made in the inguinal fossa region of the right hind leg. 50% of the saphenous nerve was tightly ligated using a size 6.0 sterile silk suture and the wound was closed with size 4.0 sterile silk suture. Sham-operated animals (n = 5) underwent anesthesia and surgery involving solely an incision in the inguinal fossa region of the right hind limb.
Electrophysiological recording of identified primary afferents in the saphenous nerve
Teased fiber electrophysiology from the saphenous nerve was carried out in 44 adult male Wistar rats (250–300 g). Properties of isolated afferents in terminally anesthetized rats (sodium pentobarbital ~ 20 mg/kg/h) and the effects of local injection of compounds into the receptive fields were determined (see section Pharmacological treatments for further information), as previously described (Dunham et al., 2008, Hulse et al., 2010b). Fine nerve filaments were dissected from the main trunk of the nerve cut centrally and differential recordings were made using bipolar platinum wire recording electrodes. Primary afferents were identified in the filaments using mechanical and/or electrical search stimulation of identified receptive fields located in the dorsomedial region of the right hind paw, the area innervated by the saphenous nerve in the rat. Filaments usually contained a single identified afferent (unit), but up to 3 units could be studied in the same filament provided the receptive fields were distinguishable. Action potentials from each fiber could be distinguished individually by offline action potential recognition and sorting. Data capture was through a micro 1401-3 (Cambridge Electronic Design) and offline action potential sorting and analysis was carried out on Spike 2 version 7 (CED).
Identified units were characterized according to their conduction velocity (CV) and response to mechanical stimulation of the receptive field. Units that could not be activated by peripheral mechanical stimulation were not studied further. Monopolar electrical stimulation was applied to the receptive field (up to 100 V, 0.5 ms duration) and 3 reproducible action potential latencies were required to calculate the conduction velocity (CV). Following CV measurement, any ongoing activity (action potential firing) was recorded for 100 s. Note that under normal conditions, the majority of afferents in the saphenous nerve do not show significant ongoing activity, as there are no muscle spindles, and very few cooling afferents in this largely cutaneous sensory nerve. Ongoing activity was defined as firing > 0.1 impulses/s occurring without any obvious initiating factor. During the period of recording of ongoing activity, no further stimulation of the receptive field was applied.
Mechanical thresholds were determined as the lowest von Frey hair applied that elicited a robust (> 3 action potentials) reproducible response (Dina et al., 2004, Dunham et al., 2008, Koltzenburg et al., 1999a, Lynn and Carpenter, 1982). Responses to light brush with a paintbrush and to a series of von Frey hairs were then recorded. Primary afferents with a CV less than 1 m·s− 1 were classified as C fibers, based on compound action potentials recorded in the same preparation in animals of a similar weight, sex and age (Dunham et al., 2008). Afferents that were not brush sensitive, with von Frey thresholds > 1 g were classified as nociceptors (Dunham et al., 2008, Lynn and Carpenter, 1982); C fiber nociceptors were those that met these criteria and had CV < 1 m·s− 1. Ongoing activity was outlined as those units with greater than 0.1 Hz (Shim et al., 2005).
Methodological note
It should be noted that hand-held von Frey hairs give an approximation of the mechanical thresholds of primary afferent units as application of a range of hairs exerts incremental, discrete forces rather than a continuous force on the receptive field. As von Frey hairs were used for behavioral tests, comparable methods of single neuronal activation were used. Single afferent mechanical thresholds are typically lower than behavioral withdrawal thresholds, as withdrawal reflexes require summation of input from multiple high threshold nociceptive afferents for activation.
Intracellular calcium measurements in primary dorsal root ganglion cells
DRG were dissected from adult Wistar rats, dissociated, and cultured as previously described (Wong et al., 2006). For TRPV1 experiments, following overnight pretreatment with VEGF-A isoforms in media or media alone, primary DRG cultures were pre-incubated with 100 μl of Ca2 +-sensitive dye (Fluo-4 direct) at 37 °C for 1 h during which time concentrated capsaicin agonist solutions at 6 × the final concentrations used were prepared and preheated to 37 °C. Fluorescence recordings were performed row by row. First the baseline fluorescence at 488 nm was read, then 20 μl capsaicin solution was added to the dye to achieve the final capsaicin concentration, and the fluorescence read within 45 s and then repeatedly every 10 s for ~ 4 min on a Wallac 1420 Victor 3™ multi-label reader (PerkinElmer Inc.). The change in fluorescence over baseline was determined for each recorded time point. Each capsaicin concentration was tested in multiple replicates (3–7). Some cultures were fixed, and stained for VEGFR2 (Cell Signaling, rabbit mAb 55B11).
Capsaicin-evoked currents in primary dorsal root ganglion cells
Primary DRG cultures were prepared as above, and grown on glass coverslips coated with poly-l-lysine and laminin. After 3–5 days coverslips were mounted into a recording chamber and visualized using Olympus BX50WI microscope (Olympus, UK) using a 60× water immersion objective. Cells were chosen for study based on cell diameter (all < 30 μm diameter). Cells were continually perfused 2–3 ml/min with an extracellular solution containing (mM): 145 NaCl, 5 KCl, 0.5 CaCl, 2 MgCl2, 10 HEPES, and 10 d-glucose, pH was adjusted to 7.4 at 310–320 mOsm. Low calcium solution was used to reduce any calcium dependent desensitization of the current. Cells were patch-clamped in the whole-cell configuration and held at − 80 mV at room temperature (18–22 °C). Patch pipettes were pulled from soda glass (Harvard Apparatus, UK) to a resistance of 3–4 mΩ and the tips were coated in surf wax (Mr Zoggs) to reduce the capacitive transient. The pipette solution contained (mM): KCl 140, NaCl 5, EGTA 5, MgCl2 2, HEPES 10, adjusted to pH 7.2 with KOH. Puff pipettes pulled from soda glass with resistances of 3–4 M were filled with 500 μM capsaicin in external solution (stock solution 10 mM dissolved in 10% DMSO, 10% Tween-80, 80% saline and diluted to working concentration in external solution) and positioned between 20 and 30 μm from the cell. A Pneumatic Picopump PV800 (WPI, Hertfordshire, UK) was used to apply a two second puff at 10 psi. Inward currents in response to the puff application were recorded using an axon 200B amplifier and pClamp 9 software (Axon Instruments, CA, USA) with a sampling rate 20 kHz and filtering at 5 kHz filter. The peak of the inward currents during puff application were measured using Clampfit 9 (Axon instruments).
TRPV1 immunoprecipitation and Western blotting
Immortalized rat embryonic DRG cells, that represent largely nociceptive neurons expressing TRPV1 and NGF receptors (50B11) (Chen et al., 2007) were grown in Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen), 10% FBS, 0.5 mM l-glutamine and an additional 2.2% glucose in 6 well plates. These neurons were used as an alternative to primary culture, to generate the amounts of protein required for phospho-immunoprecipitation, as required by the principles of the ‘3Rs’ (reduction, replacement, refinement) under UK and EU legislation. Upon reaching 80% confluence cells were differentiated for 24 h with 75 μg/mL forskolin and 0.1 nM NGF-2.5S, and then treated overnight (~ 16 h) with VEGF-A proteins (2.5 nM), NGF-2.5S (4 nM) or vehicle (PBS). Following treatment, cell lysate protein, extracted in the presence of phenylmethylsulfonyl fluoride and proteinase inhibitors, was subjected to immunoprecipitation using a TRPV1 antibody (Abcam, rabbit pAb, ab10296) and Millipore PureProteome™ Protein A Magnetic Bead System. The manufacturer's direct immunoprecipitation method was followed using 2 μg/mL antibody and 50 μg protein lysate. The eluates were separated on 10% SDS-PAGE gels blotted on PVDF membrane by wet transfer and incubated overnight with 2 μg/mL phospho-serine mouse mAb (Millipore, 4A4). After secondary antibody incubation (goat pAb HRP-anti-mouse IgG) blots were developed using Pierce ECL SuperSignal Femto reagent. Blots were stripped and re-probed using the aforementioned TRPV1 rabbit pAb. Cell lysate protein not subjected to immunoprecipitation was separated and blotted as above and TRPV1 levels detected. This indicated the TRPV1 input level into the immunoprecipitation.
VEGF-A isoform and VEGFR2 expression studies — immunofluorescence, ELISA and qRT-PCR
Total VEGF-A and VEGF-A165b were detected using validated, commercially available antibodies (Santa Cruz A-20, and AbCam MVRL56/1 respectively). The VEGF-A165b antibody detects the unique C-terminal of the alternatively spliced VEGF-Axxxb family (Woolard et al., 2004). VEGF-A165a levels were determined by subtraction of VEGF-A165b levels from total VEGF-A.
For immunofluorescence in rat DRG, animals were terminally anesthetized with sodium pentobarbital (60 mg/ml) and perfuse fixed with 4% paraformaldehyde (PFA). Ipsi- and contralateral lumbar dorsal root ganglia were removed. DRG were placed into PFA for 4 h and then cryoprotected in 30% sucrose solution overnight. 8 μM cryosections were cut from OCT embedded DRG and thaw mounted onto subbed slides. Sections were blocked in 10% fetal calf serum/5% bovine serum albumin/0.2% Triton × 100 in PBS for 2 h at room temperature. Primary antibodies were used at the concentrations below diluted in the blocking solution and incubated overnight at 4 °C. Prior to secondary antibody incubations sections were washed extensively with PBS. Sections were incubated in secondary antibodies diluted in 0.2% Triton × 100 in PBS for 2 h at room temperature. Streptavidin amplification was performed as per manufacturer's instructions (Vector, UK). Primary antibody concentrations: TrkA, 1 μg/ml; 56/1, 12 μg/ml; α-phospho-tyr1175 VEGFR2 and VEGFR2 (1 in 500 dilution). Secondary antibodies: biotinylated anti-rabbit (Jackson Immune Research, diluted 1 in 500). Alexa Fluor 488 or 594 were used at 1 in 1000 dilution. α-goat Alexa Fluor 594 (Invitrogen, UK). Hoechst (Sigma Aldrich) and goat SRSF1 (2 μg/ml, Santa Cruz). Images were visualized using a fluorescent microscope and captured before quantification.
VEGF-A165b splice variant levels were measured as a proportion of total VEGF-Axxx expression using sandwich ELISA (R&D systems Duoset VEGF ELISA DY-293) using N-terminal goat anti-human VEGF antibody against pan-VEGF (i.e. all the isoforms), and mouse anti-human antibody against VEGF-A165b (R&D systems), as previously described (Gammons et al., 2013).
Real-time qRT-PCR. 2 μg of DNase-digested total RNA was reverse transcribed using oligo (dT15) and random primers (Promega, UK). Real-time PCR was performed on a ABI 7000 thermocycler using ABsolute QPCR SYBR green mix (Thermo Scientific, UK) and 1 μM primers specific for VEGF-A165a, (forward — exon 7/8a: 5′-GTTCAGAGCGGAGAAAGCAT-3′; reverse 5′- TCACATCTGCAAGTACGTTCG-3′) and total VEGF-A (forward — exon 2/3: 5′-GGAGGGCAGAATCATCACGAAG-3′; reverse 5′-CACACAGGATGGCTTGAAGATG-3′) (Nowak et al., 2010, Woolard et al., 2004), and for the housekeeping genes (18S ribosomal, GAPDH and Microglobulin) (see (Amin et al., 2011) for primer sequences). Cycling conditions were: denaturation at 95 °C for 10 min, then 95 °C for 15 s, and 55 °C for 30 s for 40 cycles. RNA levels were estimated by (VEGF-A165a/VEGF-Atotal) = E−(CtVEGF165)/E−(CtVEGFtotal) where E is the efficiency of the qPCR reaction for the primer pair used, or for expression relative to housekeeping genes, V = 2−(ΔCt) where ΔCt is the difference between the cycle thresholds for VEGF-A and housekeeping gene. Values are expressed relative to saline treated tissues.
Experimental note
The presence and function of endogenous VEGF-A165b has been demonstrated using specific siRNA knockdown, expression in multiple cells, tissues and pathological conditions using rigorous controlled techniques (Harper and Bates, 2008) and the importance of VEGF-A165b in pathological conditions has recently been highlighted by its ability to predict response to bevacizumab in the registration trial in colorectal cancer (Bates et al., 2012). A recent study highlighted the importance of using appropriate controls to avoid artifactual detection of VEGF-Axxxb isoforms (Harris et al., 2012) in rodents. A further study (Bates et al., 2013) demonstrates clearly the importance of positive and negative controls, for example for effective PCR amplification, to eliminate non-specific antibody binding to mouse IgG and to prevent mispriming. To avoid possibilities of misinterpretation of artifactual amplification of products we used pro-angiogenic isoform specific primers (against exon 8a) and total VEGF-A primers to determine the effect of splicing inhibitors on VEGF-A splicing.
Pharmacological treatments
-
A)
VEGF neutralization and VEGF receptor block. Mechanical and thermal nociceptive behaviors were determined in adult mice before and 2 days after intraperitoneal (i.p.) injection with 6 μg/g bodyweight mouse G6-31 antibody (n = 5, both hind paws used as replicates) or vehicle (saline, n = 6).
VEGFR2 tyrosine kinase inhibitors PTK787 and ZM323881 were given systemically to rats by a single i.p. injection (PTK787: 30 mg/kg, 30 μg/g) or locally into the hind paw (rats and mice: ZM323881: 10 μl containing 100 nM). Vehicle (PTK787, saline; ZM323881, 0.001% DMSO in saline) was injected by the same route (n = 6/group). ZM323881 was given under brief isofluorane anesthesia (2–3% in O2). Nociceptive behavior was tested before and after treatment (PTK787 2 h; ZM323881 20–40 min).
-
B)Effect of exogenous VEGF-A165a and VEGF-A165b in control animals and after peripheral nerve injury:
-
i)Nociceptive behaviors — normal. Saline (200 μl), VEGF-A165a (8 ng/g body weight), VEGF-A165b (8 ng/g), VEGF-A165b (20 ng/g), VEGF-A121a (8 ng/g) and VEGF-A159 (8 ng/g) were injected i.p biweekly (n = 5 per group) and nociceptive behaviors assessed before and after administration for a period of 5 days (experiments performed in mice).
-
ii)Nociceptive behaviors — PSNI. After surgery for peripheral nerve injury, groups of mice received one of the following biweekly: VEGF-A165a (n = 6, 8 ng/g i.p. (Zheng et al., 2007)); VEGF-A165b (n = 6, 20 ng/g) or PBS vehicle (n = 16, 200 μl). Injections were given immediately after PSNI surgery, and after behavioral test sessions on days 3 and 7. Sham-operated controls (n = 5) received i.p. vehicle at the same times. Nociceptive testing was performed on days 1, 3, 5, 7 & 10 after PSNI.
-
iii)Primary afferent properties.Afferents were isolated and characterized as described above. The effect of rhVEGF-A165a injected locally into the receptive field (10 μl; 2.5 nM) on afferent activity was determined by a change in ongoing activity. In these experiments we determined that ~ 50% of identified nociceptive cutaneous afferents responded to VEGF-A165a with an increase in spontaneous action potential firing over a period of 1 h (Fig. 3E). In subsequent experiments, nociceptive afferents responding to VEGF-A165a were identified by this ongoing activity. Afferents were characterized by CV and mechanical threshold, and mechanical stimulus-evoked responses were recorded. VEGF-A was then injected locally, and these properties were recorded at set intervals for 60 min. Post-hoc analysis of mechanical threshold, mechanically evoked and ongoing activity recorded at 5, 30 and 60 min after VEGF-A administration was then done for those units that developed ongoing firing by 60 min.
-
i)
-
C)
Effect of SRPK1 inhibition in normal skin (mice) and after nerve injury (rats) on nociceptive behavior and VEGF-A isoform expression: SRPIN340 (10 μl; 10 μM), an inhibitor of SR protein kinases SRPK1 and 2 (Fukuhara et al., 2006) that are responsible for splicing control of VEGF-A isoforms (Nowak et al., 2010) or vehicle (saline) was injected into the plantar surface of one hind paw under brief isofluorane anesthesia (2–3% in O2). Nociceptive behavior was tested before and after injection (n = 6/group). Animals were killed by anesthetic overdose, and the plantar skin at the site of injection removed. Total VEGF-A and VEGF-A165b mRNA expression was determined at the site of SRPIN340 or saline injection by quantitative (q) PCR.
To investigate the effect of inhibition of splicing in the injured nerve, 8 rats underwent PSNI surgery and were treated with the SR protein kinase (SRPK1/2) inhibitor, SRPIN340 (Nowak et al., 2010) or vehicle (saline). SRPIN340 (10 μM) was incorporated into a sterile gel consisting of (2% hyproxymethylcellulose, 0.2% tyloxapol, 3.4% dextrose, 0.025% ethylenediaminetetraacetic acid (EDTA), 0.0006% benzalkonium chloride) and applied to the nerve in the area of the tight ligation (n = 4); gel without SRPIN340 was used in control animals (n = 4). Nociceptive testing was performed on days 1 and 2 after PSNI. Animals were killed by anesthetic overdose and the saphenous nerves and L3/L4 DRGs removed. VEGF-A splice variant mRNA expression was determined at the site of injury by qRT-PCR for total VEGF-A and VEGF-A165a.
-
D)Interactions between VEGF-A and TRPV1:
-
i)Effect of pharmacological TRPV1 receptor blockade (mice): Mechanical nociceptive behavior was determined every other day for 5 days. VEGF-A and SB366791 (500 μg/kg) or vehicle were given via systemic (i.p.) injection on days 1 and 3 immediately after behavioral testing (n = 3/group, both hind limbs tested and treated as replicates). To exclude an effect of pharmacological inhibition on central rather than peripheral TRPV1, 2.5 nM VEGF-A165a was injected subcutaneously into the plantar surface of the hindpaw was with either PBS or 1 μM SB366791 (n = 5 per group) followed by behavioral testing.
-
ii)VEGF-A-mediated TRPV1 sensitization (rats): To determine the effect of VEGF-A isoforms administered together with a non-sensitizing concentration of TRPV1 agonist in identified primary afferents, a bolus of 10 μM capsaicin was injected through an intra-femoral arterial cannula inserted in the mid-thigh in the opposite hind limb to the recordings, with the tip advanced to the bifurcation of the descending aorta. This allowed close arterial delivery of capsaicin (100 μl; 10 μM washed in with 400 μl saline) to the peripheral afferent receptors. The effects of VEGF-A165a and VEGF-A165b on TRPV1 agonist responses were investigated using close arterial injection of capsaicin combined with local (subcutaneous) injection of 2.5 nM VEGF-A165a, 2.5 nM VEGF-A165b or both together. When VEGF-A165b was used, it was then followed by VEGF-A165a to confirm VEGF receptor responses in the afferents studied, as described above.
-
i)
Fig. 3.

Effects of rhVEGF-A isoforms on primary afferent nociceptors. A. VEGFR2 is expressed in nociceptive sensory neurons as determined by double-labeling with the nociceptive markers TrkA (high affinity nerve growth factor receptor) and isolectin B4 (IB4). VEGFR2 expression is upregulated in TrkA + ve nociceptors ipsilateral, and in IB4-binding nociceptors contralateral, to partial saphenous nerve injury (PSNI). B. Photomicrographs of (i) TrkA positive DRG neurons, (ii) VEGFR2 positive neurons and (iii) merged images of (i) and (ii) showing TrkA and VEGFR2 colocalization (scale bar = 50 μm). (iv) Expression of VEGR2 in DRG neurons is much lower in naïve rat DRG compared to (v) animals with PSNI (scale bar = 100 μm). C. Endogenous VEGF-A moderates nociceptor sensitivity, as when VEGFR2 is inhibited by PTK787 mechanical activation threshold of individual nociceptors is reduced within 5 min and over the next 60 min, indicating sensitization. D. Digitized data trace showing the effect of vehicle (saline), VEGF-A165a and VEGF-A165b on mechanically evoked activity at 5 min, after discharge and ongoing activity in a single afferent nociceptor. rhVEGF-A165a sensitized afferents to mechanical stimulation, enhancing after discharge and ongoing activity. Vertical lines are time-compressed action potentials. E. Increased spontaneous ongoing activity was evoked by rhVEGF-A165a but not rhVEGF-A165b in ~ 50% of mechanonociceptive afferents in rats. (Saline vehicle n = 12, VEGF-A165a n = 15, VEGF-A165b n = 5). Graphs include data from all neurons, including those in which properties did not change in response to VEGF-A. F. VEGF-A165a led to increased ongoing activity in 56% of nociceptive C fibers (OA > 0.1 Hz (Shim et al., 2005)). VEGF-A165b did not alter the degree of ongoing activity or number of C fibers that demonstrated ongoing activity, and in addition blocked VEGF-A165a-induced ongoing activity. G. rhVEGF-A165a reduced primary afferent mechanical threshold 60 min after rhVEGF-A165a injection. This was not seen for rhVEGF-A165b, and was blocked by its co-administration. H rhVEGF-A165a increased primary afferent activity in response to stimulation at suprathreshold force, 5 and 60 min after the injection of rhVEGF-A165a, whereas saline and rhVEGF-A165b had no effect. * = p < 0.05, ** = p < 0.01, *** = p < 0.001 compared with saline, mean ± SEM.
Numbers of afferents included in the experiments were: ongoing and mechanically evoked activity — saline vehicle n = 12, VEGF-A165a n = 7, and VEGF-A165b n = 5; mechanical activation threshold — VEGF-A165a, saline n = 7, VEGF-A165b n = 5; capsaicin sensitization — baseline n = 16, VEGF-A165a n = 8, VEGF-A165b n = 8, VEGF-A165a + VEGF-A165b n = 7.
Experimental note: This concentration of capsaicin delivered by close arterial injection does not result in sensitization or desensitization of the TRPV1 receptors to agonist stimulation on repeated injection (Dunham, 2008, Dunham et al., 2008). A low capsaicin concentration to avoid possible desensitization of TRPV1 in the presence of a further sensitizing agent, as we hypothesized VEGF-A165a to be. Capsaicin injection resulted in a short burst of action potentials that confirmed access of the agonist to the afferent receptor terminals. It should be noted that the effective concentration of capsaicin at the primary afferent terminals when delivered by this method is approximately 1000 fold lower than that injected as a result of dilution in hind limb blood volume, and tissue penetration (Dunham, 2008).
Statistical analyses
The majority of data sets was Gaussian in nature and therefore met the requirements for parametric analyses; in a small number of cases, data sets were log transformed to render them Gaussian prior to analysis (e.g. withdrawal thresholds). Multiple groups were compared using one or two way ANOVA followed by post-hoc Bonferroni tests where appropriate, and where Gaussian assumptions were not met or log transformation did not render the samples Gaussian, non-parametric tests were used, in which case multiple groups were compared with Kruskal–Wallis or Friedman's tests followed by post-hoc Dunn's tests. Two group tests were 2 tailed Student's t-tests with Welch's correction where necessary for unequal variance, or Mann Whitney U tests for non-parametric data. Numbers of DRG neurons with TRPV1-activated currents were compared using Fisher's exact test.
Results
VEGF-A splice isoforms differentially affect pain behaviors, through direct VEGFR2-mediated effects on primary sensory nociceptive neurons
Systemic delivery of anti-mouse VEGF antibody acutely sensitized animals to mechanical (Fig. 2A) and thermal (Fig. 2B) stimulation. Neutralization of VEGF-A165b (Fig. 2C), and inhibition of VEGF receptor-2 (VEGFR2) by selective (ZM323881, Fig. 2D) and specific (PTK787, Fig. 2E) inhibitors also produced sensitization. Systemic recombinant human (rh)VEGF-A165b (up to 20 ng/g bodyweight, i.p.) had no effect on mechanical (Fig. 2F), or thermal (Fig. 2G) nociceptive behavior, whereas rhVEGF-A165a (8 ng/g bodyweight, i.p.) sensitized to mechanical (Fig. 2H) but not thermal stimuli (Fig. 2G). rhVEGF-A121a, which has the same C-terminal six amino acid sequence as the VEGF-Axxxa family but reduced affinity for neuropilin-1 (NP-1) also resulted in mechanical sensitization (Fig. 2I). rVEGF-A159, which lacks the six C terminal amino acids (Cebe Suarez et al., 2006), had no effect on pain (Fig. 2I), showing that the mechanism through which VEGF-A165a and VEGF-A121a enhance pain is C-terminal sequence dependent (summarized in Fig. 2J) .
Fig. 2.

VEGF-A isoforms differentially affect pain depending on VEGFR2 activation. A. Intraperitoneal injection of 6 μg/g anti-VEGF-A antibody induced significant mechanical allodynia in mice (n = 5; vehicle n = 6). B. Systemic injection of anti-pan-VEGF-A antibody (6 μg/g) but not vehicle lowered thermal nociceptive withdrawal latency. C. Mechanical allodynia was reproduced by an anti-VEGF-A165b antibody (n = 6), shown normalized to the data from panel A. D. Local blockade of VEGFR2 with 100 nM ZM323881 (specific for VEGFR2) resulted in mechanical allodynia (n = 6/group). E. Systemic injection of PTK787 (30 μg/g) significantly reduced mechanical withdrawal threshold in naïve rats compared to vehicle (saline, n = 6/group). F. rhVEGF-A165b (8 ng/g or 20 ng/g) was not painful in normal animals (n = 5/group). Arrowheads denote times of drug administration. G. Neither rhVEGF-A165a nor rhVEGF-A165b (both 8 ng/g bodyweight) affected thermal hyperalgesia in naïve mice compared to vehicle (saline, n = 5/group). H. rhVEGF-A165a (8 ng/g) induced mechanical allodynia. I. rhVEGF-A121a administration caused mechanical allodynia whereas rVEGF-A159 did not (n = 5/group). J. Comparison of the effects of different VEGF-A isoforms shows that rhVEGF-Axxxa-evoked allodynia is mediated by the C-terminal 6 amino acids. * = p < 0.05, ** = p < 0.01, *** = p < 0.001 compared with baseline measurements within the same group, ‡ = p < 0.05, ‡‡ = p < 0.01, ‡‡‡ = p < 0.001, between groups, NS = not significantly different. Mean ± SEM for mouse behavior, and median ± IQR for rat behavior.
We then determined the neuronal mechanism through which systemic rhVEGF-A165a might alter nociceptive behavior. VEGFR2 protein was detected in DRG neurons (Figs. 3A & B) as previously described (Lin et al., 2010, Sondell et al., 2000) by immunofluorescence in proportions of both TrkA- and isolectin B4, nociceptive neurons (Fig. 3A), with increased expression following traumatic nerve injury. Inhibition of VEGFR2 (locally applied PTK787 to the receptive field) directly sensitized nociceptors to mechanical stimulation (Fig. 3C). As endogenous VEGF-A isoforms can exert potent vascular effects, we also determined whether VEGFR2 inhibitors PTK787 and ZM323881 overtly affected local blood flow. Neither receptor blocker resulted in any reduction in local blood flow as measured by laser Doppler flowmetry, or in skin temperature, in contrast to local adrenaline injection used as a positive control (data not shown). To determine the roles of VEGF-A isoforms on sensory afferents, effects on nociceptors were determined before and after injection of vehicle, VEGF-A165a or VEGF-A165b (Fig. 3D). Injection of rhVEGF-A165a, but not rhVEGF-A165b into individual characterized sensory neuronal receptive fields resulted in the initiation of spontaneous ongoing firing (Fig. 3E) in 56% of mechano-sensitive primary afferent nociceptors tested (Fig. 3F), indicating expression of functional VEGF receptors in a large proportion of the sampled afferents. The proportion of neurons responding to VEGF-A (> 50%) was significantly higher than the proportion of VEGFR2 positive IB4/TrkA + ve neurons — posited to be nociceptors (Fang et al., 2005, Fang et al., 2006). There are a number of explanations for this including: unconscious bias in our search strategy for afferents (mechanosensitive, C-fiber nociceptors) leading to an over-representation of afferents expressing VEGFR2; detection of protein by immunofluorescence underestimating the degree to which functional VEGFR are found on sensory neurons; or a higher proportion of VEGF sensitive nociceptors in the paw than in other regions through which L4 DRG neurons receive input. As the characteristics and distribution of VEGF sensitive afferents are not fully known in any species, any bias was unavoidable, and only came to light in post-hoc analysis.
Those neurons that developed ongoing firing after VEGF-A165a administration also became more sensitive to mechanical stimulation after 5 min (Fig. 3H, evoked activity at 5 and 60 min after rhVEGF-A165a, main effect of drug p < 0.0001), had lowered mechanical activation thresholds at 5 min (thresholds were saline: 6 (9) g (median (range)); rhVEGF-A165a: 1.5 (3.9) g; rhVEGF-A165b: 4 (14.4) g, ANOVA p = 0.08) and 60 min (Fig. 3G) and increased after discharge post-stimulus at 5 (Fig. 3D) and 60 min (not shown), indicating VEGF-A165a mediated peripheral neuronal sensitization that would translate into increased sensitivity to painful mechanical stimulation. Conversely, VEGF-A165b does not lead to sensitization of nociceptor activity (Figs. 3E, F, G and H) and importantly, completely abolished VEGF-A165a induced nociceptor hyperexcitability in all instances (Figs. 3E, F, G and H). Thus both VEGFR2 inhibition and VEGFR2 activation by rhVEGF-A165a enhance nociception by sensitization of peripheral mechanosensitive nociceptors.
Alternative splicing of pre-mRNA to VEGF-Axxxa rather than VEGF-Axxxb is controlled by the constitutively active serine–arginine protein kinase SRPK1 (Nowak et al., 2010), leading to activation of the splicing factor SRSF1 and selection of the proximal splice site (Fig. 1). On activation, SRSF1 translocates to the nucleus, and therefore activation of SRSF1 and subsequent splice site choice can be assessed by the degree of nuclear localization (Ghosh and Adams, 2011). SRPK1 inhibition and block of SRSF1 function, thus enhancing distal splice site selection, results in an increased proportion of VEGF-Axxxb (Fig. 1). Subcutaneous injection of an SRPK inhibitor (SRPIN340) in normal rat hind paw switched splicing, reducing VEGF-A165a relative to total VEGF-A to 33% of control levels in skin (Fig. 4A). This was associated with a 50% increase in mechanical threshold (Fig. 4B), but no effect on thermal withdrawal latency (Fig. 4C). We then determined whether VEGF-Axxxa expression was altered in traumatic nerve injury (Hulse et al., 2008). After peripheral saphenous nerve injury (PSNI), there was a > 10 fold increase in the expression of VEGF-A165a mRNA (Fig. 4D) in the local environment at the site of injury. In the same animals, there was also increased nuclear localization of SRSF1 in the damaged L4 DRG (Fig. 4E, F), consistent with a switch in SRPK1 mediated splicing to VEGF-Axxxa in neurons. Staining of DRG for Y1175-phosphoVEGFR2 (Fig. 4G) demonstrated increased numbers of VEGFR2-pY1175 positive neurons (Figs. 4G, H), indicative of increased VEGFR2 activation in these neurons. SRPK inhibition by SRPIN340 as a depot at the site of nerve injury blocked the change in mechanical withdrawal threshold (Fig. 4I), with no effect on thermal withdrawal latencies (data not shown). It also blocked the increased expression of VEGF-A165a mRNA (Fig. 4D) and the SRSF1 activation in DRG neurons (Fig. 4J). In SRPIN340 treated animals there were no contralateral changes in either mechanical or thermal nociceptive behavior (not shown).
Fig. 4.

Splicing inhibitors that shift the balance of endogenous VEGF-A towards an excess of VEGF-Axxxb isoforms are anti-nociceptive in normal and nerve injured rats. A. Intraplantar injection of SRPK1 inhibitor SRPIN340 reduced the amount of VEGF-A165a mRNA as a proportion of the total VEGF-A mRNA in plantar skin compared to vehicle (saline). B. SRPK inhibition raised mechanical withdrawal thresholds i.e. resulted in hypoalgesia, in mice. C. SRPIN340 did not alter thermal withdrawal latencies. D. VEGF-Axxxa expression increased as a proportion of total VEGF-A after PSNI. This increase was inhibited by SRPK inhibition. E. Nuclear localization of SRSF1, indicative of SRPK1 activity, is increased in L3/4 DRG neurons following PSNI. F. SRSF1 expression (red) in the cytoplasm of naïve rat DRG sensory neurons (scale bar 50 μm) and SRSF1 expression in the nucleus (stained blue with Hoechst) of rat DRG sensory neurons following PSNI. Note blue staining of nuclei in naïve rats, but purple in PSNI (inset, arrow). G. Phosphorylated (p)Y1175-VEGFR2 (red) staining in naïve and nerve injured mice. H. The number of pY1175-VEGFR2 positive DRG neurons increased after PSNI (*p = 0.019). I. SRPIN340 prevented PSNI-induced mechanical allodynia. J. SRPIN340 reduced SRSF1 activation in DRG containing injured neurons 2 days after nerve injury. ‡, ‡‡‡, p < 0.05, 0.001 respectively compared to baseline; *, *** = p < 0.05, 0.001 respectively compared to other groups.
As nerve injury shifted the balance of VEGF-A isoforms towards VEGF-Axxxa, in both injured neurons and at the site of nerve injury, resulted in pro-nociception, and through blockade of this SPRK1–SRSF1 mediated switch with SRPIN340, VEGFxxxa mediated pro-nociceptive actions could be reversed, we hypothesized that altering the relative balance of VEGF-A isoforms with exogenous protein would have a similar effect. In contrast to normal animals (Fig. 2F), systemic rhVEGF-A165b treatment exerted anti-nociceptive effects on both mechanical (Fig. 5A) and thermal behavior (Fig. 5B) after PSNI, whereas rhVEGF-A165a was pro-nociceptive (Figs. 5B & C). Similar changes in thermal latencies but not in mechanical thresholds were also seen in the contralateral hindpaw (Fig. 5D), suggesting that central VEGF-A-dependent mechanisms may also contribute to changes in thermal nociception following nerve injury. It is possible that rhVEGF-A165b exerted little effect in uninjured animals because VEGF-A165b is the predominant VEGF-A isoform in both skin (Pritchard-Jones et al., 2007), and human and rat DRG neurons (~ 70% total, measured by ELISA, Figs. 6A & B), where it is expressed (Fig. 6C) in a proportion of TrkA-positive nociceptive neurons (Fig. 6D).
Fig. 5.

Exogenous VEGF-A165a exacerbates, and VEGF-A165b alleviates neuropathic pain. A. PSNI resulted in ipsilateral mechanical allodynia (NI + Vehicle) compared with sham and baseline. rhVEGF-A165b (20 ng/g) was anti-allodynic on days 3 (p < 0.001), 7 (p < 0.01) and 10 (p < 0.0001). Nerve injury on day 0, arrowheads denote drug injection. B. PSNI does not normally result in thermal hyperalgesia (NI + vehicle), but rhVEGF-A165a induced hyperalgesia (NI + VEGF-A165a) and rhVEGF-A165b hypoalgesia. C. rhVEGF-A165a (8 ng/g) enhanced ipsilateral mechanical allodynia (filled squares) compared to vehicle (filled circles). D. rhVEGF-A165a induced thermal hyperalgesia contralateral to PSNI. rhVEGF-A165b again resulted in hypoalgesia. ‡, ‡‡‡, p < 0.05, 0.001 respectively compared to baseline (not shown for mechanical thresholds for clarity as all significant); *, *** p < 0.05, 0.001 respectively compared to vehicle.
Fig. 6.

Expression of VEGF-A165a and VEGF-A165b in rat DRG. A. VEGF-A165b represents ~ 70% of total VEGF-A expression in DRG. B. In one human DRG VEGF-A165b represented a similar proportion of total VEGF-A expression to that seen in the rat. C. VEGF-A165b is expressed in neurons in embryonic human spinal cord and DRG. Higher magnification images are derived from the boxes in the top image and are left: DRG and right: spinal cord ventral horn. D. VEGF-A165b is expressed in a proportion of rat DRG neurons (Ai, iii, v), with overlap (arrows) with the nociceptive markers TrkA (Aii, iv, vi) and a small colocalization with IB4 (Aii, iv, vi). Scale bar = 75 μm. High power images of a single neuron showing colocalization of VEGF-A165b (green) and TrkA (red). Scale bar = 50 μm.
VEGF-A isoforms affect pain by a TRPV1-dependent mechanism
Sensitization through phosphorylation of the TRPV1 ‘capsaicin’ receptor is a common endpoint in the sensitization of many nociceptors to both thermal and mechanical stimulation in inflammation, and nerve injury (Levine and Alessandri-Haber, 2007). TRPV1 is a thermal (Caterina et al., 1997), not a mechano-transducer molecule, but TRPV1 agonists are well recognized to alter both thermal and mechanical thresholds in humans (Fluhr et al., 2009). TRPV1-expressing peripheral sensory nerves are mechanosensitive in addition to thermosensitive (Brenneis et al., 2013). There is substantial evidence of an involvement of TRPV1 in mechanical sensitization in visceral afferents (see references in Jones et al., 2005, Kiyatkin et al., 2013, Ravnefjord et al., 2009). Peripheral sensitization of afferents involving TRPV1-dependent mechanisms has also been reported in deep tissue afferents (Kelly et al., 2013, Lam et al., 2009), and importantly for these data, in skin, where TRPV1 sensitization by agonist, such as capsaicin, lowers mechanical thresholds and hence contributes to enhanced mechanonociception (Li et al., 2008, Ren et al., 2005, Ren et al., 2006). Systemic pharmacological antagonism (using SB366791 Fig. 7A) and TRPV1 knockout (Fig. 7B) both eliminated VEGF-A165a-mediated mechanical allodynia indicating that the mechanism of action of VEGF-A165a involves, at least in part, TRPV1. TRPV1 was colocalized with VEGFR2 in DRG neurons (Fig. 7C). Administration of locally applied VEGF-A165a to the plantar surface of the hindpaw led to mechanical hypersensitivity, which was blocked by local co-administration of the TRPV1 antagonist SB366791 (Fig. 7D), indicating peripheral TRPV1 in VEGF-A165a-induced mechanical hypersensitivity.
Fig. 7.

VEGF-A isoforms alter nociception in a TRPv1 dependent manner. A. Systemic TRPV1 antagonism with SB366791 in mice resulted in inhibition of rhVEGF-A165a-induced mechanical allodynia. Arrows denote time of drug administration. B. TRPV1 knockout mice did not develop rhVEGF-A165a-induced mechanical allodynia, in contrast to wild-type strain matched controls. C. TRPV1 was co-expressed with VEGFR2 in sensory dorsal root ganglia sensory neurons (scale bar = 20 μm). D. Local administration of VEGF165a + vehicle into the plantar hindpaw resulted in a reduction in mechanical withdrawal values, which was blocked by co-administration of the TRPV1 antagonist SB366791 (TRPV1 antagonist).
We then determined whether VEGF-A isoforms affected TRPV1 function in sensory neurons. Capsaicin induced a dose dependent increase in intracellular calcium in primary DRG cells (Fig. 8A). Treatment with rhVEGF-A165a enhanced TRPV1-ligand (capsaicin) stimulated calcium influx (Figs. 8B & C), confirmed by patch clamping, where rhVEGF-A165a enhanced TRPV1-ligand induced currents (Figs. 8D & E) consistent with altered pain behavior. Capsaicin induced currents were found more frequently in primary DRG neurons incubated with VEGF-A165a (10/14 responders) than control (4/16, p = 0.03). rhVEGF-A165a, but not rhVEGF-A165b, caused significant TRPV1 phosphorylation in DRG cells, with no increase in overall TRPV1 expression level (Fig. 8F). Sensitization of TRPV1 is fundamental to the development of hyperalgesia (Ferrari et al., 2010) and dependent on PKC phosphorylation (Ristoiu et al., 2011). The VEGF-A165a-enhanced calcium response was inhibited by incubation with the PKC inhibitor bisindolylmaleiamide-1 (BIM, Fig. 8G). In vivo, low dose capsaicin evoked neuronal activity in primary afferent nociceptors, which was increased by rhVEGF-A165a (Fig. 8H) and was blocked by rhVEGF-A165b (Fig. 8H). These behavioral, cellular and in vivo physiological experiments indicate that VEGF-A165a-enhanced pain is at least partly mediated by enhanced sensory neuronal properties, through mechanisms that involve activation of PKC, and TRPV1 phosphorylation.
Fig. 8.

VEGF-A modulated TRPV1-agonist evoked responses in dorsal root ganglion neurons. A. Capsaicin stimulated a concentration-dependent increase in intracellular calcium in DRG neurons. B. This was increased by rhVEGF-A165a, and reduced by rhVEGF-A165b (mean ± SEM, n = 3–7). C. Treatment of rat DRG neurons with rhVEGF-A165a increased capsaicin-stimulated calcium influx (area under the curve of the calcium responses shown in Fig. 8B) compared with capsaicin alone or rhVEGF-A165b (2 way ANOVA main effect of drug p = 0.0051). The bell shaped concentration–response curve displays TRPV1 desensitization at higher capsaicin concentrations (5 μM). D. Example of a digitized trace of raw capsaicin-evoked current in the presence (gray) and absence of capsaicin. E. Capsaicin-evoked currents in primary DRG neurons were significantly larger in neurons incubated in VEGF-A165a overnight compared to vehicle treated neurons (box and whisker plots showing median, range, min and max). F. rhVEGF-A165b treatment enhanced TRPV1 serine phosphorylation in 50B11 immortalized DRG cells. IP of protein with TRPV1 antibody followed by IB with anti-pSer antibody showed rhVEGF-A165a, but not rhVEGF-A165b-mediated phosphorylation of TRPV1. (NGF treatment = positive control). G. Whereas 0.2 μM capsaicin alone did not alter intracellular calcium itself, overnight treatment with rhVEGF-A165a + 0.2 μM capsaicin resulted in a robust sustained increase in response to capsaicin, which was blocked by treatment with the PKC inhibitor BIM1 (2 way ANOVA main effect of drug p = 0.0003). H. Low concentration capsaicin (concentration at terminals ~ 10 nM) led to evoked activity from C fiber nociceptors in vivo. Capsaicin-evoked activity was increased by rhVEGF-A165a and blocked by rhVEGF-A165b. ‡, ‡‡, ‡‡‡, p < 0.05, 0.01, 0.001 respectively compared to baseline. *, **, *** = p < 0.05, 0.01, 0.001 respectively compared to other groups.
Discussion
Clinical and experimental reports of the detrimental effects of anti-VEGF agents on neuronal integrity and pain have raised concerns over the use of such therapies as their use can result in neuronal damage, often leading to pain (Verheyen et al., 2012). VEGF-A165a is reported to have both pro- (Benton and Whittemore, 2003, Herrera et al., 2009, Liu et al., 2012, Malykhina et al., 2012) and anti-nociceptive effects (Grosios et al., 2004, Lin et al., 2010, Verheyen et al., 2013). We hypothesized that this conflict in the literature regarding findings on pain may be resolved by a more detailed understanding of the contributions of the alternatively spliced VEGF-A isoforms to nociception. We show herein that a controlled change in the repertoire of VEGF-A alternative splice variants in the environment around peripheral sensory neuronal fibers/terminals, using either exogenous protein or control of endogenous splicing in favor of VEGF-Axxxa, results in enhanced pain, and that VEGF-A165b can alleviate pain in neuropathy.
Although differential expression of several alternatively spliced growth factors has been reported after peripheral nerve injury (Amiri et al., 2009, Chen et al., 2008, Kerber et al., 2003, Kerr et al., 2010), and injured peripheral neurons show altered RNA splicing (Kiryu-Seo et al., 1998), control of pain through targeting of alternative RNA splicing has not been previously reported. We have shown, for the first time, that peripheral axotomy activates changes in alternative RNA splicing in the area of damage, where mediators in the local environment can profoundly affect neuronal properties (Djouhri et al., 2012, Obata et al., 2004), possibly through TRPV1 activation on sensory nerve fibers (Hoffmann et al., 2009), as well as in the damaged neurons themselves. Use of a specific SRPK1 inhibitor has, also for the first time, allowed RNA splicing mechanisms to be considered as a potential analgesic strategy and enabled us to identify a relationship between changes in alternative RNA splicing and pain. The serine–arginine-rich protein kinases (SRPKs) are a small kinase family with principal actions on mRNA splicing and maturation (Giannakouros et al., 2011). Of the mammalian target RNAs affected by SRPK1/2 and SRSF1-controlled splicing (Fig. 9), none have been previously implicated in pain or nociception, other than VEGF-A.
Fig. 9.

Downstream targets of the serine–arginine protein kinase SRPK1. The serine–arginine protein kinase is known to have three major downstream targets, the RNA splicing factors SRSF1 (Edmond et al., 2011), SRSF2 (Aubol and Adams, 2011, Ngo et al., 2005, Velazquez-Dones et al., 2005), and the lamin B receptor (Papoutsopoulou et al., 1999). SRPK1 activity results in Hsp90-dependent nuclear translocation of SRSF1 (Zhou et al., 2012). SRSF1 has been reported to control alternative RNA splicing of the proto-oncogene myc, BIM (BCL2L11) (Anczukow et al., 2012), the cation cotransporter SLC39A14 (Thorsen et al., 2011), the tumor suppressors MKNK2 and BIN1 (Das et al., 2012, Karni et al., 2007), the angiogenesis related genes RON (Ghigna et al., 2005) and TEAD1 (Das et al., 2012), and VEGF-A (Amin et al., 2011, Nowak et al., 2010, Nowak et al., 2008). TEAD1 activates VEGF-A expression (Teng et al., 2010). None of the downstream targets of SRPK1 has been implicated in nociception other than VEGF-A.
As pre-mRNA splicing inhibition affected the balance of endogenous VEGF-A isoforms and nociception, and exogenous VEGF-A isoforms modulated behaviors and neuronal properties in a similar fashion, we hypothesize that it is the balance of VEGF-Axxxa and VEGF-Axxxb that determines the net effect on nociception. A slight disruption in this balance can have profound effects on VEGFR2 function (Table 1) as both receptor number and intracellular signaling mechanisms are altered. VEGF-A165a and VEGF-A165b have the same binding affinities to VEGFR2. However, when the two isoforms are equimolar or VEGF-A165b is in excess (as it often is in normal tissues, data herein, (Harper and Bates, 2008, Pritchard-Jones et al., 2007)), VEGF-A165b can reduce VEGF-A165a actions by ~ 95% (Hua et al., 2010). This is brought about by competitive antagonism at VEGFR2 (Kawamura et al., 2008, Woolard et al., 2004), and reduction in receptor number (Ballmer-Hofer et al., 2011). This complex mechanism can explain why local alteration of alternative RNA splicing, with a > 60% reduction in VEGF-A165a mRNA in skin, induced hypoalgesia in normal animals whereas systemic low concentration VEGF-A165b had little effect. Conversely, increasing VEGF-A165a using systemic exogenous recombinant protein had clear pro-nociceptive effects on both behavior and neurons.
Increasing local VEGF-A165a had a robust action on a sub-population of small unmyelinated somatic nociceptors that express functional VEGF receptors and TRPV1 receptors, increasing spontaneous firing (Djouhri et al., 2006, Hulse et al., 2010b) and mechanically-evoked activity, and lowering activation thresholds, all changes indicative of peripheral sensitization of sensory neurons. All of these changes, particularly increased spontaneous firing, increase afferent barrage and induce central sensitization in the spinal cord and higher centers, leading to altered pain behaviors (hyperalgesia and allodynia) (Grubb, 1998). Peripheral administration of VEGF-A165a had rapid (within 5 min) effects on primary afferents in vivo, suggestive of direct VEGF-A effects on neurons. This is supported by our data that show increased Y1175 phosphorylation of neuronal VEGFR2 after nerve injury, and by the direct modulation of TRPV1 currents in isolated neurons. Neuronal properties in intact afferent fibers can be affected by growth factor/inflammatory mediator actions at both receptor terminals, as a result of neuroinflammation caused by degeneration of adjacent fibers (Djouhri et al., 2012, Obata et al., 2004), and by mechanical stimulus-enhancement of endothelin hyperalgesia, mediated through endothelial cell ATP release and nociceptor sensitization (Joseph et al., 2013, Joseph, 2014). VEGF effects on neurons are unlikely to be entirely mediated through indirect vascular effects, as local blood flow was unaffected by the VEGFR antagonists that reduced nociceptive thresholds. We cannot completely exclude a contribution from the vasculature in the mechanical behavioral effects of VEGF-A165a (Chen et al., 2014, Joseph et al., 2013). This mechanism could contribute only in part to the pro-nociceptive effects that we report, as the actions of VEGF-A165a on cultured neurons, and in vivo demonstrate that VEGF-A165a exerts direct sensitizing effects on neurons that are independent of any mechanical stimulation, or other cells.
PLC/PKC signaling is key in peripheral nociceptor sensitization (Ferrari et al., 2010, Joseph et al., 2007), as changes in PKC activation modulate both voltage gated sodium channels (Malykhina et al., 2012, Stamboulian et al., 2010) and other key channels such as TRPV1 (Moriyama et al., 2005, Ristoiu et al., 2011, Rosenbaum and Simon, 2007). Our results show that, at least in vitro, PKC contributes to the VEGF-A165a modulation of TRPV1 sensitivity, possibly thereby contributing to alteration of neuronal properties/excitability. VEGF-A proteins also interact with neuropeptides in other tissues, such as somatostatin and angiotensin in the retina (Mei et al., 2012, Wilkinson-Berka, 2004) and kappa opioids in tumor angiogenesis (Yamamizu et al., 2013, Yamamizu et al., 2011) often through common downstream signaling pathways (Pan et al., 2008). Interestingly, all these neuropeptides are also implicated in nociception (Pan et al., 2008, Rice et al., 2014), suggesting that VEGF-A nociceptive signaling may also involve complex interactions with other pro-nociceptive molecules, in addition to its direct effects.
VEGF-A165b has actions on nociception that involve TRPV1, a key molecule in the sensitization of neurons leading to chronic pain states (Levine and Alessandri-Haber, 2007). VEGF-A165a exerts direct effects on agonist-induced TRPV1 channel opening (Figs. 8D, E), TRPV1-evoked calcium signaling and TRPV1 phosphorylation in isolated DRG neurons (Fig. 8), and alters neuronal properties in neurons co-expressing functional TRPV1 receptors resulting in peripheral mechanical sensitization (Fig. 3) suggesting direct modulation of neuronal TRPV1. It is therefore somewhat surprising that VEGF-A165a altered mechanical but not thermal thresholds in the normal animal, given that TRPV1 is well-known as a thermal transducer molecule (Caterina et al., 1997). Local capsaicin can however cause peripheral mechanical sensitization of cutaneous (Li et al., 2008, Ren et al., 2005, Ren et al., 2006, Wang et al., 2011), deep tissue and visceral afferents (Kiyatkin et al., 2013, Lam et al., 2009). The mechanism(s) through which TRPV1-dependent peripheral mechanical sensitization of afferents occurs are not known, but may be a consequence of altered nociceptor excitability, rather than directly affecting mechanotransduction per se (Malykhina et al., 2012, Raouf et al., 2012). Heterodimerization of TRPV1 with TRPA1 (Akopian, 2011), a molecule implicated in mechanical sensitization of primary afferents (Dunham et al., 2008, Lennertz et al., 2012), may explain, in part, the TRPV1 agonist effects on mechanical nociception. Of course, we cannot exclude the possibility of a contribution of an indirect effect through TRPV1 expressed elsewhere, particularly as TRPV1 is expressed in vascular and connective tissues (Fernandes et al., 2012), but the weight of evidence suggests a direct effect is at the very least a major contributor. In addition to a peripheral sensitizing action, VEGF-A165a could exert central effects, as both TRPV1 knockout and antagonist interventions (Fernandes et al., 2011) can also affect central TRPV1 receptor function. Indeed our results suggest that this is the case under sensitized but not normal conditions as contralateral effects of VEGF-Axxxa and b were seen in nerve injured animals but not in normals, (Fig. 5) (Hulse et al., 2012).
Pain is an expected consequence of neuronal damage, as the resulting local neuro-inflammatory responses alter the properties of peripheral sensory neurons. Neuroprotective therapeutic strategies are therefore thought to be good for both analgesia, and functional loss associated with neuronal damage. VEGF is known to be neuroprotective. It has thus been suggested that anti-VEGF therapies cause pain through blockade of the neuroprotective actions of VEGF (Verheyen et al., 2013, Verheyen et al., 2012). However, while both VEGF-A165a (Rosenstein and Krum, 2004, Storkebaum et al., 2004) and VEGF-A165b are neuroprotective for peripheral and central neurons (Beazley-Long et al., 2013) our findings show that only VEGF-A165b is anti-nociceptive. Thus the pain associated with anti-VEGF and anti-VEGFR therapies is unlikely to be entirely attributable to a loss of neuroprotective effect, but probably also involves modulation of nociception by VEGF-A isoforms. A more likely explanation for the difference in the effects of VEGF-A isoforms on pain behavior, and sensory neuronal function is a multifactorial process including alteration of the balance of isoforms present, different downstream actions on VEGFR2, and/or effects on central processing of nociceptive inputs, as well as neuroprotection.
These findings have important implications for the treatment of conditions in which VEGF-A drives pathology. VEGFR2 upregulation both ipsi- and contralateral to nerve injury in nociceptive neurons, involved in the establishment of chronic pain (Ferrari et al., 2010) may imply that VEGF-A is an important molecule in the protective priming of nociceptive systems around the body that can occur as a result of peripheral nerve damage or inflammation (Donaldson, 1999, Koltzenburg et al., 1999b). Consideration will need to be given as to whether isoform-specific VEGF-A supplementation might itself be used as an analgesic therapy. Early intervention to prevent changes in VEGF-A mRNA alternative splicing in pathological conditions may contribute to the prevention of the development of pain, in addition to being valuable in the treatment of existing pain. These findings open up the possibility of developing a novel class of analgesic agents based on controlling the splice regulatory mechanisms determining the balance of VEGF-A isoforms.
Author contribution statement
NB-L, RPH, HK, JP, DOB, HB, MVG, YQ, & ESF performed research. KBH, YQ, SJH, ADdG, & SDB provided research materials (antibodies, tissues, animals). NB-L, RPH, JH, SJH, AJC, SDB, DOB, & LFD designed the research, and analyzed data. DOB and LFD wrote the manuscript with contributions and final approval from all other authors. The data reported in this manuscript are available from the corresponding authors.
Acknowledgments
This work was funded by The Wellcome Trust (79633, 69029), The University of Bristol, Diabetes UK (11/0004192), the British Heart Foundation (BS06/005) and Richard Bright VEGF Research Fund. We thank Ahmet Hoke, John's Hopkins University, and Damon Lowes, University of Aberdeen for the gift of the 50B11 cell line.
Footnotes
Conflict of interest: LFD, DOB, JH, SJH are co-inventors on patents protecting VEGF-A165b and alternative RNA splicing control for therapeutic application in a number of different conditions. LFD, DOB, SJH are founder equity holders in, and Dob and SJH are directors of Exonate Ltd, a new company with a focus on the development of alternative RNA splicing control for therapeutic application in a number of different conditions, including analgesia.
Contributor Information
D.O. Bates, Email: David.Bates@nottingham.ac.uk.
L.F. Donaldson, Email: Lucy.Donaldson@nottingham.ac.uk.
References
- Akopian A.N. Regulation of nociceptive transmission at the periphery via TRPA1–TRPV1 interactions. Curr. Pharm. Biotechnol. 2011;12:89–94. doi: 10.2174/138920111793937952. [DOI] [PubMed] [Google Scholar]
- Amin E.M. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell. 2011;20:768–780. doi: 10.1016/j.ccr.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiri S. Differential gene expression and alternative splicing of survivin following mouse sciatic nerve injury. Spinal Cord. 2009;47:739–744. doi: 10.1038/sc.2009.26. [DOI] [PubMed] [Google Scholar]
- Anczukow O. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012;19:220–228. doi: 10.1038/nsmb.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubol B.E., Adams J.A. Applying the brakes to multisite SR protein phosphorylation: substrate-induced effects on the splicing kinase SRPK1. Biochemistry. 2011;50:6888–6900. doi: 10.1021/bi2007993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballmer-Hofer K. Neuropilin-1 promotes VEGFR-2 trafficking through Rab11 vesicles thereby specifying signal output. Blood. 2011;118:816–826. doi: 10.1182/blood-2011-01-328773. [DOI] [PubMed] [Google Scholar]
- Bates D.O. Association between VEGF splice isoforms and progression-free survival in metastatic colorectal cancer patients treated with bevacizumab. Clin. Cancer Res. 2012;18:6384–6391. doi: 10.1158/1078-0432.CCR-12-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D.O. Detection of VEGF-Axxxb isoforms in human tissues. PLoS ONE. 2013;8:e68399. doi: 10.1371/journal.pone.0068399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beazley-Long N. VEGF-A165b is an endogenous neuroprotective splice isoform of vascular endothelial growth factor a in vivo and in vitro. Am. J. Pathol. 2013;183:918–929. doi: 10.1016/j.ajpath.2013.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton R.L., Whittemore S.R. VEGF165 therapy exacerbates secondary damage following spinal cord injury. Neurochem. Res. 2003;28:1693–1703. doi: 10.1023/a:1026013106016. [DOI] [PubMed] [Google Scholar]
- Brenneis C. Phenotyping the function of TRPV1-expressing sensory neurons by targeted axonal silencing. J. Neurosci. 2013;33:315–326. doi: 10.1523/JNEUROSCI.2804-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger R.A. Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: a Gynecologic Oncology Group Study. J. Clin. Oncol. 2007;25:5165–5171. doi: 10.1200/JCO.2007.11.5345. [DOI] [PubMed] [Google Scholar]
- Caterina M.J. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Caterina M.J. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Cebe Suarez S. A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell. Mol. Life Sci. 2006;63:2067–2077. doi: 10.1007/s00018-006-6254-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. Immortalization and characterization of a nociceptive dorsal root ganglion sensory neuronal line. J. Peripher. Nerv. Syst. 2007;12:121–130. doi: 10.1111/j.1529-8027.2007.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. Expression patterns in alternative splicing forms of prosaposin mRNA in the rat facial nerve nucleus after facial nerve transection. Neurosci. Res. 2008;60:82–94. doi: 10.1016/j.neures.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Chen X. Does the anti-hyperalgesic disruptor of endothelial cells, octoxynol-9, alter nociceptor function? J. Neurophysiol. 2014;112(2):463–466. doi: 10.1152/jn.00034.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D.J., Hochster H.S. Update on clinical data with regimens inhibiting angiogenesis and epidermal growth factor receptor for patients with newly diagnosed metastatic colorectal cancer. Clin. Colorectal Cancer. 2007;7(Suppl. 1):S21–S27. doi: 10.3816/ccc.2008.s.004. [DOI] [PubMed] [Google Scholar]
- Cohen M.H. FDA drug approval summary: bevacizumab (Avastin) plus Carboplatin and Paclitaxel as first-line treatment of advanced/metastatic recurrent nonsquamous non-small cell lung cancer. Oncologist. 2007;12:713–718. doi: 10.1634/theoncologist.12-6-713. [DOI] [PubMed] [Google Scholar]
- Das S. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep. 2012;1:110–117. doi: 10.1016/j.celrep.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina O.A. Integrin signaling in inflammatory and neuropathic pain in the rat. Eur. J. Neurosci. 2004;19:634–642. doi: 10.1111/j.1460-9568.2004.03169.x. [DOI] [PubMed] [Google Scholar]
- Djouhri L. Spontaneous pain, both neuropathic and inflammatory, is related to frequency of spontaneous firing in intact C-fiber nociceptors. J. Neurosci. 2006;26:1281–1292. doi: 10.1523/JNEUROSCI.3388-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouhri L. Partial nerve injury induces electrophysiological changes in conducting (uninjured) nociceptive and nonnociceptive DRG neurons: possible relationships to aspects of peripheral neuropathic pain and paresthesias. Pain. 2012;153:1824–1836. doi: 10.1016/j.pain.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson L.F. Unilateral arthritis: contralateral effects. Trends Neurosci. 1999;22:495–496. doi: 10.1016/s0166-2236(99)01481-2. [DOI] [PubMed] [Google Scholar]
- Dunham J. Physiology and Pharmacology. PhD. University of Bristol; Bristol: 2008. On the role of TRPA1 in acute and inflammatory nociception; p. 244. [Google Scholar]
- Dunham J.P. Inflammation reduces mechanical thresholds in a population of transient receptor potential channel A1-expressing nociceptors in the rat. Eur. J. Neurosci. 2008;27:3151–3160. doi: 10.1111/j.1460-9568.2008.06256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmond V. SRSF2 is required for sodium butyrate-mediated p21(WAF1) induction and premature senescence in human lung carcinoma cell lines. Cell Cycle. 2011;10:1968–1977. doi: 10.4161/cc.10.12.15825. [DOI] [PubMed] [Google Scholar]
- Fang X. trkA is expressed in nociceptive neurons and influences electrophysiological properties via Nav1.8 expression in rapidly conducting nociceptors. J. Neurosci. 2005;25:4868–4878. doi: 10.1523/JNEUROSCI.0249-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X. Intense isolectin-B4 binding in rat dorsal root ganglion neurons distinguishes C-fiber nociceptors with broad action potentials and high Nav1.9 expression. J. Neurosci. 2006;26:7281–7292. doi: 10.1523/JNEUROSCI.1072-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes E.S. A distinct role for transient receptor potential ankyrin 1, in addition to transient receptor potential vanilloid 1, in tumor necrosis factor alpha-induced inflammatory hyperalgesia and Freund's complete adjuvant-induced monarthritis. Arthritis Rheum. 2011;63:819–829. doi: 10.1002/art.30150. [DOI] [PubMed] [Google Scholar]
- Fernandes E.S. The functions of TRPA1 and TRPV1: moving away from sensory nerves. Br. J. Pharmacol. 2012;166:510–521. doi: 10.1111/j.1476-5381.2012.01851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes E.S. Superoxide generation and leukocyte accumulation: key elements in the mediation of leukotriene B4-induced itch by transient receptor potential ankyrin 1 and transient receptor potential vanilloid 1. Faseb J. 2013;27(4):1664–1673. doi: 10.1096/fj.12-221218. [DOI] [PubMed] [Google Scholar]
- Ferrari L.F. Nociceptor subpopulations involved in hyperalgesic priming. Neuroscience. 2010;165:896–901. doi: 10.1016/j.neuroscience.2009.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluhr K. Capsaicin or menthol sensitization induces quantitative but no qualitative changes to thermal and mechanical pain thresholds. Clin. J. Pain. 2009;25:128–131. doi: 10.1097/AJP.0b013e3181817aa2. [DOI] [PubMed] [Google Scholar]
- Fukuhara T. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc. Natl. Acad. Sci. U. S. A. 2006;103:11329–11333. doi: 10.1073/pnas.0604616103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammons M.V. SRPK1 inhibition modulates VEGF splicing to reduce pathological neovascularization in a rat model of retinopathy of prematurity. Invest. Ophthalmol. Vis. Sci. 2013;54:5797–5806. doi: 10.1167/iovs.13-11634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia A.A. Phase II clinical trial of bevacizumab and low-dose metronomic oral cyclophosphamide in recurrent ovarian cancer: a trial of the California, Chicago, and Princess Margaret Hospital phase II consortia. J. Clin. Oncol. 2008;26:76–82. doi: 10.1200/JCO.2007.12.1939. [DOI] [PubMed] [Google Scholar]
- Ghigna C. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell. 2005;20:881–890. doi: 10.1016/j.molcel.2005.10.026. [DOI] [PubMed] [Google Scholar]
- Ghosh G., Adams J.A. Phosphorylation mechanism and structure of serine–arginine protein kinases. FEBS J. 2011;278:587–597. doi: 10.1111/j.1742-4658.2010.07992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakouros T. Serine–arginine protein kinases: a small protein kinase family with a large cellular presence. FEBS J. 2011;278:570–586. doi: 10.1111/j.1742-4658.2010.07987.x. [DOI] [PubMed] [Google Scholar]
- Grosios K. Angiogenesis inhibition by the novel VEGF receptor tyrosine kinase inhibitor, PTK787/ZK222584, causes significant anti-arthritic effects in models of rheumatoid arthritis. Inflamm. Res. 2004;53:133–142. doi: 10.1007/s00011-003-1230-4. [DOI] [PubMed] [Google Scholar]
- Grubb B.D. Peripheral and central mechanisms of pain. Br. J. Anaesth. 1998;81:8–11. doi: 10.1093/bja/81.1.8. [DOI] [PubMed] [Google Scholar]
- Hargreaves K. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Harper S.J., Bates D.O. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat. Rev. Cancer. 2008;8:880–887. doi: 10.1038/nrc2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris S. Do anti-angiogenic VEGF (VEGFxxxb) isoforms exist? A cautionary tale. PLoS ONE. 2012;7:e35231. doi: 10.1371/journal.pone.0035231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera J.J. Reduced vascular endothelial growth factor expression in contusive spinal cord injury. J. Neurotrauma. 2009;26:995–1003. doi: 10.1089/neu.2008.0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann T. Projected pain from noxious heat stimulation of an exposed peripheral nerve — a case report. Eur. J. Pain. 2009;13:35–37. doi: 10.1016/j.ejpain.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Hua J. Recombinant human VEGF165b inhibits experimental choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 2010;51:4282–4288. doi: 10.1167/iovs.09-4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulse R. Characterization of a novel neuropathic pain model in mice. Neuroreport. 2008;19:825–829. doi: 10.1097/WNR.0b013e328300ee0a. [DOI] [PubMed] [Google Scholar]
- Hulse R. 13th World Congress on Pain International Association for the Study of Pain (IASP), Montreal, Canada. 2010. Block of VEGF receptor 2 causes mechanical allodynia and peripheral sensitisation of mechanonociceptors in vivo; p. PM280. [Google Scholar]
- Hulse R. Intact cutaneous C fibre afferent properties in mechanical and cold neuropathic allodynia. Eur. J. Pain. 2010;14 doi: 10.1016/j.ejpain.2009.10.001. (565 e1-565 e10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulse R.P. Physiology. Proc Physiol Soc; Edinburgh: 2012. Vascular endothelial growth factor splice variants VEGF165a and VEGF165b modulate spinal nociceptive processing in the rat; p. PC263. [Google Scholar]
- Jones R.C., III The mechanosensitivity of mouse colon afferent fibers and their sensitization by inflammatory mediators require transient receptor potential vanilloid 1 and acid-sensing ion channel 3. J. Neurosci. 2005;25:10981–10989. doi: 10.1523/JNEUROSCI.0703-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph E.K. PLC-beta 3 signals upstream of PKC epsilon in acute and chronic inflammatory hyperalgesia. Pain. 2007;132:67–73. doi: 10.1016/j.pain.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Joseph E.K. Vascular endothelial cells mediate mechanical stimulation-induced enhancement of endothelin hyperalgesia via activation of P2X2/3 receptors on nociceptors. J. Neurosci. 2013;33:2849–2859. doi: 10.1523/JNEUROSCI.3229-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph E.K. ATP release mechanisms of endothelial cell-mediated stimulus-dependent hyperalgesia. J Pain. 2014;15(7):771–777. doi: 10.1016/j.jpain.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karni R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007;14:185–193. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura H. Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res. 2008;68:4683–4692. doi: 10.1158/0008-5472.CAN-07-6577. [DOI] [PubMed] [Google Scholar]
- Kelly S. Increased function of pronociceptive TRPV1 at the level of the joint in a rat model of osteoarthritis pain. Ann. Rheum. Dis. 2013 doi: 10.1136/annrheumdis-2013-203413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerber G. Neuregulin-1 isoforms are differentially expressed in the intact and regenerating adult rat nervous system. J. Mol. Neurosci. 2003;21:149–165. doi: 10.1385/JMN:21:2:149. [DOI] [PubMed] [Google Scholar]
- Kerr N. The expression of ELK transcription factors in adult DRG: Novel isoforms, antisense transcripts and upregulation by nerve damage. Mol. Cell. Neurosci. 2010;44:165–177. doi: 10.1016/j.mcn.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu-Seo S. A sequence-specific splicing activator, tra2beta, is up-regulated in response to nerve injury. Brain Res. Mol. Brain Res. 1998;62:220–223. doi: 10.1016/s0169-328x(98)00255-1. [DOI] [PubMed] [Google Scholar]
- Kisko K. Structural analysis of vascular endothelial growth factor receptor-2/ligand complexes by small-angle X-ray solution scattering. Faseb J. 2011;25:2980–2986. doi: 10.1096/fj.11-185397. [DOI] [PubMed] [Google Scholar]
- Kiyatkin M.E. Combined genetic and pharmacological inhibition of TRPV1 and P2X3 attenuates colorectal hypersensitivity and afferent sensitization. Am. J. Physiol. Gastrointest. Liver Physiol. 2013;305:G638–G648. doi: 10.1152/ajpgi.00180.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltzenburg M. Neutralization of endogenous NGF prevents the sensitization of nociceptors supplying inflamed skin. Eur. J. Neurosci. 1999;11:1698–1704. doi: 10.1046/j.1460-9568.1999.00590.x. [DOI] [PubMed] [Google Scholar]
- Koltzenburg M. Does the right side know what the left is doing? Trends Neurosci. 1999;22:122–127. doi: 10.1016/s0166-2236(98)01302-2. [DOI] [PubMed] [Google Scholar]
- Lam D.K. Glutamate and capsaicin effects on trigeminal nociception I: activation and peripheral sensitization of deep craniofacial nociceptive afferents. Brain Res. 2009;1251:130–139. doi: 10.1016/j.brainres.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert R. A Jackson Laboratory Resource Manual; The Jackson Laboratory: 2009. Breeding strategies for maintaining colonies of laboratory mice. [Google Scholar]
- Langenberg M.H. Phase I evaluation of telatinib, a VEGF receptor tyrosine kinase inhibitor, in combination with bevacizumab in subjects with advanced solid tumors. Ann. Oncol. 2011 doi: 10.1093/annonc/mdq767. [DOI] [PubMed] [Google Scholar]
- Lennertz R.C. TRPA1 mediates mechanical sensitization in nociceptors during inflammation. PLoS ONE. 2012;7:e43597. doi: 10.1371/journal.pone.0043597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine J.D., Alessandri-Haber N. TRP channels: targets for the relief of pain. Biochim. Biophys. Acta. 2007;1772:989–1003. doi: 10.1016/j.bbadis.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Li D. Sensitization of primary afferent nociceptors induced by intradermal capsaicin involves the peripheral release of calcitonin gene-related peptide driven by dorsal root reflexes. J Pain. 2008;9:1155–1168. doi: 10.1016/j.jpain.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W.C. Cross-species vascular endothelial growth factor (VEGF)-blocking antibodies completely inhibit the growth of human tumor xenografts and measure the contribution of stromal VEGF. J. Biol. Chem. 2006;281:951–961. doi: 10.1074/jbc.M508199200. [DOI] [PubMed] [Google Scholar]
- Lin J. VEGF and its receptor-2 involved in neuropathic pain transmission mediated by P2X(2)(/)(3) receptor of primary sensory neurons. Brain Res. Bull. 2010;83:284–291. doi: 10.1016/j.brainresbull.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Liu S. Vatalanib decrease the positive interaction of VEGF receptor-2 and P2X2/3 receptor in chronic constriction injury rats. Neurochem. Int. 2012;60:565–572. doi: 10.1016/j.neuint.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Lynn B., Carpenter S.E. Primary afferent units from the hairy skin of the rat hind limb. Brain Res. 1982;238:29–43. doi: 10.1016/0006-8993(82)90768-5. [DOI] [PubMed] [Google Scholar]
- Malykhina A.P. VEGF induces sensory and motor peripheral plasticity, alters bladder function, and promotes visceral sensitivity. BMC Physiol. 2012;12:15. doi: 10.1186/1472-6793-12-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei S. Mechanisms underlying somatostatin receptor 2 down-regulation of vascular endothelial growth factor expression in response to hypoxia in mouse retinal explants. J. Pathol. 2012;226:519–533. doi: 10.1002/path.3006. [DOI] [PubMed] [Google Scholar]
- Miller K. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007;357:2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- Moriyama T. Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol. Pain. 2005;1:3. doi: 10.1186/1744-8069-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesic O. Vascular endothelial growth factor and spinal cord injury pain. J. Neurotrauma. 2010;27:1793–1803. doi: 10.1089/neu.2010.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo J.C. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol. Cell. 2005;20:77–89. doi: 10.1016/j.molcel.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Nowak D.G. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 2008;121:3487–3495. doi: 10.1242/jcs.016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak D.G. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J. Biol. Chem. 2010;285:5532–5540. doi: 10.1074/jbc.M109.074930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K. Contribution of degeneration of motor and sensory fibers to pain behavior and the changes in neurotrophic factors in rat dorsal root ganglion. Exp. Neurol. 2004;188:149–160. doi: 10.1016/j.expneurol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Pan H.L. Modulation of pain transmission by G-protein-coupled receptors. Pharmacol. Ther. 2008;117:141–161. doi: 10.1016/j.pharmthera.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papoutsopoulou S. SRPK1 and LBR protein kinases show identical substrate specificities. Biochem. Biophys. Res. Commun. 1999;255:602–607. doi: 10.1006/bbrc.1999.0249. [DOI] [PubMed] [Google Scholar]
- Pritchard-Jones R.O. Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma. Br. J. Cancer. 2007;97:223–230. doi: 10.1038/sj.bjc.6603839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raouf R. Sodium channels and mammalian sensory mechanotransduction. Mol. Pain. 2012;8:21. doi: 10.1186/1744-8069-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravnefjord A. Involvement of the transient receptor potential vanilloid 1 (TRPV1) in the development of acute visceral hyperalgesia during colorectal distension in rats. Eur. J. Pharmacol. 2009;611:85–91. doi: 10.1016/j.ejphar.2009.03.058. [DOI] [PubMed] [Google Scholar]
- Ren Y. Sympathetic modulation of activity in Adelta- and C-primary nociceptive afferents after intradermal injection of capsaicin in rats. J. Neurophysiol. 2005;93:365–377. doi: 10.1152/jn.00804.2004. [DOI] [PubMed] [Google Scholar]
- Ren Y. Involvement of peripheral purinoceptors in sympathetic modulation of capsaicin-induced sensitization of primary afferent fibers. J. Neurophysiol. 2006;96:2207–2216. doi: 10.1152/jn.00502.2006. [DOI] [PubMed] [Google Scholar]
- Rice A.S. EMA401, an orally administered highly selective angiotensin II type 2 receptor antagonist, as a novel treatment for postherpetic neuralgia: a randomised, double-blind, placebo-controlled phase 2 clinical trial. Lancet. 2014;383:1637–1647. doi: 10.1016/S0140-6736(13)62337-5. [DOI] [PubMed] [Google Scholar]
- Ristoiu V. Hypoxia-induced sensitization of transient receptor potential vanilloid 1 involves activation of hypoxia-inducible factor-1 alpha and PKC. Pain. 2011;152:936–945. doi: 10.1016/j.pain.2011.02.024. [DOI] [PubMed] [Google Scholar]
- Rosenbaum T., Simon S.A. In: TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. Liedtke W.B., H. S, editors. CRC Press; 2007. TRPV1 receptors and signal transduction. [PubMed] [Google Scholar]
- Rosenstein J.M., Krum J.M. New roles for VEGF in nervous tissue — beyond blood vessels. Exp. Neurol. 2004;187:246–253. doi: 10.1016/j.expneurol.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Shim B. Mechanical and heat sensitization of cutaneous nociceptors in rats with experimental peripheral neuropathy. Neuroscience. 2005;132:193–201. doi: 10.1016/j.neuroscience.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Sondell M. Vascular endothelial growth factor is a neurotrophic factor which stimulates axonal outgrowth through the flk-1 receptor. Eur. J. Neurosci. 2000;12:4243–4254. doi: 10.1046/j.0953-816x.2000.01326.x. [DOI] [PubMed] [Google Scholar]
- Stamboulian S. ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel Na(v)1.7 and alters its gating properties. J. Neurosci. 2010;30:1637–1647. doi: 10.1523/JNEUROSCI.4872-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkebaum E. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays. 2004;26:943–954. doi: 10.1002/bies.20092. [DOI] [PubMed] [Google Scholar]
- Sundberg L.M. Effect of vascular endothelial growth factor treatment in experimental traumatic spinal cord injury: in vivo longitudinal assessment. J. Neurotrauma. 2011;28(4):565–578. doi: 10.1089/neu.2010.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng A.C. IRF2BP2 is a skeletal and cardiac muscle-enriched ischemia-inducible activator of VEGFA expression. Faseb J. 2010;24:4825–4834. doi: 10.1096/fj.10-167049. [DOI] [PubMed] [Google Scholar]
- Thorsen K. Alternative splicing of SLC39A14 in colorectal cancer is regulated by the Wnt pathway. Mol. Cell. Proteomics. 2011;10(M110):002998. doi: 10.1074/mcp.M110.002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga A. Effects of the novel TRPV1 receptor antagonist SB366791 in vitro and in vivo in the rat. Neurosci. Lett. 2005;385:137–142. doi: 10.1016/j.neulet.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Velazquez-Dones A. Mass spectrometric and kinetic analysis of ASF/SF2 phosphorylation by SRPK1 and Clk/Sty. J. Biol. Chem. 2005;280:41761–41768. doi: 10.1074/jbc.M504156200. [DOI] [PubMed] [Google Scholar]
- Verheyen A. Systemic anti-vascular endothelial growth factor therapies induce a painful sensory neuropathy. Brain. 2012;135:2629–2641. doi: 10.1093/brain/aws145. [DOI] [PubMed] [Google Scholar]
- Verheyen A. Therapeutic potential of VEGF and VEGF-derived peptide in peripheral neuropathies. Neuroscience. 2013;244:77–89. doi: 10.1016/j.neuroscience.2013.03.050. [DOI] [PubMed] [Google Scholar]
- Walczak J.S. Behavioral, pharmacological and molecular characterization of the saphenous nerve partial ligation: a new model of neuropathic pain. Neuroscience. 2005;132:1093–1102. doi: 10.1016/j.neuroscience.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Wang J. Octreotide inhibits capsaicin-induced activation of C and Adelta afferent fibres in rat hairy skin in vivo. Clin. Exp. Pharmacol. Physiol. 2011;38:521–527. doi: 10.1111/j.1440-1681.2011.05542.x. [DOI] [PubMed] [Google Scholar]
- Whittles C.E. ZM323881, a novel inhibitor of vascular endothelial growth factor-receptor-2 tyrosine kinase activity. Microcirculation. 2002;9:513–522. doi: 10.1038/sj.mn.7800164. [DOI] [PubMed] [Google Scholar]
- Wilkinson-Berka J.L. Diabetes and retinal vascular disorders: role of the renin–angiotensin system. Expert Rev. Mol. Med. 2004;6:1–18. doi: 10.1017/S1462399404008129. [DOI] [PubMed] [Google Scholar]
- Wong L.F. Retinoic acid receptor beta2 promotes functional regeneration of sensory axons in the spinal cord. Nat. Neurosci. 2006;9:243–250. doi: 10.1038/nn1622. [DOI] [PubMed] [Google Scholar]
- Wood J.M. PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res. 2000;60:2178–2189. [PubMed] [Google Scholar]
- Woolard J. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004;64:7822–7835. doi: 10.1158/0008-5472.CAN-04-0934. [DOI] [PubMed] [Google Scholar]
- Yamamizu K. The kappa opioid system regulates endothelial cell differentiation and pathfinding in vascular development. Blood. 2011;118:775–785. doi: 10.1182/blood-2010-09-306001. [DOI] [PubMed] [Google Scholar]
- Yamamizu K. small ka, Cyrillic Opioids inhibit tumor angiogenesis by suppressing VEGF signaling. Sci. Rep. 2013;3, 3213 doi: 10.1038/srep03213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C. VEGF reduces astrogliosis and preserves neuromuscular junctions in ALS transgenic mice. Biochem. Biophys. Res. Commun. 2007;363:989–993. doi: 10.1016/j.bbrc.2007.09.088. [DOI] [PubMed] [Google Scholar]
- Zhou Z. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol. Cell. 2012;47:422–433. doi: 10.1016/j.molcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]