

Abstract
Prolidase gene (PEPD) encodes prolidase enzyme, which is responsible for hydrolysis of dipeptides containing proline or hydroxypro-line at their C-terminal end. Mutations in PEPD gene cause, in human, prolidase deficiency (PD), a rare autosomal recessive disorder. PD patients show reduced or absent prolidase activity and a broad spectrum of phenotypic traits including various degrees of mental retardation. This is the first report correlating PD and brain damages using as a model system prolidase deficient mice, the so called dark-like (dal) mutant mice. We focused our attention on dal postnatal brain development, revealing a panel of different morphological defects in the cerebral and cerebellar cortices, such as undulations of the cerebral cortex, cell rarefaction, defects in cerebellar cortex lobulation, and blood vessels overgrowth. These anomalies might be ascribed to altered angiogenic process and loss of pial basement membrane integrity. Further studies will be directed to find a correlation between neuroarchitecture alterations and functional consequences.
Key words: Mental retardation, prolidase deficiency, postnatal development, CNS alteration, cardiac hypertrophy, extracellular matrix
Introduction
Prolidase deficiency (PD) (OMIM 170100) is a rare autosomal recessive disorder whose incidence of 1-2:1,000,000 people is probably underestimated. PD patients have a broad and variable phenotypic spectrum, as well as a diverse clinical onset (as early as shortly after birth or as late as 22 years of age). The principal clinical feature is dermatological manifestation with chronic, recurrent, slowly healing ulcerations mainly located on the lower legs and feet.1,2 Typically, the phenotype includes moderate to severe degree of mental retardation, rarely epileptic seizures, facial dysmorphism, splenomegaly, susceptibility to recurrent respiratory infections, hypotonia, skeletal and vascular anomalies. The severity of the disease varies widely, but no relationship between genotype and phenotype has been established yet, mainly due to the limited number of mutations described.3
Prolidase is linked to the metabolism of many biologically important molecules and it has a central role in extracellular matrix (ECM) remodelling, being essential for normal physiology including embryonic development, tissue resorption, wound healing, cell migration, and cell differentiation.4-6 In particular, the ECM structure participating to the last two processes in the brain is the pial basement membrane (BM).7 One of its major constituents is laminin,8 which has adhesive properties anchoring the plasma membrane of cells adjacent to BM.6,9 Meningeal cells actively participate in ECM formation10 and BM remodelling,11 in fact, impaired meningeal integrity could be the cause of migration defects.10,12
The dark-like mutant mouse (dal), obtained through random mutagenesis (Mouse Mutant Resource Web Site, The Jackson Laboratory, 2008), could be used as animal model for PD, because of a 4bp deletion in the 14th exon of the murine PEPD gene. Moreover, mice develop congenital cardiomyocyte hypertrophy due to a significant reduction in the levels of several integrin transducers and, being collagen the major ligand of integrins, it was suggested that prolidase, through its effects on the ECM, influences cardiomyocyte growth.13 At present, no information are available on the functional correlation between PD and the mental retardation described in these patients. Therefore, this is the first neuromorphological study on postnatal brain development in dal mutant mice, representing a suitable model to study the morphological/molecular causes of brain defects. Mainly two brain regions were analyzed: cerebral and cerebellar cortices, with the challenging goal to find a parallelism between the human phenotype and brain defects we found in PD mouse model. To investigate the general neuroarchitecture organization haematoxylin and eosin staining and MAP2 immunoreaction14 were used; to evaluate a possible involvement of meningeal BM, immunoreaction for laminin was carried out (this marker labels the ECM of BM and also wall blood vessels).15 Simultaneously, GFAP antibody was used to understand the possible consequences of disorganized BM,1 since it is an important anchor point for astrocyte end-feet, resulting fundamental during brain development, because GFAP-containing fibers provide a migratory scaffold for neurons.10
Materials and Methods
Dark-like mice and their wild-type (WT) littermates (C3H/J) used for this study were provided by Prof. T. Gunn (Great Falls, MT, USA). Five animals per genotype, per age were sacrificed at 10 postnatal day (P10), 21 postnatal day (P21), 30 postnatal day (P30), and 60 postnatal day (P60). Mice were deeply anesthetized with an intraperitoneal injection of 35% chloral hydrate (100 µL/100 g b.w.; Sigma, St. Louis, MO, USA); the brain was quickly removed and isolated. The use of mice was approved by the local Pavia City Hall authorities (reference no. 7287/00) and by the Italian Ministry of Health according to Art. 12, Leg. Decree 116/92. All efforts were made to minimize the number of animals and their suffering. The samples were fixed in Carnoy’s solution (6 absolute ethanol/3 chloroform/1 glacial acetic acid) for 48 h with a change with fresh solution after 1 h, then placed in absolute ethanol, in acetone, and then embedded in Paraplast X-tra (Sigma-Aldrich, St. Louis, MO, USA). Brain sections (8 µm thick) were obtained serially in the sagittal plane and collected on silan-coated slides.
Haematoxylin and eosin staining
Paraplast embedded sections were deparaffinized in xylene and rehydrated in a decreasing ethanol series and rinsed in distilled water. Then, sections were immersed for 10 min in Carazzi’s haematoxylin at room temperature, washed in running tap water for 20 min and immersed in eosin for 2 s. Sections were washed in distilled water, dehydrated in ethanol, cleared in xylene, and mounted in Eukitt (Kindler, Freiburg, Germany). Slides were observed with an Olympus BX51 microscope, and images were recorded with an Olympus Camedia C-5050 digital camera and stored on a PC. Corrections to brightness and contrast were made with Paint Shop Pro 7 (Jasc Software Inc.).
Immunoperoxidase staining
Paraplast embedded sections were deparaffinized in xylene, rehydrated in a decreasing ethanol series and rinsed in phosphate-buffered saline (PBS; Sigma). The endogenous peroxidases were suppressed by incubation of sections with 3% H2O2 in 10% methanol in PBS for 7 min. Sections were incubated for 20 min in normal serum at room temperature. Localization of MAP2, was achieved by applying on brain sections a rabbit polyclonal anti-MAP2 (1:250 Santa Cruz Biotechnology, Santa Cruz, CA, USA) in PBS overnight in a dark moist chamber. Thereafter, the sections were sequentially incubated with biotinylated secondary antibodies (anti-rabbit, 1:200; Vector Laboratories, Burlingame, CA, USA) for 30 min and horseradish peroxidase conjugated avidin-biotin complex (Vector Laboratories) for 30 min at room temperature. Then, 0.05% 3,3‘-diaminobenzidine tetrahydrochloride (DAB; Sigma) with 0.01% H2O2 in Tris-HCl buffer (0.05M, pH 7.6) was used as a chromogen. After each reaction step, sections were washed thoroughly in PBS, then dehydrated in ethanol, cleared in xylene, and mounted in Eukitt (Kindler). The slides were observed with an Olympus BX51 microscope, and the images were recorded with an Olympus Camedia C-5050 digital camera and stored on a PC. Corrections to brightness and contrast were made with Paint Shop Pro 7 (Jasc Software Inc). For control staining, some sections were incubated with PBS instead of the primary antibody. No immunoreactivity was present in this condition.
Immunofluorescence reactions
Localization of laminin and GFAP was achieved by applying on Paraplast-embedded sections a rabbit polyclonal anti-laminin (1:100; Sigma) and a goat polyclonal anti-GFAP (1:100; Santa Cruz Biotechnology) in PBS overnight in dark moist chamber. Sections were washed in PBS and incubated for 1 h with secondary antibodies Alexa Fluor 488-conjugated anti-rabbit (1:100; Molecular Probes, Milan, Italy) and Alexa Fluor 594-conjugated anti-rabbit (1:100; Molecular Probes). After washing with PBS, nuclei were counterstained with 0.1 µg/mL Hoechst 33258 for 6 min and coverslips were lastly mounted in a drop of Mowiol (Calbiochem, San Diego, CA, USA). Slides were observed by fluorescence microscopy with an Olympus BX51 equipped with a 100W mercury lamp used under the following conditions: 450-480 nm excf, 500 nm dm, and 515 nm bf for Alexa 488 and 540 nm excf, 480 nm dm, and 620 nm bf for Alexa 594. Images were recorded with an Olympus Camedia C-5050 digital camera and stored on a PC. Images were optimized for colour, brightness and contrast by using Paint Shop Pro 7 software (Jasc Software Inc).
For control staining, some sections were incubated with PBS instead of the primary antibodies. No immunoreactivity was present in these sections.
Results
In Figure 1, a panel of the most relevant features are presented. Compared to WT, alterations here described were not strictly age-specific or genotype-specific (dal/+ and dal/dal). As it is shown, WT mice presented a smooth cerebral cortex (a,c), while the altered phenotype was characterized by undulations usually localized above the hippocampal formation, where the overall organization was compromised (b,d). In correspondence to these anomalies the normal layering seems to be damaged, as well as the dendrite organization and positioning marked by the MAP2 immunoreaction. In WT, neuron dendrites ran linear and parallel each other (insert in a), while in altered phenotype (insert in b) they followed cortex undulations. The immunopositivity for laminin remarked a completely different profile: BM was continuous in WT (c and insert), and interrupted and thinner in dal/+ and dal/dal (d and insert). Defects in cerebellar cortex were evident regarding lobulation in dal/+ and dal/dal mice (f,h) compared to WT (e,g); protruding cortical portions with a lack of laminin immunopositive BM were also found (h, arrow). These detachments of molecular layer were accompanied by penetration of large blood vessels (inserts in f and h, asterisk) and altered GFAP-positive fibers whose endfeet were not visible and did not reach the surface of the cerebellar cortex (insert in h, arrowhead). On the other hand, in WT the meningeal BM appears continuous shearing uniform thickness (insert in g) with well structured GFAP-positive fibers anchored, through their endfeet, to the laminin-positive BM layer (insert in g, arrowhead). The spectrum of alterations include also the absence of granule cells (insert in f, arrow) or their ectopic position surrounded by molecular layer (insert in f, arrowhead), fusion of cerebellar fissures, and presence of degenerating cells (not shown).
Figure 1.
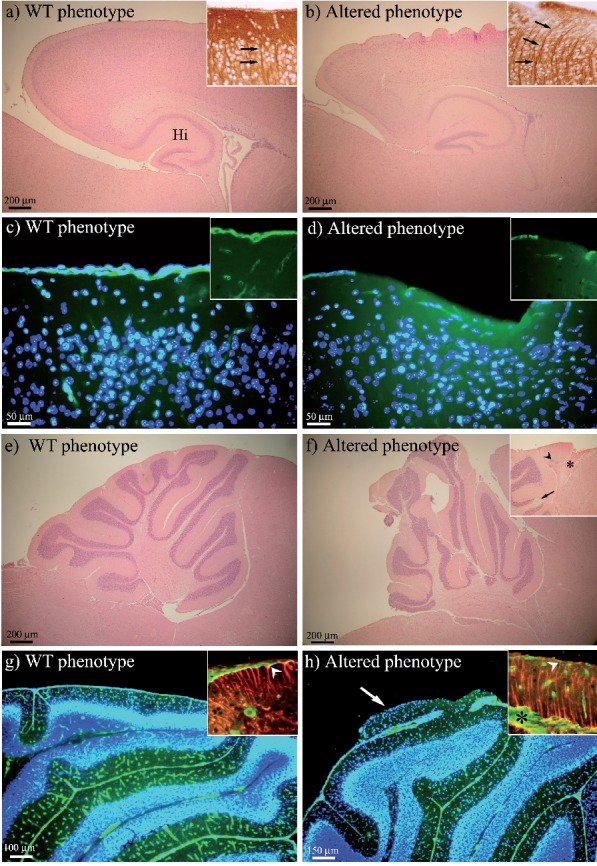
H&E staining, and MAP2, laminin (green fluorescence) and GFAP (red fluorescence) immunoreactions. In panel (a) H&E staining shows the WT lissencephaly, while the altered phenotype is shown in panel (b). In the cerebral cortex, MAP2 immunoreaction (insets of a and b, arrows) evidences the run of neuron dendrites and their positioning. Laminin immunopositivity highlights differences of cortical meningeal BM between WT (c and inset) and altered phenotype (d and inset). The WT cerebellum morphology is illustrated in panel (e), while the altered phenotype in panel (f), where the insertion points to the abnormal vascularization (asterisk), ectopic cells (arrowhead) and absence of granule cells (arrow). In the cerebellum, laminin immunopositivity brings out BM as a continuous layer in WT and small blood vessels within the molecular layer (g). The altered phenotype displays cortical abnormality with lack of BM and large blood vessels penetrating the molecular layer (h). GFAP immunopositive fibers and their endfeet are shown to reach the cerebellar surface only in WT (red fluorescence in the inset of panel g, arrowhead), while in altered phenotype (inset in h, arrowhead) they are disorganized and the endfeet are not detectable. In panels (c, d, g, h) nuclei are counterstained with Hoechst 33258 (blue fluorescence). Altered phenotype, dal/+and dal/dal, mice; Hi, hip-pocampal formation.
Discussion
PD is a rare autosomal recessive disorder caused by a mutation in the PEPD gene, which results in a broad spectrum of phenotype including mental retardation.3 Therefore, we focused our attention on brain morphology and possible factors involved in these anomalies. The presence of vascularisation defects in the cerebral and cerebellar cortices probably is linked to two different causes: i) embryonic cardiomyocyte hypertrophy in dal mutant mice;13 ii) prolidase role in angiogenic signalling processes.17 The cardiomyocyte hypertrophy results, physiologically, in net induction of angiogenesis,18 and as it is reported by previous studies,19,20 this can obstruct the normal migration of cells in the central nervous system (CNS), as we also detected. Moreover, prolidase is involved in ECM remodelling,5 which acts as scaffold for cellular support in all tissues and organs, and plays crucial roles during development of the CNS in the spatial organization of molecules and cells.2 ECM, with its constituents, is also known to contribute to other neuronal functions such as cell proliferation, migration, morphological differentiation, synaptogenesis, synaptic stability, and cell signalling cascades.4,6,22 The absence of a fully functional prolidase enzyme might result in an altered ECM remodelling leading to meningeal defects, one of the possible causes of the brain development impairments,23 the loss of pial BM integrity and the consequent disorganized cytoarchitecture in dal mutant mice.
Further studies will be directed to understand the peculiar role of prolidase in brain development, and to investigate other meningeal BM components. We will focus also on the detection of important molecules and cells involved in the proliferation, migration and differentiation pathways during neurogenesis. Our experimental findings in dal mutant mice with inherited PD underline the importance of prolidase, being relevant also for understanding PD physiology in humans.
Acknowledgments
The authors would like to thank Prof. Graziella Bernocchi and Dr. Maria Grazia Bottone for their extensive scientific mentoring and their constructive advices on the study of brain development.
References
- 1.Lupi A, Tenni R, Rossi A, Cetta G, Forlino A.Human prolidase and prolidase deficiency: an overview on the characterization of the enzyme involved in proline recycling and on the effects of its mutations. Amino Acids 2008;35:739-52 [DOI] [PubMed] [Google Scholar]
- 2.Kiratli H, Satilmiᅟ M.Prolidase deficiency associated with pathologic myopia. Ophthalmic Genet 1998;19:49-53 [DOI] [PubMed] [Google Scholar]
- 3.Royce PM, Steinmann B.Prolidase deficiency. New York, Wiley-Liss: 2002 [Google Scholar]
- 4.Berrier AL, Yamada KM.Cell–Matrix Adhesion. J Cell Physiol 2007;213:565-73 [DOI] [PubMed] [Google Scholar]
- 5.Hu CA, Phang JM, Valle D.Proline metabolism in health and disease. Preface. Amino Acids 2008;35:651-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McRae PA, Porter BE.The perineuronal net component of the extracellular matrix in plasticity and epilepsy. Neurochem Int 2012;61:963-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halfter W, Dong S, Yip YP, Willem M, Mayer U.A critical function of the pial basement membrane in cortical histogenesis. J Neurosci 2002;22:6029-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Timpl R.Structure and biological activity of basement membrane proteins. Eur J Biochem 1989;180:487-502 [DOI] [PubMed] [Google Scholar]
- 9.Aumailley M.The laminin family. Cell Adh Migr 2013;7:48-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Decimo I, Fumagalli G, Berton V, Krampera M, Bifari F.Meninges: from protective membrane to stem cell niche. Am J Stem Cells 2012;1:92-105 [PMC free article] [PubMed] [Google Scholar]
- 11.Hecht JH, Siegenthaler JA, Patterson KP, Pleasure SJ.Primary cellular meningeal defects cause neocortical dysplasia and dyslamination. Ann Neurol 2010;68:454-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zarbalis K, Choe Y, Siegenthaler JA, Orosco LA, Pleasure SJ.Meningeal defects alter the tangential migration of cortical interneurons in Foxc1hith/hith mice. Neural Dev 2012;7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung S, Silvius D, Nolan KA, Borchert GL, Millet YH, Phang JM, et al. Developmental cardiac hypertrophy in a mouse model of prolidase deficiency. Birth Defects Res A Clin Mol Teratol 2011;91:204-17 [DOI] [PubMed] [Google Scholar]
- 14.Yamaguchi S, Fujii-Taira I, Murakami A, Hirose N, Aoki N, Izawa EI, et al. Up-regulation of microtubule-associated protein 2 accompanying the filial imprinting of domestic chicks (Gallus gallus domesticus). Brain Res Bull 2008;76:282-8 [DOI] [PubMed] [Google Scholar]
- 15.Qiu Z, Cang Y, Goff SP.Abl family tyrosine kinases are essential for basement membrane integrity and cortical lamination in the cerebellum. J Neurosci 2010;27:14430-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siegenthaler JA, Pleasure SJ.We have got you ‘covered’: how the meninges control brain development. Curr Opin Genet Dev 2011; 21:249-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhodes JM, Simons M.The extracellular matrix and blood vessel formation: not just a scaffold. J Cell Mol Med 2007;11:176-205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laughlin MH, Bowles DK, Duncker DJ.The coronary circulation in exercise training. Am J Physiol Heart Circ Physiol 2012;302: 10-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akbarian S, Kim JJ, Potkin SG, Hetrick WP, Bunney WE, Jones EG.Maldistribution of interstitial neurons in prefrontal white matter of the brains of schizophrenic patients. Arch Gen Psychiatry 1996;53: 425-36 [DOI] [PubMed] [Google Scholar]
- 20.Chevassus-au-Louis N, Represa A.The right neuron at the wrong place: biology of heterotopic neurons in cortical neuronal migration disorders, with special reference to associated pathologies. Cell Mol Life Sci 1999;55:1206-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pearlman AL, Sheppard AM.Extracellular matrix in early cortical development. Prog Brain Res 1996;108:117-34 [PubMed] [Google Scholar]
- 22.Hensch TK.Controlling the critical period. Neurosci Res 2003;47:17-22 [DOI] [PubMed] [Google Scholar]
- 23.Cerri S, Piccolini VM, Bernocchi G.Postnatal development of the central nervous system: anomalies in the formation of cerebellum fissures. Anat Rec (Hoboken) 2010;293:492-501 [DOI] [PubMed] [Google Scholar]