


Abstract
Triplex-forming oligonucleotides (TFO) that bind DNA in a sequence-specific manner might be used as selective repressors of gene expression and gene-targeted therapeutics. However, many factors, including instability of triple helical complexes in cells, limit the efficacy of this approach. In the present study, we tested whether covalent linkage of a TFO to daunomycin, which is a potent DNA-intercalating agent and anticancer drug, could increase stability of the triple helix and activity of the oligonucleotide in cells. The 11mer daunomycin-conjugated GT (dauno-GT11) TFO targeted a sequence upstream of the P2 promoter, a site known to be critical for transcription of the c-myc gene. Band-shift assays showed that the dauno-GT11 formed triplex DNA with enhanced stability compared to the unmodified TFO. Band shift and footprinting experiments demonstrated that binding of dauno-GT11 was highly sequence-specific with exclusive binding to the 11 bp target site in the c-myc promoter. The daunomycin-conjugated TFO inhibited transcription in vitro and reduced c-myc promoter activity in prostate and breast cancer cells. The daunomycin-conjugated TFO was taken up by cells with a distinctive intracellular distribution compared to free daunomycin. However, cationic lipid-mediated delivery was required for enhanced cellular uptake, nuclear localization and biological activity of the TFO in cells. Dauno-GT11 reduced transcription of the endogenous c-myc gene in cells, but did not affect expression of non-target genes, such as ets-1 and ets-2, which contained very similar target sequences in their promoters. Daunomycin-conjugated control oligonucleotides unable to form triplex DNA with the target sequence did not have any effect in these assays, indicating that daunomycin was not directly responsible for the activity of daunomycin-conjugated TFO. Thus, attachment of daunomycin resulted in increased triplex stability and biological activity of the 11mer GT-rich TFO without compromising its specificity. These results encourage further testing of this approach to develop novel antigene therapeutics.
INTRODUCTION
Triplex-forming oligonucleotides (TFOs) that bind DNA in a sequence-specific manner may provide an effective way to modulate selectively gene expression via transcriptional repression, mutagenesis and recombination (1–3) This strategy has proven to be successful in various experimental models, including living cells and animals, and may provide the means for designing novel gene-targeted therapeutics (4). Binding of a TFO requires the presence of a relatively long and uninterrupted homopurine:homopyrimidine tract in DNA to ensure optimal stability and specificity of the triple helical complex (1–3). The TFO forms Hoogsteen or reverse Hoogsteen hydrogen bonds with the purine-rich strand of the duplex DNA (5,6). Purine-rich (GA) and mixed purine/pyrimidine (GT) TFOs bind preferentially antiparallel to the purine-rich strand, while pyrimidine-rich (CT) TFOs bind parallel. In both parallel and antiparallel triplex motif, the TFO is located in the major groove of the double helix. GA and GT TFOs form stable triplexes at physiological pH, whereas the ability of CT TFOs to form stable triplexes at neutral pH is severely limited (7). Studies in cell-free systems have shown that under optimal conditions (i.e. presence of Mg2+, appropriate pH and limited concentrations of monovalent cations), TFOs bind to homopurine sequences with dissociation constants (Kd) in the nanomolar range (8,9). In addition, triplex DNA, once formed, can be quite stable in vitro with half-lives of several hours or even days (9–13). The low Kd values and high stability allow TFOs to compete effectively with proteins, such as transcription regulatory factors, for binding to DNA and inhibit transcription in cell-free systems (9,14,15). However, various factors may limit the activity of TFOs in cells. Intracellular degradation of the oligonucleotide, sub-optimal ionic and pH conditions, insufficient nuclear accumulation and limited accessibility of the target site can prevent triplex formation (2,16,17). Once binding has occurred, changes in DNA and chromatin dynamics and the intracellular environment may lead to dissociation of the triple helical complex. DNA unwinding associated with replication and transcription as well as DNA repair may displace the TFO bound to DNA (18–29). Therefore, in addition to its intrinsic triplex-forming ability, the residence time of a TFO on the target is a critical factor determining the extent and duration of its biological effects. Accordingly, any approach designed to increase the half-life of the triplex DNA in physiological conditions is likely to increase the biological activity of a TFO. Direct chemical modification of TFOs by attachment of DNA-intercalating agents has been used to enhance triplex stability and increase efficacy (1,2). Non-sequence-specific DNA intercalators, such as acridine and psoralen derivatives, attached to the 5′ or 3′ ends of a TFO intercalate preferentially at duplex–triplex junctions and greatly stabilize the triple helix (30–36). The presence of the intercalator does not compromise the specificity of the TFO, since binding site recognition by acridine- and psoralen-conjugated TFOs has been shown to be determined by the oligonucleotide sequence. Furthermore, triplex-specific effects have been observed with intercalator-conjugated TFOs both in vitro and in cells (26,37–42).
In the present study, we took advantage of the unique DNA-intercalating properties of anthracycline derivatives to synthesize a modified TFO that could bind with high affinity and stability to a sequence in the promoter of the human c-myc gene (Fig. 1). Anthracyclines, such as daunomycin, are among the most commonly used and effective anticancer drugs. These compounds are well characterized DNA binding agents with estimated Kd values in the range of 10–6–10–7 M (43,44). Anthracyclines consist of two major structural elements, which are both involved in DNA binding: an aromatic aglycone chromophore or anthraquinone and the amino sugar daunosamine (44). The four-ring structure of the anthraquinone intercalates in duplex DNA with the long axis nearly perpendicular to the long axis of the base pairs adjacent to the intercalation site (43). This mode of intercalation is typical of the anthracyclines and is unlike the binding of other compounds, such as acridine and actinomycin, which bind parallel to the base pairs. Once intercalated in DNA, ring D of the anthraquinone protrudes into the major groove of the double helix, while ring A reaches out into the minor groove (43). The amino sugar, which is linked to ring A of the anthraquinone, interacts with groups in the minor groove, thus serving as an anchor that stabilizes the anthracycline–DNA complex (44). In a previous study, Garbesi et al. showed that attachment of daunomycin to the 5′ end of a 12mer CT TFO (TTTCTTCTTCTT) led to a significant increase in the affinity of the conjugated compared to the unmodified TFO (45). The daunomycin-conjugated TFO appeared to bind DNA simultaneously via triplex formation in the major groove, intercalation of the anthraquinone moiety at the duplex–triplex junction and binding of the amino sugar in the minor groove. These results were quite promising. However, it remained to be established whether this approach would work with TFOs binding in antiparallel orientation and at physiological pH and temperature. Ultimately, it needed to be determined whether conjugation with daunomycin resulted in enhanced biological activity of the TFOs, while retaining specificity.
Figure 1.


Map of the c-myc gene and structure of the daunomycin-conjugated TFO. (A) The polypurine:polypyrimidine sequence near the P2 promoter of the c-myc gene and the oligonucleotides GT11 and CO11 are shown. The sites in the target duplex complementary to GT11 and CO11 are underlined. Lines under the sequence indicate the 23, 28 and 40 bp oligonucleotides used in band shift assays and the 11 bp target site. The m40bp is a mutated duplex in which four bases, shown in italics in the target sequence, had been changed to TCTC. Brackets above the gene map indicate the inserts cloned into the pMyc-1388 and pMyc-262, respectively. K, X and R indicate KpnI, XhoI and RsrII restriction sites. (B) Oligonucleotides were modified at the 3′ end with a propanediol tail and linked at the 5′ end to the ring D of the anthraquinone via a hexamethylene bridge. The three domains of the daunomycin-conjugated oligonucleotide are expected to bind by triplex formation in the major groove (oligonucleotide), minor groove binding (amino sugar) and intercalation at the duplex-triplex junction (anthraquinone), respectively.
We have investigated the triplex-based approach as a means to downregulate expression of oncogenes, such as c-myc, in cancer cells. The c-myc gene is amplified, translocated and over-expressed in many cancers (46–49). Reducing expression of c-myc is sufficient to cause growth arrest and cell death in cancer cells, and tumor regression in mice (50–53). Therefore, compounds able to interfere with expression and function of this gene may have therapeutic applications. TFOs directed to regulatory sequences in the c-myc gene have been shown to inhibit transcription factor binding and transcription in vitro as well as promoter activity and gene expression in cells (54–58). We found previously that GT-rich TFOs directed to a sequence near the P2 promoter were particularly effective in inhibiting c-myc expression in cancer cells (59,60). In the attempt to increase the activity of TFOs by enhancing triplex stability, we have synthesized a daunomycin-conjugated GT TFO targeting this site in the c-myc promoter. The objective of the present study was to evaluate binding affinity, specificity and biological activity of the daunomycin-conjugated TFO. The antiparallel GT TFO covalently linked to daunomycin formed a stable and sequence-specific triple helical complex with duplex DNA. The daunomycin-conjugated TFO inhibited transcription in vitro, reduced c-myc promoter activity, and downregulated endogenous c-myc gene expression in cells. Daunomycin did not affect sequence-specificity of the TFO and was not directly responsible for the biological effects of the conjugated TFO. Daunomycin-conjugated control oligonucleotides unable to form triplex DNA with the target sequence did not have any effect in these assays, while the daunomycin-conjugated TFO did not inhibit expression of genes, such as ets-1 and ets-2, which had sequences similar to the c-myc target in their promoters. Thus, attachment of daunomycin resulted in increased triplex stability and biological activity of the GT-rich TFO, without apparently compromising its specificity. These studies reveal interesting properties of daunomycin-conjugated TFOs, which may represent a new class of gene-targeting agents. Furthermore, our results encourage investigation of this approach for development of novel antigene-based cancer therapeutics.
MATERIALS AND METHODS
Oligonucleotide synthesis
Daunomycin-conjugated oligonucleotides GT11 and CO11, with a hexamethylene bridge connecting the 5′ end of the oligonucleotide to the O-4 position in the D ring of the anthraquinone, were synthesized according to a previously described procedure with the following modifications (45). An iodoalkyl derivative of daunomycin was initially prepared with the amino group of daunomycin protected as trifluoroacetate. Synthesis of the daunomycin-conjugated oligonucleotides was then accomplished by reaction of the iodoalkyl derivative of daunomycin with phosphodiester oligonucleotides carrying a 5′ thiophosphate group. After extraction of the excess of iodoalkyl derivative, the trifluoroacetyl protecting group was removed from the amino group of the daunomycin by mild basic hydrolysis. The daunomycin-conjugated oligonucleotides were then purified by reverse-phase HPLC. The structure of dauno-GT11 was confirmed by ESI HPLC-MS. Oligonucleotides were also modified at the 3′ end by addition of a propanediol tail to increase resistance to 3′-5′ exonucleases (61). In agreement with the previous data (61), we observed by HPLC that more than 85% of daunomycin-conjugated TFO was intact following 24 h incubation in complete culture medium. All other phosphodiester oligonucleotides were purchased from Genset. The daunomycin-conjugated control oligonucleotide GT11C (dauno-GT11C), in which the GT11 sequence was linked to the amino sugar of daunomycin, was synthesized as described previously (45).
Electrophoresis mobility shift assay
Oligonucleotides corresponding to the pyrimidine strand of the c-myc target sequence were 5′ end-labeled with [γ-32P]ATP and then annealed with the complementary purine strand to form duplex DNA (59). Samples containing 1 nM of radiolabeled duplex DNA target were incubated with TFO or control oligonucleotide in 90 mM Tris-borate (pH 8.0) and 10 mM MgCl2 (TBM buffer). Prior to addition to the binding reactions, TFO and control oligonucleotide were heated at 65°C to reduce self-aggregation. In all the experiments binding reactions were incubated at 37°C for 18 h. Binding was then analyzed by gel electrophoresis under non-denaturing conditions using TBM as running buffer and maintaining the gel temperature at 20, 37 or 10°C as indicated in figure legends (59).
Dimethyl sulfate footprinting
A 339 bp fragment of the c-myc gene containing the TFO target sequence was isolated and radiolabeled as described previously (60). The 32P-end-labeled c-myc fragment was incubated with TFO or control oligonucleotide for 18 h at 37°C in TBM buffer. Samples were treated with 0.5% dimethyl sulfate (DMS) for 3 min at room temperature and then incubated with piperidine at 95°C for 30 min as previously described (62). Samples were analyzed by electrophoresis on a 10% polyacrylamide sequencing gel using radiolabeled 100 bp DNA ladder (Promega) as size marker (60). Sequencing of the 339 bp fragment was done using the SequiTherm Cycle sequencing system as described (60).
Luciferase reporter gene constructs
pGL3-Myc reporter plasmids, in which the firefly luciferase gene was under control of regulatory elements of the c-myc gene, were derived from the pHSR-1 plasmid that contained a genomic insert of the gene (59). A 3507 bp fragment was first excised from pHSR-1 using HindIII and XbaI and subcloned into pGL3 basic vector (Promega) to generate the p3507-Myc plasmid. An 849 bp RsrII–XbaI fragment, downstream of the c-myc P2 promoter, was then excised from p3507-Myc using HindIII and RsrII. The resulting p2658-Myc plasmid was digested with KpnI or MluI and XhoI, then religated to give p1388- and p262-Myc reporter plasmids, respectively. The P1 promoter region, upstream of P2, was retained in p1388-Myc, but excised from p262-Myc. Plasmids used for transfections into cells were purifed using the Qiagen Maxiprep kit (Qiagen, Valencia, CA).
In vitro transcription assay
The in vitro transcription assays were performed using the HeLa Scribe Nuclear Extract Transcription System (Promega) and the p1388-Myc, p262 or pGL3control plasmid (Promega) as template. Each plasmid (250 ng/sample) was incubated without or with TFO and control oligonucleotide for 18 h at 37°C in a buffer consisting of 20 mM HEPES, pH 7.2 and 10 mM MgCl2. HeLa cell nuclear extract, rNTPs and 1× transcription assay buffer (Promega) were then added to the binding reactions and the samples incubated at 30°C for 1 h. Samples were subjected to phenol/chloroform extraction and ethanol precipitation. The 1.2 kb kanamycin positive control RNA (Promega) was added as an internal control to monitor sample recovery throughout the procedure. RNA pellets were dissolved in nuclease-free water. To detect the RNA transcripts generated from the P2 promoter, samples were subjected to primer extension using the Primer Extension System with AMV Reverse Transcriptase and 32P-end-labeled GL2 primer (Promega). Primer extension products were analyzed by electrophoresis on denaturating polyacrylamide gels containing 8% acrylamide, 7 M urea and 1× TBE buffer.
Cellular uptake of daunomycin-conjugated oligonucleotide
Human prostate cancer (DU145) and breast cancer (MCF-7 and MDA-MB-231) cells were grown in RPMI-1640 and DMEM medium, respectively, supplemented with 10% heat-inactivated fetal bovine serum (Life Technologies) under standard conditions. Daunomycin (Sigma) was dissolved in sterile water and stored at –20°C. Cells were plated in 6 well plates at 1.5–3 × 105 cells/well, grown overnight and then were incubated with daunomycin and dauno-GT11. Cells were washed three times with OptiMEM (Gibco-BRL) and then kept in 2 ml of OptiMEM. Live cells were examined by phase-contrast and fluorescence microscopy (63). Digital images were captured using Image Pro software. For flow cytometry, cells were plated and incubated with daunomycin and dauno-GT11 as described above. At the indicated times, cells were washed three times with OptiMEM before harvesting by trypsinization. Cells were recovered by centrifugation, washed once with PBS, and then analyzed using a FACSCalibur (Becton Dickinson) equipped with a 488 nm argon laser. Fluorescence emission was measured using a 575/42 nm band-pass filter. Data were analyzed using Cell Quest software. In additional experiments, uptake of dauno-GT11 was examined with or without delivery of the TFO with cationic lipids (DOTAP; Roche). Cellular uptake and intracellular distribution were assessed by flow cytometry and fluorescence microscopy as described above.
Luciferase reporter assay
Cells were plated in 96 well plates at a density of 8–10 × 103 cells/well and grown for 24 h prior to transfection. Cells were transfected with 100 ng of pGL3-Myc reporter vectors, 50 ng of pRL-TK or pRL-SV40 (Promega) control vector and various concentrations of oligonucleotides using the transfection reagent DOTAP. pGL3-Myc and control plasmids were mixed with TFO or control oligonucleotide in 20 mM HEPES, pH 7.2, and incubated in the presence of DOTAP at a mass ratio of 5:1 (DOTAP:DNA) for 15 min at room temperature (64). The DOTAP/DNA complexes were diluted in serum-containing medium and added to the cells (100 µl/well). After 6 h, the transfection medium was removed and replaced with fresh medium. Cells were harvested 24 h later. Firefly and Renilla luciferase activities were measured in cell extracts using the Dual-luciferase assay system (Promega) according to the manufacturer’s instructions. Either the pRL-TK or pRL-SV40 control vectors were used to monitor transfection efficiency. Data were expressed as percent of the relative luciferase activity in TFO-treated cells compared to cells treated with an equal concentration of control oligonucleotide.
RNA and protein analysis
DU145 cells were seeded in 6 well plates at a density of 7 × 104 cells/well and transfected 24 h later with TFO or control oligonucleotide using DOTAP as described above. Total RNA was extracted from control and TFO-treated cells after 24 h using Trizol (Invitrogen). RNA concentrations were determined by spectrophotometry. RT–PCR was performed using the SuperScript One Step RT–PCR system (Invitrogen) and optimized conditions as described previously (64). Forward and reverse primers for c-myc were 5′-GGTCTTCCCC TACCCTCTCAACGA-3′ and 5′-GGCAGCAGGATAGT CCTTCCGAGT-3′, respectively, which amplified a fragment of 386 bp. Primers for β-actin were 5′-AAGAGAGG CATCCTCACCCT-3′ and 5′-TACATGGCTGGGGTGTT GAA-3′ and amplified a fragment of 218 bp. RNA (100 ng) was reverse-transcribed for 30 min at 50°C and then subjected to 26 cycles of PCR (94°C, 15 s; 55°C, 30 s; 72°C, 15 s). Samples were analyzed on 2% agarose gels and quantified using the AlphaImager and AlphaEase software (Alpha Innotech, San Leandro, CA). To determine protein content, cells were transfected with TFO and control oligonucleotides as indicated above. After 24 h cells were harvested and the level of selected proteins was examined by western blot as described previously (60). c-myc, ets-1, ets-2 and α-tubulin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and peroxidase-conjugated secondary antibodies from Amersham (Piscataway, NJ).
RESULTS
Selection of target site and design of daunomycin-conjugated TFOs
The selected target site is within a region that is highly conserved and known to regulate the activity of the P2 promoter of the c-myc gene (Fig. 1A). The P2 promoter is the major transcription initiation site in the c-myc gene, giving rise to 75–90% of transcripts in most cell types (48). A purine-rich sequence extends from –39 to –61 relative to the P2 start site and overlaps the binding sites of transcription factors, such as Sp1/Sp3, MAZ, Ets, E2F and Stat 3 (65–70). This region is required for transcription of both human and murine c-myc genes (66,71,72). TFOs directed to this polypurine sequence were found to inhibit binding of transcription activating factors, c-myc transcription in vitro and promoter activity in cells (57,58). Furthermore, an antiparallel 23mer GT TFO directed to this sequence inhibited c-myc expression and proliferation of cells that expressed high levels of the gene (59,60). For the present study, an 11mer GT TFO (dauno-GT11) directed to the 3′ portion of the polypurine sequence was synthesized and conjugated to daunomycin. The 11 bp homopurine target sequence (GGAGGGAGGGA) was optimal for triplex formation in the antiparallel motif (59,60,62). However, the complex formed by the unmodified 11mer TFO was unstable at physiological temperatures, thereby offering an ideal system for testing the effects of daunomycin on triplex stability. As shown in Figure 1B, the oligonucleotides, synthesized with a phosphodiester backbone, were covalently linked via a hexamethylene bridge to the O-4 position on the ring D of a daunomycin molecule. Oligonucleotides were also modified at the 3′ end by addition of a propanediol tail to increase nuclease resistance (61). A second oligonucleotide (dauno-CO11) with similar sequence was synthesized and conjugated to daunomycin. Dauno-CO11 matched in parallel orientation a sequence in the 5′ portion of the 23 bp target sequence (Fig. 1A). However, like the non-conjugated CO11, dauno-CO11 was unable to bind to a significant extent to the target duplex in physiological conditions (see below). Therefore, dauno-CO11 was used as a control oligonucleotide to assess the specificity of the effects of dauno-GT11 both in vitro and in cells.
Binding affinity and stability of triplex DNA formed by daunomycin-conjugated TFO
Binding of daunomycin-conjugated and non-conjugated oligonucleotides to the target sequence was assessed by gel shift assays. In all these experiments radiolabeled duplex DNA was mixed with increasing concentrations of third strand oligonucleotide and incubated at 37°C for 18 h. Binding was then examined by gel electrophoresis under non-denaturating conditions. Electrophoresis was first carried out under the most common assay conditions, which we use routinely, to examine triplex DNA formation: i.e. gel temperature maintained at 20°C or room temperature (57–60). As shown in Figure 2A, we could not detect triplex formation with the unmodified GT11 in these conditions. This was not due to the inability of GT11 to bind to the duplex DNA. In agreement with our previous data (62), triplex DNA formation was clearly seen with this TFO, when electrophoresis was done at a lower gel temperature (10°C) following the overnight incubation at 37°C (Fig. 2C). We attributed this to the instability of the triple helical complex formed by the unmodified oligonucleotide during the electrophoresis and reasoned that addition of an intercalating agent, such as daunomycin, might increase triplex stability and the likelihood that it would persist at physiological or near physiological temperature. As shown in Figure 2B, triplex DNA was detected as a distinct band in the autoradiogram following overnight incubation of the target duplex at 37°C with concentrations of dauno-GT11 as low as 100 nM. Approximately 50% of triplex DNA was seen at concentrations between 250 and 500 nM of dauno-GT11 (i.e. 250–500-fold molar excess compared to target DNA). Thus, the 11mer daunomycin-conjugated TFO had an affinity for the target sequence comparable to that of the longer TFOs directed to the P2 site that had been tested previously under similar conditions (57–60). Triplex DNA formed by dauno-GT11 was clearly more stable than the triplex formed by the unmodified GT11, since it persisted when electrophoresis was carried not only at 20°C (Fig. 2B) but also at 37°C (see Fig. 3 below). Even the 23mer GT TFO formed a triplex that was partially unstable under these conditions (59). As shown in Figure 2D, incubation of duplex DNA with 0.5–2 µM of dauno-CO11 produced a shift of <5% of the target. The limited binding of dauno-CO11 was in agreement with the presence of a matching sequence in the c-myc promoter fragment (62). The complex formed by dauno-CO11 was highly unstable and almost completely dissociated when electrophoresis was carried out at 37°C (see Fig. 3 below). Thus, unlike dauno-GT11, dauno-CO11 bound less and very weakly to the target duplex. Taken together, these results indicated that daunomycin enhanced binding of the TFO by increasing triplex stability, but binding was clearly dependent on the sequence of the oligonucleotide and the matching target.
Figure 2.

Triplex DNA formation by non-conjugated and daunomycin-conjugated TFOs. Oligonucleotides corresponding to the pyrimidine-rich strands of the duplex targets were 5′ end-labeled with [γ-32P]ATP and annealed to the complementary oligonucleotides. Duplex DNA at a concentration of 1 nM was incubated for 18 h at 37°C with the indicated concentrations of non-conjugated GT11, dauno-GT11 or dauno-CO11. Gel electrophoresis was carried out under non-denaturating conditions at a gel temperature of 20 (A, B, D, E and F) or 10°C (C). Duplex targets were the 28 (A–D), 23 (E) and 40 bp (F) double-stranded oligonucleotides (see Fig. 1). Positions of duplex and triplex DNA are indicated.
Figure 3.

Sequence-specific binding and inhibition of transcription initiation by the daunomycin-conjugated TFO. (A) Two 40 bp duplex oligonucleotides with either the wild-type (top) or a mutated (bottom) target sequence were incubated with dauno-GT11 or dauno-CO11 for 18 h at 37°C. Samples were then analyzed under non-denaturating conditions at a gel temperature of 37°C. The mutated site is shown in Figure 1. (B) A 32P-end-labeled 339 bp fragment of the c-myc promoter was incubated with or without dauno-GT11 and dauno-CO11 for 18 h at 37°C. DNA was then treated with DMS and piperidine to cleave unprotected guanines and samples were analyzed by gel electrophoresis under denaturating conditions. The position of the 11 nt target sequence is shown. (C) The p1388-Myc (top) and the pGL3control plasmid (bottom), which contained the SV40 promoter, were incubated without or with 2.5 µM of dauno-GT11 and dauno-CO11 for 18 h at 37°C. HeLa cell nuclear extract was added and samples were incubated for 1 h at 30°C. Following phenol/chloroform extraction and ethanol precipitation, samples were subjected to primer extension with a 32P-labeled primer. A radiolabeled internal control RNA (i.c.) was added to monitor sample recovery throughout the procedure. Samples were analyzed on a denaturating polyacrylamide gel. The positions of the transcripts generated from the c-myc and the SV40 promoter, respectively, are indicated.
To confirm that the daunomycin conjugated to the TFO intercalated at the duplex–triplex junction and was important for stabilization of the triplex, we performed additional assays using a shorter DNA duplex as target. The 23 bp duplex lacked any flanking sequence 3′ to the GT11 binding site (see Fig. 1A). In this case, because of the loss of a suitable intercalation site, the presence of daunomycin should not affect the stability of the triplex formed by dauno-GT11. As shown in Figure 2E, binding of dauno-GT11 to the 23 bp duplex was considerably reduced compared to the 28 bp duplex. Less than 10% of the target was bound at 500 nM of TFO, a concentration that gave more than 50% binding on the 28 bp duplex. These data strongly suggested that increased stability of the triplex formed by dauno-GT11 depended upon the intercalation of daunomycin at the duplex–triplex junction. Binding was significantly reduced but not completely abolished probably because of the persistence of some favorable stacking interactions between the anthraquinone moiety and the duplex. We also examined binding of the daunomycin-conjugated oligonucleotides on a 40 bp duplex to rule out that the limited binding of dauno-CO11 was due to the absence of an appropriate flanking sequence for intercalation of daunomycin in the shorter 28 bp duplex. The longer duplex had 8 and 10 nt flanking the 3′ and 5′ side of the target sequence, respectively (see Fig. 1A). The results obtained with the 40 bp duplex were identical to those observed with the 28 bp duplex, showing again that dauno-GT11 bound effectively to the target sequence, while dauno-CO11 was unable to bind (Fig. 2F).
Sequence-specific recognition of target DNA by the daunomycin-conjugated TFO
To provide evidence that binding of dauno-GT11 required the presence of perfectly matching sequence in the target duplex, we determined by gel shift assay whether dauno-GT11 was able to bind to a duplex in which the 11 bp target sequence had been mutated. As indicated in Figure 1, the mutated duplex contained four mismatched bases in the center of the target sequence. The presence of these centrally located mismatches would be highly destabilizing for binding of the oligonucleotide, but would not affect intercalation of daunomycin. Wild-type and mutated duplexes were incubated with the oligonucleotides overnight at 37°C and binding was analyzed by gel electrophoresis at 37°C. As shown in Figure 3A, under these conditions dauno-GT11 was clearly able to form triplex DNA with the wild-type duplex, while it was unable to bind to the mutated duplex even at a concentration of 5 µM. Dauno-CO11 did not bind to either duplex. This indicated that binding required the intact target sequence and the presence of daunomycin did not affect the discriminating power of the daunomycin-conjugated TFOs. DMS footprinting was used to further examine the triplex-forming ability and sequence-specificity of dauno-GT11. Although far from the complexity of the intracellular environment and genomic DNA, the footprinting assay is a more stringent test of the ability of a TFO to recognize and bind to a specific sequence in DNA than gel shift assays. In this study, a 339 bp fragment of the c-myc promoter was end-labeled and incubated without or with dauno-GT11 and dauno-CO11 at 37°C overnight. Following this incubation, binding reactions were treated with DMS and piperidine. Triplex formation with almost complete protection of the 11 bp target sequence was observed in samples incubated with 1 and 10 µM of dauno-GT11, whereas no protection was seen in the presence of equal concentrations of dauno-CO11 (Fig. 3B). The extent of protection achieved with dauno-GT11 was determined by densitometric analysis. More than 90% of the target DNA was protected in the presence of 1 and 10 µM of dauno-GT11, again indicating high binding affinity of the short daunomycin-conjugated TFO. It is worth noting that a similar degree of protection was achieved only with concentrations ≥10 µM of the 23mer GT-TFO under identical conditions (60). The interaction of dauno-GT11 with duplex DNA was highly sequence-specific. Protection was seen exclusively in correspondence with the 11 bp target sequence and no other region of the 339 bp fragment showed any alteration of the DMS cleavage pattern. This is remarkable since the c-myc promoter fragment used for footprinting contained at least two additional sites (GAACGGAGGGA and AGAGGGAGCGA), which differ only at two positions from the target sequence and, thus, could have allowed binding of the TFO. Binding specificity of the daunomycin-conjugated oligonucleotides was also demonstrated by the absence of interaction of dauno-CO11 with the 339 bp fragment. Thus, footprinting studies provided direct evidence that binding of daunomycin-conjugated TFOs depended strictly on the nucleotide sequence and was limited to the specific target site.
Inhibition of transcription initiation by daunomycin-conjugated TFO in a cell-free system
Dauno-GT11 is directed to a sequence in the c-myc promoter known to regulate transcription of the gene. To determine whether the dauno-TFO was able to inhibit transcription, we examined its effects in an in vitro transcription assay using c-myc reporter constructs as template. The p1388-Myc plasmid was incubated without and with dauno-GT11 or dauno-CO11 overnight at 37°C under conditions that allowed triplex DNA formation. Oligonucleotides were present in the binding reactions at a concentration of 2.5 µM (approximately equivalent to a 500-fold molar excess compared to template DNA). HeLa cell nuclear extract was added to the samples that were further incubated for 1 h at 30°C. RNA transcripts from the P2 promoter were detected by primer extension and gel electrophoresis (Fig. 3C, top). In the presence of dauno-GT11, transcription was reduced more than 50% compared to control samples. The control oligonucleotide dauno-CO11, which failed to form a stable triplex as documented by gel shift and DMS footprinting, had no effect on transcription. Similar results were observed when the p262-Myc plasmid was used as template in the assay (data not shown). Both dauno-GT11 and dauno-CO11 did not affect transcription from the SV40 promoter of the PGL3control vector (Fig. 3C, bottom). These results supported the conclusion that sequence-specific triplex formation at the c-myc promoter site by the daunomycin-conjugated TFO resulted in transcription inhibition and ruled out alternative decoy mechanisms.
Cellular uptake of the daunomycin-conjugated TFO
Prior to investigating the biological activity of dauno-GT11, we wanted to determine whether the presence of daunomycin had any effect on cellular uptake and intracellular distribution of the oligonucleotide. Because of the presence of the anthraquinone chromophore, uptake of the daunomycin-conjugated TFO could be examined by flow cytometry and fluorescence microscopy. The intensity and distribution of fluorescence signals in MCF-7 cells incubated with 0.2 and 1 µM of dauno-GT11 or daunomycin was first examined by flow cytometry. As shown in Figure 4, ∼24% of cells incubated with 0.2 µM of dauno-GT11 exhibited fluorescence signals above background levels compared to 62% of cells incubated with the same concentration of daunomycin. A higher percentage of cells took up daunomycin and dauno-GT11 at 1 µM (99 and 80%, respectively). However, at this concentration the mean fluorescence intensity in cells incubated with dauno-GT11 was only a third of that in cells incubated with daunomycin (16 versus 44). Similar data were obtained in three separate experiments, which confirmed that ∼3-fold less daunomycin-conjugated oligonucleotide was taken up by the cells compared to free daunomycin. Thus, these results suggest a differential uptake of dauno-GT11 and daunomycin, with a decreased ability of the oligonucleotide to enter cells compared to free drug. Intracellular distribution of the daunomycin-conjugated TFO was also examined by fluorescence microscopy. MCF-7 cells were incubated with 1 µM of daunomycin or dauno-GT11 for 6 h, washed and examined under a fluorescence microscope. As shown in Figure 4B, daunomycin and dauno-GT11 localized to distinct cellular compartments. Daunomycin was predominantly nuclear, whereas dauno-GT11 was localized primarily in the cytoplasm. When MCF-7 cells incubated with dauno-GT11 were examined after 18 h, the fluorescence was still mainly in the cytoplasm with a very limited amount in nuclear and perinuclear foci (data not shown). Previous studies have shown that anthracyclines, such as daunomycin, accumulate rapidly in the nucleus and to a minor extent in cytoplasmic organelles (63). In contrast, oligonucleotides are known to accumulate initially in endosomes and localize only later to the nucleus after being released in the cytoplasm (73). Thus, our results suggest that the oligonucleotide component, rather than the anthracycline moiety, determine both cellular uptake and intracellular trafficking of dauno-GT11. Based on these findings, we examined next whether a cationic lipid, such as DOTAP, which is commonly used to improve delivery of oligonucleotides into cells, could increase intracellular accumulation of dauno-GT11. As shown in Figure 5A, the mean fluorescence intensity determined by flow cytometry 24 h post-transfection of DU145 cells with 0.5 µM of dauno-GT11 was ∼2-fold higher with TFO delivered with DOTAP compared to TFO alone (27 versus 17). Also a higher percentage of cells took up the oligonucleotide when it was delivered with DOTAP (95 versus 75%). Fluorescence microscopy confirmed that overall fluorescence intensity was higher in cells incubated with TFO and DOTAP compared to TFO alone (Fig. 5A). Furthermore, when delivered as a complex with DOTAP, dauno-GT11 became concentrated in intensely fluorescent foci with greater staining of nuclear and perinuclear regions compared to TFO alone (Fig. 5B). These results confirmed that cationic lipid-mediated uptake produced a larger intracellular pool of dauno-GT11 with a corresponding increase in nuclear accumulation.
Figure 4.

Cellular uptake of free daunomycin and daunomycin-conjugated TFO. (A) MCF-7 cells were incubated with 0.2 and 1 µM of daunomycin or dauno-GT11. After 4 h, cells were washed, harvested and collected by centrifugation. Uptake was then determined by flow cytometry. Untreated cells (control) were used to determine background fluorescence levels. (B) MCF-7 cells were incubated with 1 µM of daunomycin (a and b) or dauno-GT11 (c and d) for 6 h and then examined by fluorescence (top) and phase-contrast (bottom) microscopy.
Figure 5.

Uptake of daunomycin-conjugated TFO in the presence of cationic lipids. (A) DU145 cells were incubated with dauno-GT11 (0.5 µM) alone or combined with the transfection reagent DOTAP for 6 h. After 24 h, cells were harvested and analyzed by flow cytometry. Untreated cells (control) were used to determine background fluorescence levels. (B) DU145 cells incubated with dauno-GT11 complexed with DOTAP (a, b and c) or dauno-GT11 alone (d, e and f) were examined by fluorescence and phase-contrast microscopy. Fluorescence (top), phase-contrast (middle) and merged (bottom) images are shown for each treatment group.
Inhibition of c-myc promoter activity by daunomycin-conjugated TFO in cells
To determine whether the dauno-GT11 was able to affect transcription in cells, we investigated its effects on c-myc promoter activity using luciferase reporter plasmids, in which the firefly luciferase gene had been put under the control of the P1 and P2 promoters or the P2 promoter alone (see Fig. 1A). The pGL3-Myc reporter plasmids were transfected along with a control vector and either dauno-GT11 or dauno-CO11 in prostate and breast cancer cells. Oligonucleotides were present in the medium during transfection at concentrations of 0.25–1 µM. These concentrations corresponded approximately to a 500- and 2000-fold molar excess of oligonucleotide relative to the reporter plasmid. After 24 h, luciferase activity was measured in extracts of control and oligonucleotide-treated cells. As shown in Figure 6A, incubation of DU145 cells with dauno-GT11 resulted in a significant reduction of the activity of the p1388-Myc reporter, which contained both P1 and P2 promoters, compared to cells incubated with identical concentrations of dauno-CO11. Inhibition of c-myc promoter activity by the TFO was dose-dependent with ∼30, ∼50 and ∼60% reduction at 0.25, 0.5 and 1 µM, respectively. To ensure that the effects of the TFO were due to a block of transcription from the P2 promoter, similar studies were repeated with the p262-Myc reporter construct. This reporter plasmid contained only the P2 promoter and the immediate upstream region that included the homopurine sequence targeted by dauno-GT11. Activity of the p262-Myc reporter in DU145 cells was inhibited by dauno-GT11 to a degree similar to that of the p1388-Myc reporter (Fig. 6B). Similar results were obtained with both reporter vectors in MCF-7 and MDA-231 breast cancer cells. Data with the p262-Myc reporter are shown in Figure 6C and D. It should be noted that dauno-CO11, which was directed to a site in the c-myc promoter and was potentially capable of forming triplex DNA, did not inhibit the activity of the reporter constructs in these assays. This was consistent with the fact that binding of this short oligonucleotide was unstable in physiological conditions even after conjugation with daunomycin and conversely suggested that the effects of dauno-GT11 were sequence-specific and not due to the cationic lipid-mediated delivery of daunomycin into cells. To further rule out other non-triplex-mediated mechanisms of inhibition of c-myc promoter activity, such as a decoy-like mechanism, we synthesized an oligonucleotide, dauno-GT11C, which had the same sequence of GT11 but was conjugated to the amino sugar instead of the aglycone moiety of daunomycin (Fig. 7A). As shown previously, this mode of attachment should eliminate any stabilizing contribution of daunomycin to the triple helix formed by the oligonucleotide (45). Gel shift assays confirmed our prediction, showing no binding of dauno-GT11C, which essentially acted similar to the non-conjugated GT11, in physiological conditions (Fig. 7B). As shown in Figure 7C, luciferase reporter assays demonstrated that dauno-GT11C was unable to inhibit c-myc promoter activity in cells, consistent with its inability to form stable triplex DNA. Taken together, these results strongly confirm that inhibition of c-myc promoter activity by dauno-GT11 required triplex formation and ruled out alternative sequence- and non-sequence-specific mechanisms that might be responsible for the biological effects of the daunomycin-conjugated TFO.
Figure 6.

Inhibition of c-myc promoter activity by daunomycin-conjugated TFO in prostate and breast cancer cells. Cells were transfected for 6 h either with p1388-Myc or p262-Myc reporter plasmid, a pRL control plasmid and the indicated concentrations of dauno-GT11 (empty bars) or dauno-CO11 (filled bars). Luciferase activity was measured after 24 h. Data are presented as percent of luciferase activity in TFO-treated cells compared to cells incubated with an equal concentration of control oligonucleotide. (A) DU145 cells transfected with p1388-Myc; (B) DU145 cells transfected with p262-Myc; (C) MCF-7 cells transfected with p262-Myc; (D) MDA-MB-231 cells transfected with p262-Myc.
Figure 7.

Inhibition of c-myc promoter activity is dependent on the triplex-forming ability of daunomycin-conjugated TFO. (A) Structure of the daunomycin-conjugated control oligonucleotide dauno-GT11C. (B) Gel mobility shift assay with duplex DNA incubated without (control) or with dauno-GT11 (D-GT11), dauno-GT11C (D-GT11C) or non-conjugated GT11. (C) DU145 cells were transfected with p262-Myc reporter plasmid and pRL control vector in the presence of 1 µM of dauno-GT11 (empty bars), dauno-CO11 (filled bars) or dauno-GT11C (gray bars). Luciferase activity was measured as described in the legend to Figure 6.
Dauno-GT11 inhibits endogenous c-myc gene expression
Dauno-GT11 inhibited in vitro transcription and promoter activity in cells suggesting that it could form a stable triplex complex with the targeted sequence and inhibit transcription of the endogenous c-myc gene in cells. To investigate this aspect, dauno-GT11 was delivered into DU145 cells using DOTAP. The level of c-myc RNA was then determined by RT–PCR. At 24 h post-transfection, c-myc RNA was reduced ∼70% in cells treated with 1 µM dauno-GT11 compared to untreated and control oligonucleotide-treated cells (Fig. 8A). To confirm that reduction of c-myc transcription would result in a lower level of c-myc protein, we performed western blot analysis. As shown in Figure 8B, c-myc protein level was reduced in cells treated with dauno-GT11 compared to control cells (∼40 and ∼75% at 0.5 and 1 µM, respectively). The control oligonucleotide had no effect on c-myc expression. In addition, we measured ets-1 and ets-2 protein expression level in cells treated with dauno-GT11 (Fig. 8B). Both ets-1 and ets-2 have promoter elements with significant sequence and functional similarity to the site in the c-myc gene targeted by dauno-GT11. The ets-2 promoter has a sequence (GGAAGGAGGGA) corresponding to a critical Sp1 binding site with a single mismatch (underlined base) compared to the c-myc site (64). The ets-1 gene has also in its promoter a similar sequence (AGAGGGAGGGA) with a single mismatch nucleotide (underlined base) compared to the c-myc site and adjacent to an Sp1 site (74). Expression of both ets-1and ets-2 was not affected by dauno-GT11, suggesting that the antigene activity of the daunomycin-conjugated oligonucleotide was selective for the targeted sequence and gene.
Figure 8.
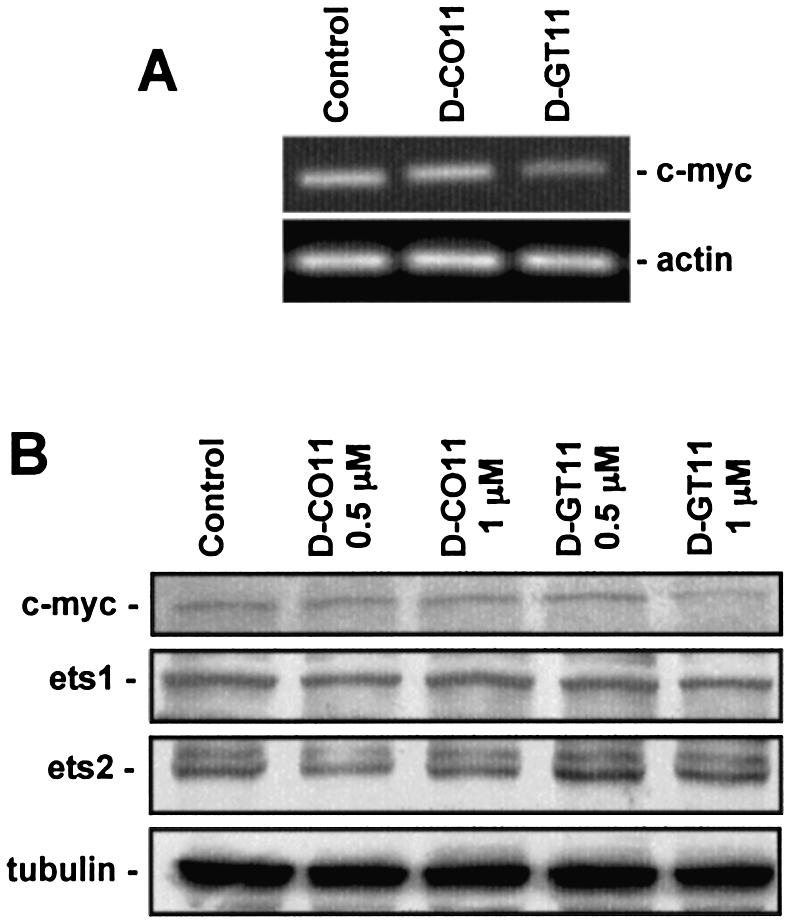
Inhibition of endogenous c-myc gene expression by daunomycin-conjugated TFO. (A) DU145 cells were transfected for 6 h with 1 µM of dauno-GT11 or dauno-CO11 using DOTAP. After 24 h, total RNA was extracted from untreated (Control), control oligonucleotide- (D-CO11) and TFO- (D-GT11) treated cells. c-myc and β-actin RNA levels were determined by RT–PCR. (B) DU145 cells were transfected with 0.5 and 1 µM of dauno-GT11 or dauno-CO11 as indicated above. Western blot analysis was performed after 24 h to determine c-myc, ets-1, ets-2 and α-tubulin protein level.
DISCUSSION
Covalent attachment of a non-sequence-specific DNA-intercalating agent, such as acridine and psoralen, to a TFO has been shown to be a useful approach to stabilize the triple helical complex. We have explored the possibility of increasing DNA binding and antigene activity of TFOs by linking the oligonucleotide to an anthracycline moiety. Anthracyclines may represent an alternative to the more commonly used acridine and psoralen derivatives and may offer additional advantages due to their complex mode of interaction with DNA. Attachment of an anthraquinone aglycone or an intact daunomycin molecule to a parallel CT TFO was previously shown to lead to a significant increase in triplex stability (45). We now provide evidence that this approach works also in the context of an antiparallel triple helix formed by a GT TFO and in physiological pH and temperature conditions. Moreover, our study provides the first evidence that daunomycin-conjugated TFOs are active as antigene compounds in cells. The activity of the dauno-GT TFO, both in vitro and in cells, was consistent with a triplex-mediated mechanism and was observed at relatively low oligonucleotide concentrations.
Using gel mobility shift assays we found that both conjugated and non-conjugated 11mer GT TFOs were able to bind the target duplex following overnight incubation at 37°C. However, there was a considerable increase in stability of the triplex formed by GT11 upon conjugation with daunomycin. The complex formed by the unmodified GT11 was not stable during electrophoresis at 20°C (Fig. 2A), while the triplex DNA formed by dauno-GT11 was clearly stable at gel temperatures of both 20 (Fig. 2B) and 37°C (Fig. 3A). This was consistent with the observations made in the previous study with daunomycin-conjugated CT TFOs (45). The Tm of triplex formed with a modified TFO was ∼23°C higher (increasing from 13 to 36°C) than that of the unmodified TFO at pH 6.8, and was >55°C at pH 5.5. Together with our present data, these results indicate that anthracyclines can have a stabilizing effect on TFOs binding both in the parallel and antiparallel orientation. The potency of daunomycin as DNA binding agent and the high degree of stability conferred to triplex DNA upon conjugation to a TFO can be explained by its mode of interaction with double-stranded DNA. Virtually every part of a daunomycin molecule, particularly the anthraquinone and the amino sugar, is in contact with DNA at the binding site. The design of the daunomycin-conjugated TFOs took this into account and tried to increase the opportunity for cooperative binding. Attachment of the TFO to position O4 of the anthraquinone preserved the orientation of the intercalating moiety in the double helix and maintained the oligonucleotide and the amino sugar in the major and minor groove, respectively (45). Indeed, modifying the site of attachment, as we did with the dauno-GT11C, eliminated completely any triplex stabilizing effect of the anthracycline. Daunomycin-conjugated TFOs can be seen as DNA ligands optimized for binding to homopurine sequence in double-stranded DNA. On the other hand, TFOs do not bind to single-stranded oligonucleotides containing the target sequence and anthracyclines do not bind significantly to single- and double-stranded RNA. Thus, conjugation with daunomycin should not increase the likelihood of undesired binding of TFOs to nucleic acids other than double-stranded DNA.
Attachment of a non-sequence-specific DNA binding agent could affect the specificity of the interaction of the TFO with its duplex DNA target. Previous studies indicated that binding of acridine- and psoralen-conjugated TFOs was highly sequence-specific even in the presence of these potent DNA intercalators. In a previous study, random intercalation of daunomycin-conjugated TFOs was excluded by fluorescence quenching experiments (45). Intercalation and quenching of the chromophore occurred only when the DNA contained the appropriate target sequence, thus proving that site recognition and binding were dictated exclusively by the oligonucleotide component of the intercalator-TFO conjugate. Here, we addressed this issue by performing footprinting studies and showed that dauno-GT11 was able to bind exclusively to its unique target sequence in the c-myc promoter. Dauno-GT11 was also able to discriminate against targets that contained a limited number of mismatched nucleotides and thus could serve as alternative binding sites. Gel shift assays also confirmed the inability of dauno-GT11 to bind to duplex DNA, in which the 11 bp target sequence had been mutated. As was proposed for acridine- and psoralen-linked TFOs, it is possible that random intercalation of daunomycin into DNA is abolished by the presence of the TFO. Repulsion between the oligonucleotide tail attached to the daunomycin and double-stranded DNA might prevent intercalation at sites that do not present the complementary target sequence. Therefore, binding of the intercalator would take place only at those sites where the TFO finds the matching target sequence.
The increased stability of the triple helical complex formed by daunomycin-conjugated TFOs suggests that these compounds might be used to target even relatively short homopurine sequences in genomic DNA. This might considerably increase the number of potential target sites for triplex formation and expand the applications of the triplex-mediated gene targeting strategy. In agreement with this hypothesis, dauno-GT11 was able to inhibit c-myc transcription in vitro and promoter activity in prostate and breast cancer cells. These effects were not observed with daunomycin-conjugated control oligonucleotides and were not associated with general toxicity associated with equimolar concentrations of daunomycin. In addition, experiments with dauno-GT11C, a daunomycin-conjugated oligonucleotide with the GT11 sequence but unable to form triplex DNA, demonstrated that the effects of dauno-GT11 on the c-myc promoter could not be attributed to a decoy-like mechanism (e.g. trapping of transcription activators, such as Sp1, required for c-myc promoter activity). We further demonstrated the ability of dauno-GT11 to block transcription of the endogenous c-myc gene in prostate cancer cells, which indicated that the TFO could reach its target sequence in chromosomal DNA. Expression of genes, such as ets-1 and ets-2, which had very similar sequences in their promoters with only single mismatches compared to the c-myc target, was not affected by dauno-GT11, suggesting that the antigene activity of daunomycin-conjugated TFO was also selective for the targeted gene. Thus, in the present study, we provide indirect evidence of the ability of dauno-GT11 to form triplex DNA under physiological conditions and in cells (i.e. inhibition of promoter activity in vitro and in cells, inhibition of the endogenous gene expression, and absence of activity of an identical oligonucleotide unable to form a stable triplex). In the absence of direct evidence of triplex formation in cells, however, we cannot exclude conclusively that other mechanisms, although unlikely, are involved. In line with our data, efficient and selective targeting of a homopurine site in a chromosomal reporter gene was recently achieved using a 10mer TFO synthesized with cationic phosphoroamidate linkages to increase binding affinity (75). However, one must consider that short TFOs, like dauno-GT11, might not be optimal if one wants to achieve truly gene-specific inhibition of transcription in cells. The shorter the target sequence, the higher the probability that other genes might have identical sequences. Longer oligonucleotides of at least 16–20 nt in length would be more likely to achieve selective gene targeting.
In general, the greater stability of triplex DNA achieved with the addition of an intercalating agent should translate into increased half-life of the triple helical complex and biological activity of a TFO in cells. The gain in triplex stability obtained by conjugation with daunomycin would certainly be important when the objective is to inhibit transcription at the initiation or elongation stage by preventing either formation or progression of the transcription complex. Alternative mechanisms of gene repression might also operate in the case of daunomycin-conjugated TFOs. The presence of the intercalating agent might induce bending or distortion of the DNA and alter chromatin structure. Other possibilities due to the presence of the daunomycin moiety are recruitment of DNA topoisomerase II and DNA repair enzymes, which could result in cleavage of DNA and/or induction of mutations in the targeted gene. Indeed, conjugation of topoisomerase-interactive compounds to TFOs could be used to direct the DNA cleaving activity of these enzymes to specific genomic targets (76,77). Future studies will evaluate further the mechanisms of antigene activity and the pharmacological properties of daunomycin-conjugated TFOs. Novel antigene-based cancer therapeutics could be developed using this approach.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to thank Dr Anna Garbesi (ISOF-CNR, Bologna, Italy) for her advice, Angela Collier and Rick Peppler (MUSC, Charleston, SC) for their assistance, and Dr Fabrizio Dal Piaz (University of Bologna, Bologna, Italy) for the MS analysis. This work was supported by grants from National Cancer Institute (CA70735 to C.V.C.), Swiss Cancer League (to C.V.C.), Ticino Cancer League (to G.M.C.), Biofin Laboratories and MUSRT-Italy (to M.L.C.). E.M.M. was supported by a postdoctoral award DAMD17-00-1-0339 from US Army Breast Cancer Research Program.
REFERENCES
- 1.Chan P.P. and Glazer,P.M. (1997) Triplex DNA: fundamentals, advances and potential applications for gene therapy. Mol. Med., 75, 267–282. [DOI] [PubMed] [Google Scholar]
- 2.Praseuth D., Guieysse,A.L. and Helene,C. (1999) Triple helix formation and the antigene strategy for sequence-specific control of gene expression. Biochim. Biophys. Acta, 1489, 181–206. [DOI] [PubMed] [Google Scholar]
- 3.Casey B.P. and Glazer,P.M. (2001) Gene targeting via triple-helix formation. Prog. Nucleic Acid Res. Mol. Biol., 67, 163–192. [DOI] [PubMed] [Google Scholar]
- 4.Winters T.A. (2000) Gene targeted agents: new opportunities for rational drug development. Curr. Opin. Mol. Ther., 2, 670–681. [PubMed] [Google Scholar]
- 5.Moser H.E. and Dervan,P.B. (1987) Sequence-specific cleavage of double helical DNA by triple helix formation. Science, 238, 645–650. [DOI] [PubMed] [Google Scholar]
- 6.Beal P.A. and Dervan,P.B. (1991) Second structural motif for recognition of DNA by oligonucleotide-directed triple-helix formation. Science, 251, 1360–1363. [DOI] [PubMed] [Google Scholar]
- 7.Singleton S.F. and Dervan,P.B. (1992) Influence of pH on the equilibrium association constants for oligodeoxyribonucleotide-directed triple helix formation at single DNA sites. Biochemistry, 31, 10995–11003. [DOI] [PubMed] [Google Scholar]
- 8.Durland R.H., Kessler,D.J., Gunnell,S., Duvic,M., Pettitt,B.M. and Hogan,M.E. (1991) Binding of triple helix forming oligonucleotides to sites in gene promoters. Biochemistry, 30, 9246–9255. [DOI] [PubMed] [Google Scholar]
- 9.Maher L.J. III (1996) Prospects for the therapeutic use of antigene oligonucleotides. Cancer Invest., 14, 66–82. [DOI] [PubMed] [Google Scholar]
- 10.Maher L.J. III, Dervan,P.B. and Wold,B.J. (1990) Kinetic analysis of oligodeoxyribonucleotide-directed triple-helix formation on DNA. Biochemistry, 29, 8820–8826. [DOI] [PubMed] [Google Scholar]
- 11.Svinarchuk F., Bertrand,J.R. and Malvy,C. (1994) A short purine oligonucleotide forms a highly stable triple helix with the promoter of the murine c-pim-1 proto-oncogene. Nucleic Acids Res., 22, 3742–3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Svinarchuk F., Paoletti,J. and Malvy,C. (1995) An unusually stable purine(purine-pyrimidine) short triplex. The third strand stabilizes double-stranded DNA. J. Biol. Chem., 270, 14068–14071. [DOI] [PubMed] [Google Scholar]
- 13.Alunni-Fabbroni M., Pirulli,D., Manzini,G. and Xodo,L.E. (1996) (A,G)-oligonucleotides form extraordinary stable triple helices with a critical R.Y sequence of the murine c-Ki-ras promoter and inhibit transcription in transfected NIH 3T3 cells. Biochemistry, 35, 16361–16369. [DOI] [PubMed] [Google Scholar]
- 14.Maher L.J., Wold,B. and Dervan,P.B. (1989) Inhibition of DNA binding proteins by oligonucleotide-directed triple helix formation. Science, 245, 725–730. [DOI] [PubMed] [Google Scholar]
- 15.Maher L.J.D., Dervan,P.B. and Wold,B. (1992) Analysis of promoter-specific repression by triple-helical DNA complexes in a eukaryotic cell-free transcription system. Biochemistry, 31, 70–81. [DOI] [PubMed] [Google Scholar]
- 16.Guieysse A.L., Praseuth,D., Grigoriev,M., Harel-Bellan,A. and Helene,C. (1996) Detection of covalent triplex within human cells. Nucleic Acids Res., 24, 4210–4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giovannangeli C., Diviacco,S., Labrousse,V., Gryaznov,S., Charneau,P. and Helene,C. (1997) Accessibility of nuclear DNA to triplex-forming oligonucleotides: the integrated HIV-1 provirus as a target. Proc. Natl Acad. Sci. USA, 94, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin F.L., Majumdar,A., Klotz,L.C., Reszka,A.P., Neidle,S. and Seidman,M.M. (2000) Stability of DNA triplexes on shuttle vector plasmids in the replication pool in mammalian cells. J. Biol. Chem., 275, 39117–39124. [DOI] [PubMed] [Google Scholar]
- 19.Maine I.P. and Kodadek,T. (1994) Efficient unwinding of triplex DNA by a DNA helicase. Biochem. Biophys. Res. Commun., 204, 1119–1124. [DOI] [PubMed] [Google Scholar]
- 20.Kopel V., Pozner,A., Baran,N. and Manor,H. (1996) Unwinding of the third strand of a DNA triple helix, a novel activity of the SV40 large T-antigen helicase. Nucleic Acids Res., 24, 330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandor Z. and Bredberg,A. (1994) Repair of triple helix directed psoralen adducts in human cells. Nucleic Acids Res., 22, 2051–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Degols G., Clarenc,J.P., Lebleu,B. and Leonetti,J.P. (1994) Reversible inhibition of gene expression by a psoralen functionalized triple helix forming oligonucleotide in intact cells. J. Biol. Chem., 269, 16933–16937. [PubMed] [Google Scholar]
- 23.Sandor Z. and Bredberg,A. (1995) Deficient DNA repair of triple helix-directed double psoralen damage in human cells. FEBS Lett., 374, 287–291. [DOI] [PubMed] [Google Scholar]
- 24.Wang G. and Glazer,P.M. (1995) Altered repair of targeted psoralen photoadducts in the context of an oligonucleotide-mediated triple helix. J. Biol. Chem., 270, 22595–22601. [DOI] [PubMed] [Google Scholar]
- 25.Raha M., Wang,G., Seidman,M.M. and Glazer,P.M. (1996) Mutagenesis by third-strand-directed psoralen adducts in repair-deficient human cells: high frequency and altered spectrum in a xeroderma pigmentosum variant. Proc. Natl Acad. Sci. USA, 93, 2941–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang G., Seidman,M.M. and Glazer,P.M. (1996) Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science, 271, 802–805. [DOI] [PubMed] [Google Scholar]
- 27.Musso M., Wang,J.C. and Van Dyke,M.W. (1996) In vivo persistence of DNA triple helices containing psoralen-conjugated oligodeoxyribonucleotides. Nucleic Acids Res., 24, 4924–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barre F.X., Asseline,U. and Harel-Bellan,A. (1999) Asymmetric recognition of psoralen interstrand crosslinks by the nucleotide excision repair and the error-prone repair pathways. J. Mol. Biol., 286, 1379–1387. [DOI] [PubMed] [Google Scholar]
- 29.Barre F.X., Giovannangeli,C., Helene,C. and Harel-Bellan,A. (1999) Covalent crosslinks introduced via a triple helix-forming oligonucleotide coupled to psoralen are inefficiently repaired. Nucleic Acids Res., 27, 743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asseline U., Delarue,M., Lancelot,G., Toulme,F., Thuong,N.T., Montenay-Garestier,T. and Helene,C. (1984) Nucleic acid-binding molecules with high affinity and base sequence specificity: intercalating agents covalently linked to oligodeoxynucleotides. Proc. Natl Acad. Sci. USA, 81, 3297–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asseline U., Toulme,F., Thuong,N.T., Delarue,M., Montenay-Garestier,T. and Helene,C. (1984) Oligodeoxynucleotides covalently linked to intercalating dyes as base sequence-specific ligands. Influence of dye attachment site. EMBO J., 3, 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun J.S., Francois,J.C., Montenay-Garestier,T., Saison-Behmoaras,T., Roig,V., Thuong,N.T. and Helene,C. (1989) Sequence-specific intercalating agents: intercalation at specific sequences on duplex DNA via major groove recognition by oligonucleotide-intercalator conjugates. Proc. Natl Acad. Sci. USA, 86, 9198–9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takasugi M., Guendouz,A., Chassignol,M., Decout,J.L., Lhomme,J., Thuong,N.T. and Helene,C. (1991) Sequence-specific photo-induced cross-linking of the two strands of double-helical DNA by a psoralen covalently linked to a triple helix-forming oligonucleotide. Proc. Natl Acad. Sci. USA, 88, 5602–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Orson F.M., Kinsey,B.M. and McShan,W.M. (1994) Linkage structures strongly influence the binding cooperativity of DNA intercalators conjugated to triplex forming oligonucleotides. Nucleic Acids Res., 22, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gasparro F.P., Havre,P.A., Olack,G.A., Gunther,E.J. and Glazer,P.M. (1994) Site-specific targeting of psoralen photoadducts with a triple helix-forming oligonucleotide: characterization of psoralen monoadduct and crosslink formation. Nucleic Acids Res., 22, 2845–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lacoste J., Francois,J.C. and Helene,C. (1997) Triple helix formation with purine-rich phosphorothioate-containing oligonucleotides covalently linked to an acridine derivative. Nucleic Acids Res., 25, 1991–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grigoriev M., Praseuth,D., Robin,P., Hemar,A., Saison-Behmoaras,T., Dautry-Varsat,A., Thuong,N.T., Helene,C. and Harel-Bellan,A. (1992) A triple helix-forming oligonucleotide-intercalator conjugate acts as a transcriptional repressor via inhibition of NF kappa B binding to interleukin-2 receptor alpha-regulatory sequence. J. Biol. Chem., 267, 3389–3395. [PubMed] [Google Scholar]
- 38.Duval-Valentin G., Thuong,N.T. and Helene,C. (1992) Specific inhibition of transcription by triple helix-forming oligonucleotides. Proc. Natl Acad. Sci. USA, 89, 504–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grigoriev M., Praseuth,D., Guieysse,A.L., Robin,P., Thuong,N.T., Helene,C. and Harel-Bellan,A. (1993) Inhibition of gene expression by triple helix-directed DNA cross-linking at specific sites. Proc. Natl Acad. Sci. USA, 90, 3501–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Havre P.A. and Glazer,P.M. (1993) Targeted mutagenesis of simian virus 40 DNA mediated by a triple helix-forming oligonucleotide. J. Virol., 67, 7324–7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Havre P.A., Gunther,E.J., Gasparro,F.P. and Glazer,P.M. (1993) Targeted mutagenesis of DNA using triple helix-forming oligonucleotides linked to psoralen. Proc. Natl Acad. Sci. USA, 90, 7879–7883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang G., Levy,D.D., Seidman,M.M. and Glazer,P.M. (1995) Targeted mutagenesis in mammalian cells mediated by intracellular triple helix formation. Mol. Cell. Biol., 15, 1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frederick C.A., Williams,L.D., Ughetto,G., van der Marel,G.A., van Boom,J.H., Rich,A. and Wang,A.H. (1990) Structural comparison of anticancer drug–DNA complexes: adriamycin and daunomycin. Biochemistry, 29, 2538–2549. [PubMed] [Google Scholar]
- 44.Chaires J.B., Satyanarayana,S., Suh,D., Fokt,I., Przewloka,T. and Priebe,W. (1996) Parsing the free energy of anthracycline antibiotic binding to DNA. Biochemistry, 35, 2047–2053. [DOI] [PubMed] [Google Scholar]
- 45.Garbesi A., Bonazzi,S., Zanella,S., Capobianco,M.L., Giannini,G. and Arcamone,F. (1997) Synthesis and binding properties of conjugates between oligodeoxynucleotides and daunorubicin derivatives. Nucleic Acids Res., 25, 2121–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dang C.V. (1999) c-Myc target genes involved in cell growth, apoptosis and metabolism. Mol. Cell. Biol., 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coller H.A., Grandori,C., Tamayo,P., Colbert,T., Lander,E.S., Eisenman,R.N. and Golub,T.R. (2000) Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling and adhesion. Proc. Natl Acad. Sci. USA, 97, 3260–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spencer C.A. and Groudine,M. (1991) Control of c-myc regulation in normal and neoplastic cells. Adv. Cancer Res., 56, 1–48. [DOI] [PubMed] [Google Scholar]
- 49.Grandori C., Cowley,S.M., James,L.P. and Eisenman,R.N. (2000) The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell. Dev. Biol., 16, 653–699. [DOI] [PubMed] [Google Scholar]
- 50.Skorski T., Nieborowska-Skorska,M., Campbell,K., Iozzo,R.V., Zon,G., Darzynkiewicz,Z. and Calabretta,B. (1995) Leukemia treatment in severe combined immunodeficiency mice by antisense oligodeoxynucleotides targeting cooperating oncogenes. J. Exp. Med., 182, 1645–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leonetti C., D’Agnano,I., Lozupone,F., Valentini,A., Geiser,T., Zon,G., Calabretta,B., Citro,G.C. and Zupi,G. (1996) Antitumor effect of c-myc antisense phosphorothioate oligodeoxynucleotides on human melanoma cells in vitro and and in mice (see comments). J. Natl Cancer Inst., 88, 419–429. [DOI] [PubMed] [Google Scholar]
- 52.Skorski T., Perrotti,D., Nieborowska-Skorska,M., Gryaznov,S. and Calabretta,B. (1997) Antileukemia effect of c-myc N3′→P5′ phosphoramidate antisense oligonucleotides in vivo. Proc. Natl Acad. Sci. USA, 94, 3966–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Felsher D.W. and Bishop,J.M. (1999) Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell, 4, 199–207. [DOI] [PubMed] [Google Scholar]
- 54.Cooney M., Czernuszewicz,G., Postel,E.H., Flint,S.J. and Hogan,M.E. (1988) Site-specific oligonucleotide binding represses transcription of the human c-myc gene in vitro. Science, 241, 456–459. [DOI] [PubMed] [Google Scholar]
- 55.Postel E.H., Flint,S.J., Kessler,D.J., Hogan,M.E. (1991) Evidence that a triplex-forming oligodeoxyribonucleotide binds to the c-myc promoter in HeLa cells, thereby reducing c-myc mRNA levels. Proc. Natl Acad. Sci. USA, 88, 8227–8231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomas T.J., Faaland,C.A., Gallo,M.A. and Thomas,T. (1995) Suppression of c-myc oncogene expression by a polyamine-complexed triplex forming oligonucleotide in MCF-7 breast cancer cells. Nucleic Acids Res., 23, 3594–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim H.G. and Miller,D.M. (1995) Inhibition of in vitro transcription by a triplex-forming oligonucleotide targeted to human c-myc P2 promoter. Biochemistry, 34, 8165–8171. [DOI] [PubMed] [Google Scholar]
- 58.Kim H.G., Reddoch,J.F., Mayfield,C., Ebbinghaus,S., Vigneswaran,N., Thomas,S., Jones,D.E.,Jr and Miller,D.M. (1998) Inhibition of transcription of the human c-myc protooncogene by intermolecular triplex. Biochemistry, 37, 2299–2304. [DOI] [PubMed] [Google Scholar]
- 59.Catapano C.V., McGuffie,E.M., Pacheco,D. and Carbone,G.M. (2000) Inhibition of gene expression and cell proliferation by triple helix-forming oligonucleotides directed to the c-myc gene. Biochemistry, 39, 5126–5138. [DOI] [PubMed] [Google Scholar]
- 60.McGuffie E.M., Pacheco,D., Carbone,G.M. and Catapano,C.V. (2000) Antigene and antiproliferative effects of a c-myc-targeting phosphorothioate triple helix-forming oligonucleotide in human leukemia cells. Cancer Res., 60, 3790–3799. [PubMed] [Google Scholar]
- 61.Pandolfi D., Rauzi,F. and Capobianco,M.L. (1999) Evaluation of different types of end-capping modifications on the stability of oligonucleotides toward 3′- and 5′-exonucleases. Nucl. Nucl., 18, 2051–2069. [DOI] [PubMed] [Google Scholar]
- 62.McGuffie E.M. and Catapano,C.V. (2002) Design of a novel triple helix-forming oligonucleotide directed to the major promoter of the c-myc gene. Nucleic Acids Res., 30, 2701–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rutherford A.V. and Willingham,M.C. (1993) Ultrastructural localization of daunomycin in multidrug-resistant cultured cells with modulation of the multidrug transporter. J. Histochem. Cytochem., 41, 1573–1577. [DOI] [PubMed] [Google Scholar]
- 64.Carbone G.M., McGuffie,E.M., Collier,A. and Catapano,C.V. (2003) Selective inhibition of transcription of the Ets2 gene in prostate cancer cells by a triplex-forming oligonucleotide. Nucleic Acids Res., 31, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Majello B., De Luca,P., Suske,G. and Lania,L. (1995) Differential transcriptional regulation of c-myc promoter through the same DNA binding sites targeted by Sp1-like proteins. Oncogene, 10, 1841–1848. [PubMed] [Google Scholar]
- 66.Bossone S.A., Asselin,C., Patel,A.J. and Marcu,K.B. (1992) MAZ, a zinc finger protein, binds to c-MYC and C2 gene sequences regulating transcriptional initiation and termination. Proc. Natl Acad. Sci. USA, 89, 7452–7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roussel M.F., Davis,J.N., Cleveland,J.L., Ghysdael,J. and Hiebert,S.W. (1994) Dual control of myc expression through a single DNA binding site targeted by ets family proteins and E2F-1. Oncogene, 9, 405–415. [PubMed] [Google Scholar]
- 68.Hiebert S.W., Lipp,M. and Nevins,J.R. (1989) E1A-dependent trans-activation of the human MYC promoter is mediated by the E2F factor. Proc. Natl Acad. Sci. USA, 86, 3594–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thalmeier K., Synovzik,H., Mertz,R., Winnacker,E.L. and Lipp,M. (1989) Nuclear factor E2F mediates basic transcription and trans-activation by E1a of the human MYC promoter. Genes Dev., 3, 527–536. [DOI] [PubMed] [Google Scholar]
- 70.Kiuchi N., Nakajima,K., Ichiba,M., Fukada,T., Narimatsu,M., Mizuno,K., Hibi,M. and Hirano,T. (1999) STAT3 is required for the gp130-mediated full activation of the c-myc gene. J. Exp. Med., 189, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Asselin C., Nepveu,A. and Marcu,K.B. (1989) Molecular requirements for transcriptional initiation of the murine c-myc gene. Oncogene, 4, 549–558. [PubMed] [Google Scholar]
- 72.Albert T., Wells,J., Funk,J.O., Pullner,A., Raschke,E.E., Stelzer,G., Meisterernst,M., Farnham,P.J. and Eick,D. (2001) The chromatin structure of the dual c-myc promoter P1/P2 is regulated by separate elements. J. Biol. Chem., 276, 20482–20490. [DOI] [PubMed] [Google Scholar]
- 73.Gewirtz A.M., Sokol,D.L. and Ratajczak,M.Z. (1998) Nucleic acid therapeutics: state of the art and future prospects. Blood, 92, 712–736. [PubMed] [Google Scholar]
- 74.Jorcyk C.L., Watson,D.K., Mavrothalassitis,G.J. and Papas,T.S. (1991) The human ETS1 gene: genomic structure, promoter characterization and alternative splicing. Oncogene, 6, 523–532. [PubMed] [Google Scholar]
- 75.Vasquez K.M., Dagle,J.M., Weeks,D.L. and Glazer,P.M. (2001) Chromosome targeting at short polypurine sites by cationic triplex-forming oligonucleotides. J. Biol. Chem., 276, 38536–38541. [DOI] [PubMed] [Google Scholar]
- 76.Arimondo P., Bailly,C., Boutorine,A., Asseline,U., Sun,J.S., Garestier,T. and Helene,C. (2000) Linkage of a triple helix-forming oligonucleotide to amsacrine-4-carboxamide derivatives modulates the sequence-selectivity of topoisomerase II-mediated DNA cleavage. Nucl. Nucl. Nucleic Acids, 19, 1205–1218. [DOI] [PubMed] [Google Scholar]
- 77.Arimondo P.B., Boutorine,A., Baldeyrou,B., Bailly,C., Kuwahara,M., Hecht,S.M., Sun,J.S., Garestier,T. and Helene,C. (2002) Design and optimization of camptothecin conjugates of triple helix-forming oligonucleotides for sequence-specific DNA cleavage by topoisomerase I. J. Biol. Chem., 277, 3132–3140. [DOI] [PubMed] [Google Scholar]