


Abstract
In our previous study, we found that a human F-box DNA helicase, named hFBH1, interacted with SKP1 to form an SCF (SKP1–Cul1–F-box protein) complex together with CUL1 and ROC1 in an F-box-dependent manner. The complex immunoprecipitated from crude cell extracts catalyzed polyubiquitin formation in the presence of the ubiquitin-activating and ubiquitin-conjugating enzymes, E1 and E2, respectively. In this report, we characterized the enzymatic properties of the recombinant SCFhFBH1 complex purified from insect cells expressing hFBH1, SKP1, CUL1 and ROC1. The SCFhFBH1 complex was isolated as a single tight complex that retained DNA helicase, DNA-dependent ATPase and E3 ubiquitin ligase activities. The helicase and ATPase activities residing in the SCFhFBH1 complex were indistinguishable from those of the hFBH1 protein alone. Moreover, the ubiquitin ligase activity of the SCFhFBH1 complex was hardly affected by single-stranded or double-stranded DNA. The multiple activities present in this complex act independently of each other, suggesting that the SCFhFBH1 complex can catalyze a ubiquitination reaction while acting as a DNA helicase or translocating along DNA. The potential roles of the SCFhFBH1 complex in DNA metabolism based upon the enzymatic activities associated with this complex are discussed.
INTRODUCTION
We have previously isolated and characterized the enzymatic activity of DNA helicase I from Schizosaccharomyces pombe (1). The DNA-unwinding activity of this enzyme was markedly stimulated by the S.pombe replication protein A (RPA) particularly at low levels of ATP, although its ATPase activity was unaffected (1). This result suggested that S.pombe DNA helicase I may play a role in DNA transactions involving RPA, such as DNA replication, recombination and repair. In addition to well-defined helicase motifs, this helicase contained a highly conserved F-box motif in its N-terminal region (1,2). A protein with an F-box motif (collectively called F-box proteins) is an essential component of the SCF [SKP1–CUL1 (Cdc53)-Rbx1–F-box protein] complex that catalyzes the polyubiquitination of proteins, a modification required for their degradation by the 26S proteasome [reviewed in Peters (3), Laney and Hochstrasser (4), Tyers and Jorgensen (5), Hershko and Ciechanover (6), Craig and Tyers (7) and Deshaies (8)]. Therefore, protein degradation by SCF complexes has been shown to influence a variety of cellular processes such as cell cycle progression, signal transduction and gene expression (9–16). In most instances, a specific protein is recruited to an SCF complex by interacting with the F-box protein, which is linked by the F-box motif to SKP1 of the complex.
Homologs of the S.pombe helicase are found in mammals including human, monkey and mouse, but not in other organisms such as frog, fruit fly, fish, plant and budding yeast (2). The S.pombe and human enzymes were named SpFBH1 and hFBH1, respectively (S.pombe and human F-box DNA helicase I). Both hFBH1 and SpFBH1 helicases translocated in the 3′ to 5′ direction and were not absolutely dependent on substrates containing forked structures for unwinding activity (1,2).
Consistent with the presence of an F-box motif in hFBH1, hFBH1 co-immunoprecipitated with all other subunits of the SCF complex (SKP1, ROC1 and CUL1) from crude extracts prepared from cells expressing these proteins (2). The mutant hFBH1 protein deleted of the F-box motif, however, did not interact with any of the SCF proteins (2). Moreover, immunocomplexes containing hFBH1 catalyzed polyubiquitin chain formation (2), supporting the notion that hFBH1 contains a functionally active F-box motif that forms an SCF complex in vivo. These findings raised the possibility that the hFBH1–SCF complex (SCFhFBH1) can regulate the stability of proteins involved in DNA transactions in a ubiquitin-dependent manner. Unlike any known F-box protein, hFBH1 and SpFBH1 are enzymes containing intrinsic helicase and ATPase activities, raising the possibility that the helicase activity of hFBH1 may assist the function of the SCFhFBH1 complex during ubiquitination of a substrate. In light of this view, it is essential to characterize the multiple enzymatic activities associated with SCFhFBH1 in order to gain more insight into its function and mode of action in vivo. It is also critical to investigate influences of protein–protein interactions within the complex or cofactors that may affect one of the activities. For example, it is likely that the helicase activity of hFBH1 can be altered when it is integrated into the complex since it is not uncommon that specific protein–protein interactions either stimulate or inhibit one of the enzymatic activities encoded by different polypeptides in the complex. It is also possible that the E3 ubiquitin ligase activity of the complex is inhibited by DNA that binds hFBH1 in the complex and stimulates its ATPase activity.
Therefore, we have examined the biochemical properties of the activities associated with the SCFhFBH1 complex. For this purpose, we have assembled the complex in vivo and in vitro and purified the recombinant SCFhFBH1. This has permitted us to examine and compare the enzymatic properties of hFBH1 alone and of the SCF hFBH1 complex. Our results indicate that the recombinant SCFhFBH1 complex retained ATPase, helicase and ubiquitin ligase activities. The helicase and ATPase activities of the SCFhFBH1 complex are nearly identical to those of hFBH1 alone, and the ubiquitin ligase activity was hardly affected by the presence of DNA. Our findings suggest that the SCFhFBH1 complex can act as a DNA helicase and E3 ubiquitin ligase simultaneously. We discuss the potential role of the SCFhFBH1 complex in DNA metabolism based upon its enzymatic properties.
MATERIALS AND METHODS
Enzymes, antibodies, DNA and nucleotides
φX174 single-stranded circular (ssc) DNA was purchased from New England Biolabs (Beverly, MA). The oligonucleotides used were commercially synthesized by Genotech (Daejeon, Korea). Nucleoside triphosphates, nucleoside diphosphates and AMP were obtained from Sigma (St Louis, MO). [γ-32P]ATP (>5000 Ci/mmol), [α-32P]dideoxy-ATP (>5000 Ci/mmol) and [α-32P]dCTP (>6000 Ci/mmol) were purchased from Amersham Biosciences (Piscataway, NJ), while ATPγS and AppNp were obtained from Boehringer Mannheim (Germany).
The following proteins were obtained commercially: Escherichia coli single-stranded binding protein (SSB; Amersham Biosciences), cAMP kinase (Sigma), restriction endonucleases and polynucleotide kinase (KOSCO Inc., Korea). Antibodies were obtained commercially: mono clonal anti-GST–horseradish peroxidase (HRP) and rabbit polyclonal anti-human SKP1 from Santa Cruz Biotechnology (Santa Cruz, CA) and mouse monoclonal anti-His antibody from Qiagen (Valencia, CA). All secondary antibodies were from Amersham Biosciences. The ubiquitin-activating enzyme E1 was purchased from Calbiochem (La Jolla, CA), while human RPA (hRPA), Sacchromyces cerevisiae (yRPA) and S.pombe (SpRPA) enzymes were purified as described (1,17,18)
Preparation of helicase substrates
The DNA substrate used for standard helicase assays was prepared as previously described (1). The helicase substrate consists of φX174 sscDNA with an oligonucleotide annealed to it. Three oligonucleotides (5′-CGG ACG CTC GAC GCC ATT AAT AAT GTT TTC-3′; 5′-CGA ACA ATT CAG CGG CTT TAA CCG GAC GCT CGA CGC CAT TAA TAA TGT TTT C-3′; and 5′-GAA CGT CAG TGT TTC CTG CGC GTA CAC GCA AGG TAA ACG CGA ACA ATT CAG CGG CTT TAA CCG GAC GCT CGA CGC CAT TAA TGT TTT C-3′) were used for the construction of fully complementary helicase substrates. They are complementary to φX174 sscDNA (New England Biolabs Inc.) at nucleotides 702–731, 670–731 and 641–731, respectively. Four oligonucleotides (5′-TTT TTT TTT TCG GAC GCT CGA CGC CAT TAA TAA TGT TTT C-3′; 5′-TTT TTT TTT TTT TTT TTT TTT TTT TCG GAC GCT CGA CGC CAT TAA TAA TGT TTT C-3′; 5′-CGG ACG CTC GAC GCC ATT AAT AAT GTT TTC TTT TTT TTT T-3′; and 5′-CGG ACG CTC GAC GCC ATT AAT AAT GTT TTC TTT TTT TTT TTT TTT TTT TTT TTT T-3′) were used to construct helicase substrates with either a 5′ or a 3′ tail of different size. The underlined sequence is complementary to φX174 sscDNA at nucleotides 702–731. The oligonucleotide was labeled at its 5′ end with [γ-32P]ATP and polynucleotide kinase and then annealed to φX174 sscDNA. The substrates were purified as previously reported (1).
Helicase and ATPase assays
Standard ATPase assays were carried out in reaction mixtures (20 µl) containing 25 mM Tris–HCl pH 7.8, 2 mM dithiothreitol (DTT), 2 mM MgCl2, 0.25 mg/ml bovine serum albumin (BSA), 250 µM cold ATP, 20 nM [γ-32P]ATP (>5000 Ci/mmol) and 50 ng of φX174 sscDNA or other polynucleotides as indicated. After incubation at 37°C for 10 min, aliquots (2 µl) were spotted onto a polyethyleneimine-cellulose plate (J. T. Baker Inc.) and developed in 0.5 M LiCl/1.0 M formic acid. The products were analyzed using a Phosphorimager (Molecular Dynamics, Inc.).
Standard helicase assays were performed in reaction mixtures (20 µl) containing 25 mM Tris–HCl pH 7.8, 2 mM MgCl2, 2 mM DTT, 2 mM ATP, 0.25 mg/ml BSA and the 5′-32P-labeled 30 bp partial duplex DNA substrate (15 fmol). Reactions were incubated at 37°C for 10 min, followed by the addition of 4 µl of 6× stop solution [60 mM EDTA pH 8.0, 40% (w/v) sucrose, 0.6% SDS, 0.25% bromophenol blue and 0.25% xylene cyanol]. The reaction products were subjected to electrophoresis for 1 h at 150 V through 10% polyacrylamide gels containing 0.1% SDS in 0.5× TBE (45 mM Tris base, 45 mM boric acid, 1 mM EDTA). The gels were dried on DEAE–cellulose paper and autoradiographed. Labeled DNA products were quantitated with the use of a PhosphoImager. The background level detected in the absence of added DNA helicase was <2% of the input substrate, and this value was subtracted from the amount of displaced products formed in the presence of DNA helicase.
Purification of recombinant hFBH1 and recombinant SCFhFBH1
The hFBH1 enzyme was expressed as a recombinant protein (GST–hFBH1) with GST fused to its N-terminus, and purified as previously described (2). Baculoviruses expressing hexahistidine-tagged (HT-) CUL1, HT-ROC1 and SKP1 were infected into Sf9 cells at a multiplicity of infection of 10 and incubated for 72 h. The infected cells (4 × 106 cells/ml, 2 l) were harvested, resuspended in the 40 ml of lysis buffer, T1000 [25 mM Tris-HCl pH 7.5, 1 M NaCl, 1 mM EDTA, 10% glycerol, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.1 mM benzamidine, 1.25 µg/ml leupeptin and 0.63 µg/ml pepstatin A; the subscript number indicates NaCl in mM], and disrupted by sonication (three cycles of a 30 s pulse and a 2 min cooling interval). The extract was cleared by centrifugation at 37 000 r.p.m. for 1 h in a Beckman 45 Ti rotor, and the supernatant (20.1 mg/ml, 40 ml) was diluted with 40 ml of T0. The diluted extract was incubated with 2.5 ml of glutathione–Sepharose 4B beads (Amersham Biosciences) for 8 h at 4°C, and the mixture was packed into a column. The column was washed with 20 column volumes of T500, and eluted with glutathione elution buffer (10 mM glutathione, 25 mM Tris–HCl pH 7.5, 300 mM NaCl, 1 mM EDTA, 10% glycerol, 1 mM DTT, 1 mM PMSF). The peak protein fractions (0.33 mg/ml, 17 ml) were pooled, adjusted to T200 and then loaded onto a Hitrap-heparin column (1 ml, Amersham Biosciences). The column was eluted with T600 (10 ml), and the peak protein fractions (>90% in purity) were pooled, and stored at –70°C. This fraction was used in all of the experiments carried out to characterize the enzymatic properties of the SCFhFBH1 complex unless noted otherwise. Glycerol gradient sedimentation (5 ml, 15–35% in buffer T500) was performed at 45 000 r.p.m. for 24 h in a Beckman SW 55Ti rotor to further confirm that all four polypeptides co-sedimented during centrifugation.
The HT-CUL1–HT-ROC1 heterodimeric complex was purified from crude extracts prepared from insect cells infected with baculoviruses expressing HT-CUL1 and HT-ROC1. The extracts were prepared as described above. The supernatant (6 mg/ml, 60 ml) was loaded onto an SP-Sepharose column (15 ml). After washing with 75 ml of T100, bound proteins were eluted with 150 ml of linear gradient (NaCl 100–1000 mM) in T buffer. The peak fractions, eluting at ∼150 mM NaCl, were pooled and directly loaded onto a 1 ml Hitrap Ni2+ column (Amersham Biosciences). After washing with 5 ml of T500 (–DTT, –EDTA) plus 50 mM imidazole, proteins were eluted with T500 plus 500 mM imidazole. Eluted proteins were analyzed by glycerol gradient sedimentation as described above.
HT-human SKP1 was purified as follows: a cDNA coding for the human SKP1 was cloned into pET28 (Novagen, Madison, WI) and transformed into E.coli BL21 cells. The bacterial culture (1 l) was propagated at 37°C, and induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h. The bacterial pellets were resuspended in T500 (–EDTA, –DTT) plus 10 mM imidazole, and disrupted by sonication (three cycles of a 30 s pulse and a 2 min cooling interval). Extracts were cleared by centrifuging at 37 000 r.p.m. for 1 h in a Beckman 45 Ti rotor, and the supernatant (1.2 mg/ml, 41 ml) was loaded onto 2 ml of Ni2+-NTA resin (Qiagen). After extensive washing with T500 (–DTT, –EDTA) plus 20 mM imidazole, the column was eluted with T500 (–EDTA, –DTT) plus 500 mM imidazole. The peak protein fractions were pooled, adjusted to T100 (0.36 mg/ml, 5.3 ml), and then loaded onto a Hitrap Q column (Amersham Biosciences). After washing with 10 column volume of T100, proteins were eluted with a linear gradient of 100–600 mM NaCl. Peak protein fractions eluting at 300 mM NaCl were pooled and then dialyzed against 500 ml of T100 for 6 h. The dialyzate (0.60 mg/ml, 3 ml) was then loaded onto a Hitrap SP column (Amersham Biosciences) and the flow-through fractions were pooled and used in all experiments.
Isolation of E2 and PK-ubiquitin, and preparation of 32P-labeled PK-ubiquitin
The human Cdc34 protein (hCdc34), an E2 ubiquitin-conjugating enzyme, was purified from E.coli harboring a pET3a plasmid expressing the wild-type Cdc34 protein, which was kindly provided by Dr. Z.-Q. Pan (The Mount Sinai School of Medicine, New York). The purification of E2 was carried out as reported previously (19). PK-ubiquitin (PK-Ub) was prepared as previously described (20). Phosphorylation of PK-Ub (7 µg) was carried out in a reaction mixture (20 µl) containing 20 mM Tris–HCl pH 7.4, 12 mM MgCl2, 2 mM NaF, 50 mM NaCl, 25 µM ATP, 5 µCi of [γ-32P]ATP, 0.1 mg/ml BSA and 1 U of cAMP kinase that was incubated at 37°C for 30 min. Reaction mixtures were heated at 70°C for 3 min to terminate the reaction.
Ub-ligation assay
Ub-ligation assays were carried out as described (20). Reaction mixtures (30 µl) contained 50 mM Tris–HCl pH 7.4, 5 mM MgCl2, 2 mM NaF, 10 nM okadaic acid, 2 mM ATP, 0.6 mM DTT, 5 µg of [32P]PK-Ub, E1 (2 pmol), hCdc34 (10 pmol) and the purified SCFhFBH1 complex. The mixtures were incubated at 37°C for 60 min and terminated with 7.5 µl of 5× SDS loading buffer, and then boiled for 3 min prior to loading onto 7.5% SDS–PAGE for analysis. The amounts of polyubiquitin chains larger than E2 were measured using a Phosphorimager.
In vitro reconstitution of SCFhFBH1
In order to obtain the heterotrimeric SKP1–CUL1–ROC1 complex, HT-CUL–HT-ROC1 (320 pmol) was mixed with HT-SKP1 (320 pmol) on ice for 30 min. This mixture, in parallel with HT-CUL1–HT-ROC1 and HT-SKP1, was subject to glycerol gradient centrifugation (5 ml, 15–35%) in buffer T500. After centrifugation at 45 000 r.p.m. for 24 h in a Beckman SW 55Ti rotor, fractions (∼200 µl) were collected from the top of the gradient and proteins were examined by silver staining. For the formation of SCF hFBH1 holocomplex, HT-CUL1–HT-ROC1 (3 pmol) and HT-SKP1 (3 pmol) were mixed and pre-incubated on ice for 30 min. This mixture was then supplemented with 3 pmol of GST–hFBH1, and incubated on ice or at 37°C for varying periods. Glutathione beads (30 µl of 50% slurry) and binding buffer (500 µl; T100 plus 0.1% NP-40 and 0.2 mg/ml BSA) were added to this mixture. After incubation for 3 h at 4°C, the mixtures were centrifuged and the beads were washed three times with T500. Proteins bound to the beads were eluted with glutathione and then analyzed by immunoblotting with anti-GST and anti-His antibodies. For ubiquitin ligase assay, proteins adsorbed to the glutathione beads were used to measure polyubiquitin chain formation.
RESULTS
Preparation of the SCFhFBH1 complex
Since the F-box protein is linked to the SCF complex through its interaction with SKP1, we initially determined whether the helicase and ATPase activities present in the uncomplexed hFBH1 were unaffected by its inclusion in the SCFhFBH1 complex. In order to address this question, we prepared and isolated the recombinant SCFhFBH1 complex from insect cells expressing GST–hFBH1, HT-CUL1, HT-ROC1 and SKP1. Expression of these proteins was monitored by western blot analyses using three antibodies: α-GST for the detection of hFBH1; α-His antibody for CUL1 and ROC1; and α-SKP1 for SKP1. When blots were probed with each of these antibodies, all four proteins were detected in baculovirus-infected cell extracts (Fig. 1A and B, lane 2), but not in extracts from uninfected cells (Fig. 1A and B, lane 1). The SCFhFBH1 complex was purified as described in Materials and Methods. Peak fractions obtained from the Hitrap-heparin column were subjected to a 12% SDS–PAGE (Fig. 1A and B, lane 3). The peak areas obtained from densitometric analysis were 1086, 920, 239 and 190 for GST–hFBH1 (135 kDa), HT-CUL1 (95 kDa), SKP1 (23 kDa) and HT-ROC1 (19 kDa), respectively. The stoichiometric ratio calculated was 1:1.2:1.3:1.2 when normalized with respect to the molecular weight of each protein, indicating that all four polypeptides were present in an equimolar ratio in the purified complex (Fig. 1A, lane 3). After the GST affinity column step, all four polypeptides co-purified during subsequent purification steps that included Hitrap-heparin Sepharose and glycerol gradient sedimentation (data not shown). These findings indicate that all four polypeptides form a stable complex.
Figure 1.
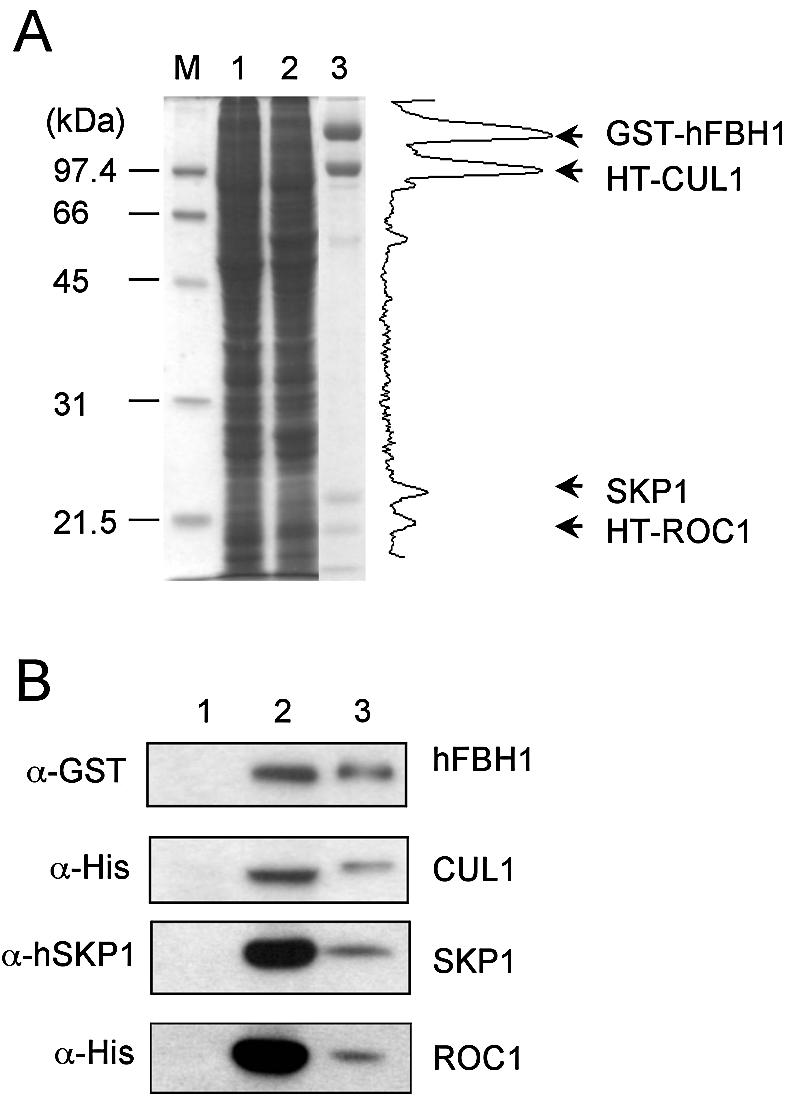
Purification of an SCFhFBH1 complex. Crude extracts prepared from Sf9 cells infected and uninfected with baculoviruses expressing GST–hFBH1, HT-CUL1, HT-ROC1 and SKP1, as well as purified SCFhFBH1, were subjected to 12% SDS–PAGE. The gels were Coomassie stained (A) and analyzed by western blot analyses using anti-GST (α-GST), anti-His (α-His) and anti-hSKP1 (α-hSKP1) antibodies (B). Shown are crude extracts from uninfected cells (lane 1) and infected cells (lane 2), and purified SCFhFBH1 complex (lane 3). Molecular size markers (M) are shown (in kDa). Lane 3 of (A) was scanned for the determination of peak areas of each protein, and the positions of each polypeptide are indicated by arrows.
Enzyme titration and kinetic experiments with the SCFhFBH1 complex and hFBH1
We showed previously that the immunoprecipitated SCFhFBH1 complex obtained from transfected human cell extracts exhibited ubiquitin ligase activity (2). However, attempts to measure helicase activity with the immunoprecipitate were unsuccessful due to the presence of nuclease that degraded labeled DNA substrates. In contrast to these results, we detected DNA-dependent ATPase and helicase activities in the recombinant SCFhFBH1 complex isolated from insect cells. This permitted us to characterize the helicase properties of the complex and compare them with those of free hFBH1. We first carried out enzyme titration experiments with equivalent amounts of hFBH1 and SCFhFBH1. Reactions were carried out with increasing amounts (15–720 fmol) of enzyme in the presence of 15 fmol of a 30mer oligonucleotide annealed to φX174 sscDNA as a substrate. Under the standard assay conditions described in Materials and Methods, oligonucleotides were displaced in proportion to the amount of enzyme added up to 240 fmol (Fig. 2A). At saturating levels of both proteins (>240 fmol), nearly all of the DNA substrate added was unwound (Fig. 2A). Quantitatively, the DNA-unwinding activities of SCFhFBH1 and hFBH1 alone were indistinguishable (Fig. 2B). DNA-unwinding activities of both SCFhFBH1 and hFBH1 were dependent on both ATP and Mg2+ (Fig. 2A, lanes 3 and 4).
Figure 2.

SCFhFBH1 unwinds duplex DNA as well as hFBH1. (A) Standard helicase assays were preformed as described in Materials and Methods with omissions indicated above the lanes (–ATP, –MgCl2). The amounts of hFBH1 (left) and SCFhFBH1 (right) added (fmol) are indicated at the top of each lane. B = boiled substrate control; S = substrate only control. (B) Quantitation of the results obtained in (A). (C) Kinetic analysis of unwinding reactions of hFBH1 and SCFhFBH1. The reaction mixture (180 µl) contained 990 fmol of hFBH1 (110 fmol per reaction); aliquots (20 µl) were withdrawn at the indicated times and the reaction products were analyzed by non-denaturing 10% PAGE in 0.5× TBE.
On the basis of the enzyme titration results shown in Figure 2B, the rate of unwinding in the presence of 110 fmol of enzyme per reaction was examined (Fig. 2C). The rate of unwinding was very rapid for both enzymes without an apparent lag period. The reaction proceeded linearly only during the earliest incubation period (<1 min), and plateaued (>10 min) after 60% of the input substrate (15 fmol per reaction) had been unwound (Fig. 2C). Thus, both enzymes displaced duplex DNA substrate with a nearly identical rate.
We also compared ATPase activities of SCFhFBH1 and hFBH1 in the presence of single-stranded DNA (ssDNA) or double-stranded DNA (dsDNA) (Fig. 3). We measured the amount of ATP hydrolyzed by 75 fmol of each enzyme in the presence of φX174 sscDNA and two forms of dsDNA, replicative form I (RFI) and RFIII of φX174. φX174 sscDNA supported ATP hydrolysis most efficiently, followed by RFI and then RFIII dsDNA. Both the extent and the rate of ATP hydrolysis by SCFhFBH1 and hFBH1 were nearly the same (Fig. 3). These results demonstrate that the SCFhFBH1 complex retains helicase and ATPase activities and both activities are not significantly altered by the interaction of hFBH1 with other proteins present in the SCF complex.
Figure 3.

Influence of the different polynucleotides on the ATPase activity. Kinetic analysis of polynucleotide-dependent ATPase activity. ATP hydrolysis was measured using the condition described in Materials and Methods. Reaction mixtures contained 75 fmol of either GST–hFBH (closed symbols) or SCFhFBH1 (open symbols) and 50 ng of DNA as indicated. The mixtures were incubated for the period of time indicated. RFI = replicative form I; RFIII = replicative form III.
Polyubiquitin chain formation by the SCFhFBH1 complex
Since the SCF complex co-immunoprecipitated with hFBH1 from transfected human cells exhibited ubiquitin ligase activity (2), we decided to confirm and extend this result with the purified recombinant SCFhFBH1 complex. For this purpose, we examined the ability of the complex to catalyze polyubiquitin chain formation in the presence of ATP, 32P-labeled ubiquitin, E1 and E2. High molecular polymers of the 32P-labeled ubiquitin were formed in proportion to the amount (0.1, 0.5 and 2 pmol) of the purified SCFhFBH1 complex added (Fig. 4A, lanes 4–6). The amounts of ubiquitin polymers formed by 0.5 and 2 pmol of SCFhFBH1 were 2.9- and 8.3-fold, respectively, more than that produced by 0.1 pmol of the complex. Polyubiquitin formation was dependent on E1 and E2, since omission of any of these two proteins did not support formation of the ubiquitin polymer (Fig. 4A, lanes 1 and 2).
Figure 4.

The purified SCFhFBH1 complex contains ubiquitin ligase activity, which is unaffected by DNA. (A) Ubiquitin ligase assay was performed as described in Materials and Methods with omissions indicated above the lanes (–). The amounts of SCFhFBH1 added (pmol) are noted at the top of the figure. The fold increases in products formed relative to the amount formed in lane 4 are indicated in italics below the lanes. (B) The indicated levels of φX174 DNAs (ng) were pre-incubated with 1 pmol of SCFhFBH1 for 30 min. Pre-incubated mixtures were then used in the ubiquitin ligase assay. The symbol (–) indicates the absence of DNA. The fold changes in products formed relative to those formed in lane 2 are indicated in italics below the lanes.
Influence of DNA on E3 ligase activity of SCFhFBH1
Because the SCFhFBH1 complex contains ATPase activity that is stimulated by DNA cofactors, we examined whether DNA affected its ability to form polyubiquitin chains. SCFhFBH1 (1 pmol) was incubated in the absence (Fig. 4B, lane 2) or presence of φX174 sscDNA (Fig. 4B, lanes 3–5) or dsDNA (RFI of φX174) (Fig. 4B, lanes 6–8). The level of ubiquitin polymers formed was reduced slightly (0–13 and 23–27%) only in presence of excess (200 and 1000 ng, respectively) sscDNA or dsDNA (Fig. 4B, lanes 4 and 5, and 7 and 8). These levels of DNA markedly exceed the DNA concentrations (5 ng of φX174 sscDNA per reaction) that maximally stimulate ATP hydrolysis (Table 2). The lower levels of ssDNA (1, 5, 10 and 20 ng) in the reaction neither stimulated nor inhibited the E3 ligase activity of the SCFhFBH1 (data not shown). Thus, these results demonstrate that the ubiquitin ligase activity of the complex is hardly affected by concentrations of DNA that maximally stimulate ATPase activity.
Table 2. Requirements for ATPase activity of hFBH1.
| Additions or omissions | Amount added | Relative activity (%)a |
|---|---|---|
| Completeb |
|
100 |
| Add human RPA |
0.2, 0.4, 0.6 µg |
105, 104, 98 |
| Add E.coli ssb |
0.2, 0.4, 0.6 µg |
67, 38, 47 |
| Add NaCl |
0.05, 0.1, 0.2 M |
122, 112, 107 |
| Omit MgCl2 |
|
<1 |
| Omit DTT + NEMc |
10 mM |
<1 |
| Omit ATP |
|
|
| Add GTP or CTP or UTP |
250 µM each |
15, 40, 52 |
| Add dATP |
250 µM |
110 |
| Add dGTP or dCTP or dTTP |
250 µM each |
83, 49, 5 |
| Omit φX174 sscDNA |
|
9 |
| Add φX174 sscDNA |
1, 5, 20 ng |
36, 98, 102 |
| Add poly(dI–dC) double strand |
50 ng |
89 |
| Add tRNA |
50 ng |
16 |
| Add φX174 RFI |
50 ng |
67 |
| Add φX174 RFIII | 50 ng | 34 |
aThe value of 100% in this experiment corresponded to the hydrolysis of 1620 pmol ATP.
bThe complete reaction contained 75 fmol of GST–hFBH1 in the standard reaction mixture described in Materials and Methods. ATP hydrolysis was measured as described in Materials and Methods.
cThe enzyme was first incubated for 10 min at 37°C in the presence of the indicated amount of NEM.
Reconstitution of SCFhFBH1
In order to examine the order of assembly of the SCFhFBH1 complex, we reconstituted the SCFhFBH1 complex in vitro with purified proteins. We first prepared HT-CUL1–HT-ROC1 as a heterodimeric complex and HT-SKP1 and GST–hFBH1 as separate proteins. When HT-SKP1 and HT-CUL1–HT-ROC1 were mixed and incubated on ice, SKP1–CUL1–ROC1 readily formed a stable complex since all three proteins co-sedimented upon glycerol gradient sedimentation, consistent with previous reports (20,21). We added GST–hFBH1 to the pre-assembed SKP1–CUL1–ROC1 complex the same way as above in a 1:1 molar ratio, and incubated the mixture on ice. However, we failed to observe the association of GST–hFBH1 with the CUL1–ROC1–SKP1 subcomplex using glycerol gradient sedimentation analysis (data not shown). This is in contrast to the efficient assembly of the SCFhFBH1complex in vivo as shown above.
In order to solve this discrepancy, a more systematic approach was undertaken to find conditions required for an efficient assembly of the complex in vitro. We first tested whether varying salt concentrations may assist complex assembly with hFBH1 and the pre-assembled SKP1–CUL1–ROC1 subcomplex, but this effort also failed (data not shown). We then tested whether incubation at elevated temperatures facilitated complex assembly. We incubated hFBH1 (3 pmol) and the SKP1–CUL1–ROC1 subcomplex (3 pmol) together at 37°C for varying periods (15, 30 and 60 min) of time. In this experiment, HT-CUL, HT-ROC1 and HT-SKP1 were used to facilitate the simultaneous detection and subsequent quantification of each protein after precipitation with glutathione beads. Using α-His monoclonal antibodies, we examined the levels of HT-CUL1, HT-ROC1 and HT-SKP1 precipitated with GST–hFBH1 with glutathione beads (Fig. 5A). After 30 and 60 min of incubation, detectable levels (>25 and >40%, respectively) of input SKP1–CUL1–ROC1 co-precipitated with GST–hFBH1, suggesting that the association of hFBH1 with SKP1–CUL1–ROC1 is a slow process that requires thermal activation (Fig. 5A and B). We noted that the amounts of CUL1 precipitated most efficiently with hFBH1 after 30 and 60 min of incubation (Fig. 5B). This was unexpected since the purified CUL1–ROC1 complex was used in this experiment. However, we noted that the recombinant GST–hFBH1 isolated from insect cells complexed with insect SKP1, which we could detect by cross-reactivity with human SKP1 antibody (data not shown). The level of insect SKP1 in our hFBH1 preparation was estimated to be ∼10–20% of that isolated from insect cells, judged from silver-stained protein gel and western blot analysis (data not shown). This may account for our observations that the higher levels of CUL1 were bound to hFBH1 after 30–60 min of incubation. It is not clear, however, why the levels of ROC1 did not concomitantly increase with CUL1. One possibility is that the binding of CUL1–ROC1 complex to the hFBH1–insect SKP1 complex caused ROC1 to dissociate from CUL1.
Figure 5.

In vitro reconstitution of SCFhFBH1. (A) HT-SKP1–HT-CUL1–HT-ROC1 complex (3 pmol) was incubated with GST–hFBH1 (3 pmol) at 37°C for different time periods. The incubated proteins were bound to glutathione-coupled beads, and then analyzed by western blot using anti-GST (α-GST) and anti-His (α-His) antibodies as described in Materials and Methods. Lane 1: input proteins. The band intensities were measured with Quantity One (Molecular Dynamics Inc.). The positions of purified polypeptides are indicated by arrows. (B) Quantitation of the results in (A). (C) GST–hFBH1 (5 pmol) and/or HT-SKP1–HT-CUL1–HT-ROC1 (5 pmol) were incubated on ice (lane 3) or at 37°C (lanes 1, 2 and 4) for 30 min in various combinations as indicated. Incubated proteins were precipitated with glutathione-coupled beads, and the bound proteins were used as the enzyme source in the ubiquitin ligase assay as described in Materials and Methods. Purified SCFhFBH1 complex (2 pmol) was also precipitated and analyzed as a positive control (lane 5).
Since the in vitro reconstitution of the complex depended on incubation at 37°C, we also investigated whether temperature affected the E3 ubiquitin ligase activity of hFBH1 and the pre-assembled SKP1–CUL1–ROC1. The hFBH1 enzyme (10 pmol) and the pre-assembled SKP1–CUL1–ROC1 complex (10 pmol) were mixed and divided into two aliquots. One aliquot was kept on ice, while the other was pre-incubated at 37°C for 10 min, followed by adsorption of the mixtures to glutathione beads. Following washing, all components (E1, E2, ATP and 32P-labeled ubiquitin) required to support polyubiquitin chain formation were added to the washed beads. The aliquot stored on ice displayed marginal synthesis of polyubiquitin chains (Fig. 5C, lane 3), whereas the other aliquot incubated at 37°C contained 3- to 5-fold more activity (Fig. 5C, lanes 4), consistent with the above finding that incubation at 37°C facilitated SCFhFBH1 complex formation. However, the ubiquitin ligase activity was relatively low compared with the activity of the recombinant SCFhFBH1 purified from insect cells (Fig. 5C, compare lanes 4 and 5). These results are in keeping with our observations that the assembly of the SCFhFBH1 complex in vitro is inefficient.
Both hFBH1 and SCFhFBH1 act in a distributive manner and do not require pre-formed fork structures for unwinding activity
In order to define further the helicase properties of the recombinant SCFhFBH1 complex, we determined whether it can act processively. To this end, we carried out substrate challenge experiments as shown in Figure 6. Reaction mixtures were pre-incubated for 3 min on ice with either hFBH1 or SCFhFHB1 (110 fmol each) and labeled oligonucleo tide-based substrates (15 fmol) in the absence of ATP. Reactions were then supplemented with 10-, 25-, 50- and 100-fold excess of the unlabeled competitor DNA substrate along with ATP, followed by incubation for 10 min at 37°C. The addition of unlabeled substrates (10-, 25-, 50- and 100-fold excess) resulted in the immediate decrease (33–37, 54–72, 79–91 and 94–98%, respectively) in the efficiency of unwinding (Fig. 6A–C). The addition of a 10-fold molar excess of φX174 sscDNA virtually abolished the unwinding activities (>95%) of both enzymes (data not shown). These results demonstrate that the helicase activities of hFBH1 and SCFhFHB1 act in a distributive manner. We also examined the influence of a fork-like structure on DNA unwinding by hFBH1 and SCFhFBH1 with five different DNA substrates. These included a blunt end substrate with a 30 bp duplex, two substrates with a 10 or 25 nt 5′ tail, and two substrates with a 10 or 25 nt 3′ tail (Fig. 7). All five substrates were displaced to similar extents in the presence of two different levels (110 and 220 fmol) of hFBH1 and SCFhFBH1 (Fig. 7A and B, respectively). These findings demonstrate that the helicase activities of hFBH1 and SCFhFBH1 are comparable and show no preference for a fork-like structure.
Figure 6.

hFBH1 is a distributive helicase. (A and B) Helicase assays were performed in reaction mixtures described in Materials and Methods with 110 fmol of GST–hFBH1 (A) and SCFhFBH1 (B) and 15 fmol of oligonucleotide-based substrates. Mixtures containing enzymes and DNA substrates were pre-incubated on ice for 3 min in the absence of ATP, followed by the addition of ATP and the indicated amounts of unlabeled cold substrates. The oligonucleotide DNA substrate used consisted of a 90mer template with 21mer oligonucleotides annealed to the 5′ end region of the template. The sequences of the oligonucleotides were as described (18). The fold excess of unlabeled DNA substrate added is noted at the top of the figure. (C) Quantitation of results in (A) and (B).
Figure 7.

The DNA helicase activities of hFBH1 and SCFhFBH1 do not require fork-like structure. Five different 30 bp duplex DNA substrates were used in this experiment; a blunt end substrate with 30 bp duplex, two substrates with a 10 or 25 nt 5′ tail, and two substrates with a 10 or 25 nt 3′ tail. The duplex region in each substrate was 30 bp long and identical in nucleotide sequence. The structure of each substrate used is as illustrated. The asterisks indicate the position of radiolabel. Reactions were carried out with 110 (indicated as +) and 220 fmol (indicated as ++) of hFBH1 (A) and SCFhFBH1 (B) in the presence of 15 fmol of each substrate shown at the top of each set of experiments. B = boiled substrates; S = substrate only control.
We also examined the ability of SCFhFBH1 to unwind longer duplex DNAs. For this purpose, three DNA substrates with different duplex sizes, 30, 52 and 91 bp, were prepared. As shown in Figure 8, the 30 bp duplex DNA was unwound most efficiently, but to a similar extent, by both enzymes. The 52 and 91 bp duplex DNA substrates were utilized ∼2-fold less efficiently than the 30 bp duplex substrate. These results suggest that both enzymes unwind longer duplex DNAs identically.
Figure 8.
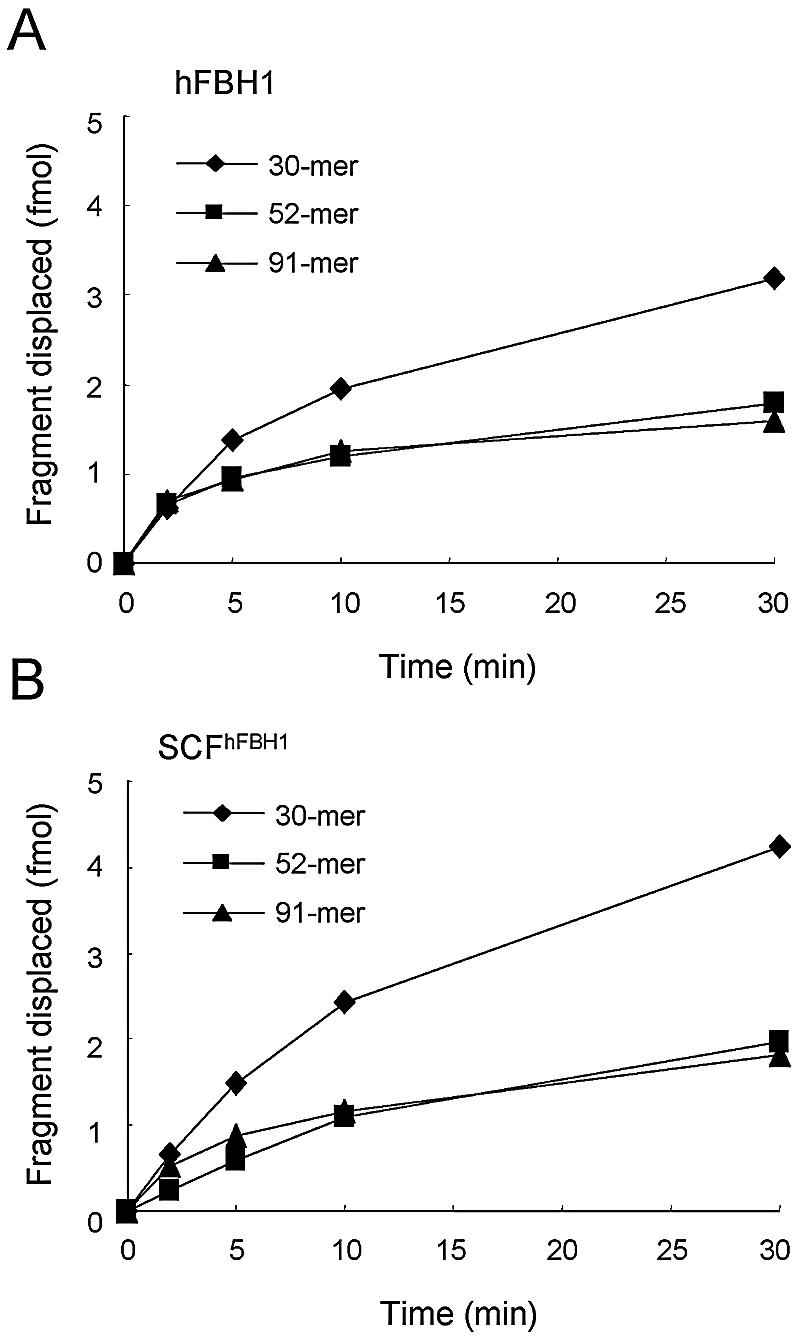
hFBH1 and SCFhFBH1 unwind longer duplex DNAs with similar efficiency. Three different partial duplex DNA substrates (30, 52 and 91 bp) were prepared as described in Materials and Methods. Reaction mixtures containing 15 fmol of each substrate and 110 fmol of either hFBH1 (A) or SCFhFBH1 (B) were incubated for the period of time indicated in the figure.
Other requirements for DNA helicase activity
Since the helicase and ATPase activities of hFBH1 and SCFhFBH1 are comparable in every test used above, we determined other properties of hFBH1 alone. As shown in Table 1, the displacement of duplex DNA catalyzed by hFBH1 required ATP and Mg2+. Its helicase activity was stimulated ∼2-fold by NaCl (50–200 mM) and the divalent cation Mn2+ completely substituted for Mg2+, while Ca2+ was only partially (11%) active. The hFBH1 unwinding activity was inactivated by treatment with N-ethylmaleimide (NEM), suggesting that a sulfhydryl group(s) may be important for its function. Non-hydrolyzable ATP analogs such as ATPγS and AppCp did not support the unwinding activity of hFBH1. Interestingly, AppNP and ADP partially (26 and 45%, respectively) supported the unwinding activity of hFBH1 (Table 1). This is unlikely to be due to the contamination of other nucleotides in ADP, since AppNp, a synthetic ATP analog, reproducibly supported DNA unwinding by hFBH1. Consistent with this, the addition of 0.5–1 mM ADP to complete reaction mixtures containing 2 mM ATP did not inhibit unwinding activity. We were not able to measure the hydrolysis of these two nucleotides by hFBH1 since their radioisotopic forms are not available. The nucleotide required for a helicase activity was also determined. All eight nucleotide triphosphates supported hFBH1 helicase activity, but their efficiency varied. dATP was as efficient as ATP at a concentration of 0.5 mM, but less efficient (71%) at 2 mM. Other nucleotides including GTP, CTP, UTP and dCTP partially (57–80%) substituted for ATP at concentrations of 0.5 or 2 mM (Table 1). The nucleotides dGTP and dTTP were poor (13–25% at 0.5 mM and 32–39% at 2 mM). Although hFBH1 could utilize ADP, AMP did not support unwinding of duplex DNA (Table 1). The helicase activity of hFBH1 was hardly affected by eukaryotic RPAs and E.coli ssb (Table 1).
Table 1. Requirements for hFBH1 DNA helicase activity.
| Additions or omissions | Amount added | Relative activity (%)a |
|---|---|---|
| Completeb |
|
100 |
| Add NaCl |
0.025, 0.05, 0.1, 0.2 M |
122, 181, 225, 205 |
| Add ADP |
0.5 or 1 mM |
116, 110 |
| Add E.coli SSB |
0.2, 0.4, 0.6 µg |
110, 110, 100 |
| Add hRPA |
0.2, 0.4, 0.6 µg |
97, 88, 88 |
| Add yRPA |
0.2, 0.4, 0.6 µg |
96, 100, 84 |
| Add SpRPA |
0.2, 0.4, 0.6 µg |
93, 96, 93 |
| Omit MgCl2 |
|
<1 |
| MnCl2 |
2 mM |
101 |
| CaCl2 |
2 mM |
11 |
| Omit DTT + NEMc |
10 mM |
<1 |
| Omit ATP |
2 mM |
<1 |
| +ATPγS |
2 mM |
<1 |
| +AppNp |
2 mM |
26 |
| +AppCp |
2 mM |
<1 |
| +ADP |
2 mM |
45 |
| +AMP |
2 mM |
<1 |
| Omit ATP |
|
<1 |
| +GTP |
0.5 or 2 mM |
57, 67 |
| +CTP |
0.5 or 2 mM |
68, 80 |
| +UTP |
0.5 or 2 mM |
63, 78 |
| +dATP |
0.5 or 2 mM |
96, 71 |
| +dGTP |
0.5 or 2 mM |
25, 39 |
| +dCTP |
0.5 or 2 mM |
64, 69 |
| +dTTP | 0.5 or 2 mM | 13, 32 |
aThe value of 100% in this experiment corresponded to the displacement of 6.4 fmol of DNA substrate.
bThe complete reaction contained 15 fmol of partial duplex sscDNA substrate containing a 30 base oligonucleotide and 75 fmol of GST–hFBH1 in the standard reaction mixture described in Materials and Methods.
cThe enzyme was first incubated for 10 min at 37°C in the presence of the indicated amount of NEM.
Characterization of the DNA-dependent ATPase activity
The DNA-dependent ATPase activity of hFBH1 was also characterized. As observed with its helicase activity, ATP hydrolysis required Mg2+ and was inhibited by NEM treatment. The ATPase activity of hFBH1 was unaffected by hRPA, but inhibited significantly (33–62%) by E.coli ssb (Table 2). Consistent with their ability to support the unwinding activity of hFBH1 (Table 1), all other nucleoside triphosphates were hydrolyzed by hFBH1 to varying extents (Table 2). Among them, dATP was most efficient (110% with respect to ATP), while hydrolysis of dTTP was the least efficient (5%). The Km values for ATP and dATP were 44 and 102 µM, respectively (data not shown). In the absence of φX174 sscDNA, ATP hydrolysis was significantly reduced (∼10 fold compared with that observed in the presence of φX174 sscDNA) (Table 2). The extent of ATP hydrolysis by hFBH1 became maximal in the presence of >5 ng of φX174 sscDNA. Form I and form III dsDNAs of φX174 supported ATP hydrolysis at a rate approximately two-thirds (67%) and one-third (34%), respectively, of that observed with φX174 sscDNA. A synthetic dsDNA, poly (dI–dC), supported ATP hydrolysis more efficiently (89%) than φX174 dsDNA (Table 2).
DISCUSSION
In this report, we have described the isolation of the recombinant SCFhFBH1 complex and demonstrated that hFBH1 forms a functional complex with SKP1, CUL1 and ROC1. In the presence of E1, E2 and monomeric ubiquitin, the recombinant complex catalyzed formation of a polyubiquitin chain. Polyubiquitin chain formation by the recombinant SCFhFBH1 complex was unaffected by ssDNA or dsDNA, that markedly stimulated ATP hydrolysis by the complex (Fig. 4B). These results indicate that the E3 ligase activity of the complex is not changed when it interacts with DNA and that the recombinant SCFhFBH1 complex interacts with the ubiquitination machinery.
We also showed that the helicase and DNA-dependent ATPase activities of SCFhFBH1 are nearly identical to those of hFBH1 in every aspect we examined. For example, both enzymes acted in a distributive manner in the unwinding reaction (Fig. 6), and their unwinding efficiencies were unaffected by the presence of fork structures in the substrate (Fig. 7). DNA helicase substrates of different duplex lengths were unwound with comparable efficiencies by both enzymes (Fig. 8). The SCFhFBH1 complex is distributive and incapable of unwinding long duplex DNA and, thus, it is inferred that the complex is most likely to act in a catalytic fashion and may be involved in a DNA transaction that requires unwinding of short stretches of DNA such as DNA repair or recombination (see also below).
The properties of the helicase and ATPase of hFBH1 are similar to those of SpFBH1, the S.pombe homolog. We noted two differences between these two enzymes; one is that hFBH1 is neither stimulated nor inhibited by hRPA, whereas SpFHB1 is stimulated by its cognate RPA particularly at low ATP concentrations. However, we found that RPA and hFBH1 interact in the yeast two-hybrid assays (data not shown). At present, the significance of this interaction is not clear. The other difference noted is that hFBH1 can utilize ADP and AppNp as an energy source, although the efficiencies are relatively poor (25 and 45%, respectively) compared with ATP. SpFHB1, however, used neither ADP nor AppNP as energy source (J.Kim and Y.S.Seo, unpublished observation). Our findings suggest that both hFBH1 and SpHBH1 can act as a DNA helicase even when the ATP supply is inadequate. The activity of SpFBH1 is stimulated by RPA, while hFBH1 utilizes ADP when the level of ATP is low.
Since hFBH1 contains an F-box motif critical for its association with SKP1, we tested whether deletion of the F-box had any effect on its helicase activity. The F-box-deleted mutant protein (ΔF-hFBH1), which was purified as a soluble protein, contained substantially lower ATPase and helicase activities (data not shown). Both activities of ΔF-hFBH1 were reduced >95%. It is believed that the deletion of the F-box motif impaired the integrity of the enzyme required for the catalytic function of enzyme, since deletion did not include any part of the helicase motifs. The fact that the efficiency of the SCFhFBH1 complex formation in vitro is very low suggests that the in vivo assembly of SCFhFBH1 may be a regulated process, requiring an additional factor(s) such as a chaperone. If this were the case, the assembly step could constitute a key step controlling the level of the complex in cells.
At present, it is unclear whether cells contain free hFBH1, a mixture of hFBH1 and SCFhFBH1 or the SCFhFBH1 complex only. We previously isolated SpFBH1 as a single polypeptide during its purification (1), and recently we found that SpFBH1 was co-immunoprecipiated from crude extracts prepared from S.pombe cells (H.Y.Kang and Y.S.Seo, unpublished observation). These findings suggest that in S.pombe, SpFBH1 and SCFSpFBH1 are present within the same cell. However, we cannot rule out the possibility that the SCFSpFBH1 complex is dissociated during the purification procedure. We speculate that this is unlikely since the SCFhFBH1 complex is very stable and does not dissociate even in the presence of 1 M NaCl (data not shown). Since both enzymes are almost identical in their ability to unwind duplex DNA, it is possible that both hFBH1 and SCFhFBH1, although differing in their architecture, can function similarly as a DNA helicase. However, SCFhFBH1 may have additional roles in vivo. We currently do not understand what DNA transactions require hFBH1 and SCFhFBH1, making it difficult to assign the role(s) of hFBH1 and hence the SCFhFBH1 complex in reactions involving DNA. Identification of the protein(s) targeted by the SCFhFBH1 complex may contribute to this solution. Since the two different activities, DNA unwinding and ubiquitin ligase, of SCFhFBH1 reside in one complex, these two activities may act in a coupled manner rather than acting in two seemingly unrelated functions. One scenario is that SCFhFBH1 can catalyze polyubiquitination of a DNA-bound substrate protein that it encounters while it unwinds duplex DNA. This possibility is consistent with our finding that unwinding occurs when ssDNA is available for binding, which suggests that the natural target is pre-existing ssDNA. This possibility is consistent with our finding that the enzyme did not unwind blunt-ended duplex DNA despite its ability to stimulate ATP hydrolysis (data not shown). The other scenario is that the energy derived from ATP hydrolysis may allow the complex to translocate along the duplex DNA rather than unwinding the duplex, since dsDNA efficiently stimulated hydrolysis of ATP by SCFhFBH1 (Table 2). The chromatin-remodeling factor SWI2/SNF2 that is devoid of helicase activity despite the presence of helicase motifs, hydrolyzes ATP in a dsDNA-dependent manner (22–24). ATP hydrolysis may provide the energy required for SWI2/SNF2 to translocate along chromatin DNA and/or alter protein–DNA structure when necessary, rather than directly altering the structure of duplex DNAs. If this is true for SCFhFBH1, the helicase activity associated with the SCFhFBH1 complex may help the enzyme move along the chromatin DNA, resulting in its interaction with proteins on chromatin, which are ubiquitinated and targeted for degradation by 26S proteasome.
Recently, Shinagawa and his colleagues discovered that the S.pombe homolog of hFBH1 plays a role in post-replicational recombination events in their genetic studies (personal communication). This result is in keeping with the helicase properties we showed, since this process does not require extensive unwinding of duplex DNA and processive helicase activity of the SCFhFBH1 complex. The strategy above used by SCFhFBH1 may be essential to remove proteins associated with DNA and/or alter nucleoprotein structures, which can then facilitate subsequent DNA transactions such as post-replication recombination. For a clearer definition of a function(s) of SCFhFBH1 in vivo, we are currently attempting to identify a substrate protein that is specifically ubiquitinated by the complex.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Jerard Hurwitz (Sloan-Kettering Institute, USA) for his critical reading of the manuscript. This work was supported by a grant from the Creative Research Initiatives of the Korean Ministry of Science and Technology given to Y.-S.S.
REFERENCES
- 1.Park J.S., Choi,E., Lee,S.H., Lee,C. and Seo,Y.S. (1997) A DNA helicase from Schizosaccharomyces pombe stimulated by single-stranded DNA-binding protein at low ATP concentration. J. Biol. Chem., 272, 18910–18919. [DOI] [PubMed] [Google Scholar]
- 2.Kim J., Kim,J.H., Lee,S.H., Kim,D.H., Kang,H.Y., Bae,S.H., Pan,Z.Q. and Seo,Y.S. (2002) The novel human DNA helicase hFBH1 is an F-box protein. J. Biol. Chem., 277, 24530–24537. [DOI] [PubMed] [Google Scholar]
- 3.Peters J.M. (1998) SCF and APC: the Yin and Yang of cell cycle regulated proteolysis. Curr. Opin. Cell Biol., 10, 759–768. [DOI] [PubMed] [Google Scholar]
- 4.Laney J.D. and Hochstrasser,M. (1999) Substrate targeting in the ubiquitin system. Cell, 97, 427–430. [DOI] [PubMed] [Google Scholar]
- 5.Tyers M. and Jorgensen,P. (2000) Proteolysis and the cell cycle: with this RING I do thee destroy. Curr. Opin. Genet. Dev., 10, 54–64. [DOI] [PubMed] [Google Scholar]
- 6.Hershko A. and Ciechanover,A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- 7.Craig K.L. and Tyers,M. (1999) The F-box: a new motif for ubiquitin dependent proteolysis in cell cycle regulation and signal transduction. Prog. Biophys. Mol. Biol., 72, 299–328. [DOI] [PubMed] [Google Scholar]
- 8.Deshaies R.J. (1999) SCF and cullin/ring H2-based ubiquitin ligases. Annu. Rev. Cell. Dev. Biol., 15, 435–467. [DOI] [PubMed] [Google Scholar]
- 9.Bai C., Sen,P., Hofmann,K., Ma,L., Goebl,M., Harper,J.W. and Elledge,S.J. (1996) SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell, 86, 263–274. [DOI] [PubMed] [Google Scholar]
- 10.Mathias N., Johnson,S., Byers,B. and Goebl,M. (1999) The abundance of cell cycle regulatory protein Cdc4p is controlled by interactions between its F box and Skp1p. Mol. Cell. Biol., 19, 1759–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willems A.R., Lanker,S., Patton,E.E., Craig,K.L., Nason,T.F., Mathias,N., Kobayashi,R., Wittenberg,C. and Tyers,M. (1996) Cdc53 targets phosphorylated G1 cyclins for degradation by the ubiquitin proteolytic pathway. Cell, 86, 453–463. [DOI] [PubMed] [Google Scholar]
- 12.Feldman R.M., Correll,C.C., Kaplan,K.B. and Deshaies,R.J. (1997) A complex of Cdc4p, Skp1p and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell, 91, 221–230. [DOI] [PubMed] [Google Scholar]
- 13.Skowyra D., Craig,K.L., Tyers,M., Elledge,S.J. and Harper,J.W. (1997) F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell, 91, 209–219. [DOI] [PubMed] [Google Scholar]
- 14.Skowyra D., Koepp,D.M., Kamura,T., Conrad,M.N., Conaway,R.C., Conaway,J.W., Elledge,S.J. and Harper,J.W. (1999) Reconstitution of G1 cyclin ubiquitination with complexes containing SCFGrr1 and Rbx1. Science, 284, 662–665. [DOI] [PubMed] [Google Scholar]
- 15.Kamura T., Koepp,D.M., Conrad,M.N., Skowyra,D., Moreland,R.J., Iliopoulos,O., Lane,W.S., Kaelin,W.G.Jr, Elledge,S.J., Conaway,R.C., Harper,J.W. and Conaway,J.W. (1999) Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science, 284, 657–661. [DOI] [PubMed] [Google Scholar]
- 16.Ohta T., Michel,J.J., Schottelius,A.J. and Xiong,Y. (1999) ROC1, a homolog of APC11, represents a family of cullin partners with an associated ubiquitin ligase activity. Mol. Cell, 3, 535–541. [DOI] [PubMed] [Google Scholar]
- 17.Henricksen L.A., Umbricht,C.B. and Wold,M.S. (1994) Recombinant replication protein A: expression, complex formation and functional characterization. J. Biol. Chem., 269, 11121–11132. [PubMed] [Google Scholar]
- 18.Bae S.H., Bae,K.H., Kim,J.A. and Seo,Y.S. (2001) RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature, 412, 456–461. [DOI] [PubMed] [Google Scholar]
- 19.Wu K., Chen,A., Tan,P. and Pan,Z.Q. (2002) The Nedd8-conjugated ROC1–CUL1 core ubiquitin ligase utilizes Nedd8 charged surface residues for efficient polyubiquitin chain assembly catalyzed by Cdc34. J. Biol. Chem., 277, 516–527. [DOI] [PubMed] [Google Scholar]
- 20.Tan P., Fuchs,S.Y., Chen,A., Wu,K., Gomez,C., Ronai,Z. and Pan,Z.Q. (1999) Recruitment of a ROC1–CUL1 ubiquitin ligase by Skp1 and HOS to catalyze the ubiquitination of I kappa B alpha. Mol. Cell, 3, 527–533. [DOI] [PubMed] [Google Scholar]
- 21.Jackson P.K. and Eldridge,A.G. (2002) The SCF ubiquitin ligase: an extended look. Mol. Cell, 9, 923–925. [DOI] [PubMed] [Google Scholar]
- 22.Eisen J.A., Sweder,K.S. and Hanawalt,P.C. (1995) Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res., 23, 2715–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cairns B.R., Kim,Y.J., Sayre,M.H., Laurent,B.C. and Kornberg,R.D. (1994) A multisubunit complex containing the SWI1/ADR6, SWI2/SNF2, SWI3, SNF5 and SNF6 gene products isolated from yeast. Proc. Natl Acad. Sci. USA, 91, 1950–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cote J., Quinn,J., Workman,J.L. and Peterson,C.L. (1994) Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science, 265, 53–60. [DOI] [PubMed] [Google Scholar]