

Abstract
Despite major improvements concerning its diagnosis and treatment, pancreatic ductal adenocarcinoma (PDAC) remains an aggressive disease with an extremely poor prognosis. Pathology, as interface discipline between basic and clinical medicine, has substantially contributed to the recent developments and has laid the basis for further progress. The definition and classification of precursor lesions of PDAC and their molecular characterization is a fundamental step for the potential identification of biomarkers and the development of imaging methods for early detection. In addition, by integrating findings in humans with the knowledge acquired through the investigation of transgenic mouse models for PDAC, a new model for pancreatic carcinogenesis has been proposed and partially validated in individuals with genetic predisposition for PDAC. The introduction and validation of a standardized system for pathology reporting based on the axial slicing technique has shown that most pancreatic cancer resections are R1 resections and that this is due to inherent anatomical and biological properties of PDAC. This standardized assessment of prognostic relevant parameters represents the basis for the successful conduction of multicentric studies and for the interpretation of their results. Finally, recent studies have shown that distinct molecular subtypes of PDAC exist and are associated with different prognosis and therapy response. The prospective validation of these results and the integration of molecular analyses in a comprehensive pathology report in the context of individualised cancer therapy represent a major challenge for the future.
Keywords: Pancreatic ductal adenocarcinoma, Precursor lesions, Atypical flat lesion, Molecular subtypes, R1, Pancreatic cancer
Core tip: Despite recent progresses, pancreatic ductal adenocarcinoma (PDAC) remains a disease with poor prognosis. Pathology has given fundamental contributions to these developments. In particular, precursor lesions have been identified and a model for PDAC development has been proposed and validated by molecular studies, which represent the basis for the identification of biomarkers for early diagnosis. A standardized protocol for the post-operative assessment of prognostic relevant parameters, such as the resection margin status, has been developed and has shown a high degree of interlaboratory reproducibility. Finally, the genome-wide analysis of PDAC has led to the identification of distinct molecular subtypes with different therapy response and clinical courses.
INTRODUCTION
In the last two decades major improvements in the understanding of pancreatic ductal adenocarcinoma (PDAC) have been achieved by the scientific community, with extraordinary contributions from all disciplines of life sciences, from genetics and molecular biology to molecular imaging and oncology[1,2]. Milestones of this continuing evolving process are for instance the definition and classification of the precursor lesions of PDAC, such as the microscopic pancreatic intraepithelial neoplasia (PanIN)[3] and the larger intraductal papillary mucinous neoplasm (IPMN), which have paved the way for further studies concerning the natural history[4] and the molecular characterization of these lesions, leading to the exploitation of new and more sensitive imaging methods for the purpose of early diagnosis[5]. The isolation and characterization of pancreatic stellate cells[6,7] and hereby the identification of the cancer-associated desmoplastic reaction as an active player in affecting deleterious properties of PDAC, such as its migratory and invasive ability and its capability to adapt to a hypoxic microenvironment[8-10] have also been subjects of intense research activities in the last years. In 2003 the first transgenic mouse model that faithfully recapitulates the development of PDAC from low-grade precursors (so-called PanIN1) to metastatic cancer has been generated and rendered available to the scientific community[11]. Numerous further mouse models have been generated and characterized ever since[12]. These models represent useful instruments to improve our knowledge of the molecular mechanisms and the cellular interactions regulating PDAC initiation and progression, as well as providing an indispensable platform for the development of molecular tracers[13] and for drug testing[14,15]. Following the complete sequencing of the human genome[16] and the advancement of high-throughput molecular methods, the genetic complexity of PDAC has been addressed, culminating in the sequencing of its genome and the identification of the most relevant genetic alterations and molecular pathways of this disease[17]. Pathology (with its branches of anatomical, clinical and molecular pathology) represents the interface between basic research and clinical medicine and facilitates translational research. As such, pathology has been playing a major role in all the above described relevant steps and achievements. In this review, the major contributions of pathology to the improvement of our knowledge and understanding of PDAC in recent years will be addressed, with special focus on clinically relevant, innovative aspects and future challenges.
PRECURSOR LESIONS: EARLY DETECTION OF PANCREATIC CANCER
Morphology and genetics
The classical and well-characterized precursor lesions of PDAC show a ductal phenotype, suggesting a ductal cell of origin of this tumor. The most frequent precursors are PanIN, followed by IPMN and mucinous cystic neoplasms (MCN). PanIN are microscopical (< 5 mm) mucinous-papillary lesions, which lead to invasive carcinoma through an adenoma-carcinoma sequence, in analogy to the Vogelstein’s model of colon carcinogenesis[18]. Accordingly, using modern methods of pyrosequencing and high-resolution melt-curve analysis, it has been shown that virtually all PanIN, including more than 90% of low-grade PanIN, harbor mutations in the KRAS gene locus, followed by CDKN2A/p16, SMAD4 and TP53 mutations in intermediate and later stages of pancreatic carcinogenesis[19-21]. Similarly, both IPMN and MCN give rise to invasive PDAC by stepwise gene alterations. IPMN are the most frequent cystic neoplasms in surgical series and show a different malignant potential depending on their site of origin (main pancreatic duct vs side-branch duct) and their histological subtype[22,23]. The main histopathological, immunophenotypical and clinical characteristics of IPMN are summarized in Table 1. KRAS and GNAS1 mutations[24], represent early genetic alterations whereas TP53 mutations represent late changes in the adenoma-carcinoma sequence of IPMN leading to invasive cancer[20,22,25]. Different histological IPMN subtypes have been associated with different frequency of mutations, with KRAS mutations being particularly associated with the gastric subtype and GNAS1 mutations with the intestinal subtype[24,26]. These different molecular changes probably reflect different pathways of cancer progression. For example it has been reported that KRAS-mutated PDAC often arises in pancreata with low-grade gastric type IPMN, whereas GNAS1-associated intestinal type IPMN are often high-grade lesions which can develop into invasive carcinoma of colloid type[27]. Despite these differences, altogether these data indicate the ductal phenotype and the presence of KRAS mutations as common characteristics of PDAC precursors.
Table 1.
Histopathological, immunophenotypical and clinical characteristics of intraductal papillary mucinous neoplasm (adapted from [22])
| Types and subtypes | Main localization | Sex | Frequency | Microscopic findings |
Immunophenotype |
Association with PDAC | ||||
| MUC1 | MUC2 | MUC5AC | MUC6 | CDX2 | ||||||
| Main-duct type IPMN | ||||||||||
| Intestinal type | Pancreatic head | M(>)F | 36% | Papillae lined by columnar cells | - | + | + | - | + | 34% |
| Pancreatobiliary type | Pancreatic head | M(>)F | 7% | Papillae lined by cuboidal cells | + | - | + | + | - | 58% |
| Oncocytic type | Pancreatic head | M(>)F | 9% | Complexe papillae lined by multiple layer of cells with eosinophilic cytoplasma | + | - | + | + | - | 25% |
| Branch-duct type IPMN | ||||||||||
| Gastric type | Uncinate process | M(>)F | 49% | Gastric foveolar epithelium | - | - | + | - | - | 6% |
IPMN: Intraductal papillary mucinous neoplasm; PDAC: Pancreatic ductal adenocarcinoma.
The generation of transgenic mouse models that closely reproduce the human PanIN/IPMN-PDAC sequence have on one side confirmed the relevance of KRAS as driver gene in PDAC (for reviews about mouse models for PDAC and the role of KRAS[12,28]). On the other side, the ductal origin of PDAC has been challenged, since targeting the ductal compartment by genetic manipulation has failed to generate PanIN and PDAC so far, with the exception of a few PanIN1-like lesions[29-31]. Early progenitor cells, as well as exocrine progenitors and even adult acinar and insulin-producing cells can instead be targeted to generate PanIN lesions thus closely reproducing the development of human PDAC[32]. These studies have raised the hypothesis of an alternative model of pancreatic carcinogenesis, which starts from the centroacinar-acinar compartment and develops into PanIN and PDAC through a metaplasia-dysplasia sequence[33] (Figure 1). According to recent data, it seems now plausible that PDAC can directly originate from the centroacinar-acinar compartment without the intermediate step of PanIN, according to the so-called AFL- (or acinar-ductal) carcinogenesis model[34,35] (Figure 2). The morphological correlate of this model is the atypical flat lesion (AFL). AFL represents the most probable precursor lesion of PDAC in the KrasG12D/+; Ptf1a-Creex1/+ mouse model. AFL are localized in the centroacinar-acinar compartment and display a ductal phenotype, thus potentially originating through a process of acinar-ductal metaplasia. AFL consists of a CK19-positive flat to cuboidal epithelium with enlarged nuclei, an increased nuclear to cytoplasmic ratio, prominent nucleoli and evident mitotic figures. The Ki-67 index is elevated; CDKN2A/p16 is altered through hypermethylation of the corresponding promoter or by intragenic deletion. The surrounding stroma shows a selective overexpression of α-smooth muscle actin, indicating a local activation like that seen in invasive carcinomas[34].
Figure 1.
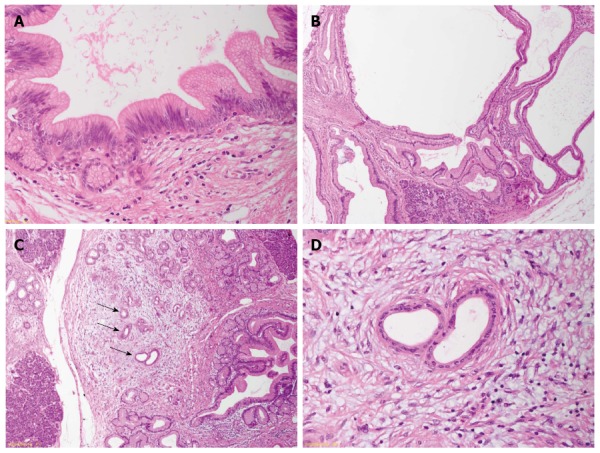
Precursor lesions of pancreatic ductal adenocarcinoma in familial pancreatic cancer-patients. A: Low-grade pancreatic intraepithelial neoplasia; B: Gastric-type intraductal papillary mucinous neoplasm showing a ductal phenotype; C: Acinar-ductal metaplasia in areas of lobulocentric atrophy. Arrows indicate an atypical flat lesion; D: Atypical flat lesion with cellular atypia and typical stromal reaction.
Figure 2.
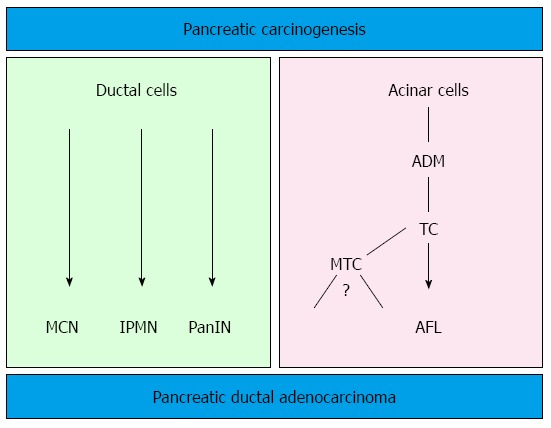
Dual model of pancreatic carcinogenesis. The well-known precursor lesions (PanIN, IPMN and MCN) show a ductal phenotype. However, it seems now plausible that pancreatic ductal adenocarcinoma can directly originate from the centroacinar-acinar compartment through atypical flat lesions without the intermediate step of pancreatic intraepithelial neoplasia (PanIN) (acinar-ductal carcinogenesis). MCN: Mucinous cystic neoplasm; IPMN: Intraductal papillary mucinous neoplasm; ADM: Acinar ductal metaplasia; TC: Tubular complexes; MTC: Mucinous tubular complexes; AFL: Atypical flat lesion.
Relevance for the clinic
The relevance of the above described carcinogenesis models has been confirmed by studying the pancreata of individuals with an elevated risk of developing PDAC during their life. An estimated 10% of all PDAC show a familial background. Several genetic syndromes (Table 2) are known to be associated with an elevated life-time risk for the development of PDAC[25,36-38]. However, specific gene mutations that account for the majority of cases, grouped under the term “familial pancreatic cancer” (FPC), have not been identified. FPC has been described as an autosomal dominant inheritance with high penetrance[37] and the pathology of resection specimens of FPC individuals has been recently described. Ductal precursor lesions such as PanIN and IPMN, especially gastric type IPMN, are a common finding. In comparison with sporadic disease, the number of precursor lesions is higher, they are usually multifocal throughout the organ and they display a higher grade of dysplasia[36,39,40]. Another important finding associated with multifocal PanIN and IPMN is the lobulocentric atrophy[25], a multifocal change consisting of acinar atrophy, fibrosis and acinar-ductal metaplasia. Lesions similar to murine AFL have been recently described in areas of lobulocentric atrophy of FPC individuals undergoing prophylactic pancreatectomy and represent the first evidence of the existence of an AFL-carcinogenesis pathway in humans in addition to the established PanIN/IPMN pathway (Figure 1)[34]. Since lobulocentric atrophy can be identified by endoscopic ultrasound and magnetic resonance tomography, its strong association with PDAC precursors can be used to identify high-risk individuals and to monitor FPC patients in future screening programs.
Table 2.
Genetic syndromes that are associated with an elevated lifetime risk for the development of pancreatic ductal adenocarcinoma (adapted from [25])
| Syndrome | Gene |
| Familial breast cancer | BRCA2 |
| FAMMM syndrome | CDKN2A/p16 |
| Lynch syndrome | MSH2, MLH1, others |
| Heriditary pancreatitis | PRSS1 |
| Peutz-Jeghers syndrome | STK11 |
| Ataxia teleangiectatica | ATM |
Future challenges
The relevance of the described changes for sporadic PDAC has yet to be fully investigated, although it is known that not only PanIN, but also acinar-ductal metaplasia is a common finding in the pancreas of adult patients, independently from the underlying disease[41].
Future studies should aim at further characterizing AFL from the molecular point of view, in order to define specific genetic signatures that may on one side strengthen the model of AFL-carcinogenesis and on the other side help in identifying biomarkers for early detection.
ANATOMICAL AND SURGICAL PATHOLOGY: STANDARDIZED PATHOLOGICAL REPORTING FOR A MORE ACCURATE DEFINITION OF PROGNOSTIC RELEVANT PARAMETERS
Axial slicing technique and the 1-mm rule
The pathology examination procedure plays an important role in the diagnostic and classification of PDAC. The axial slicing technique includes a standardized inking of the fresh or fixed resection specimen according to a pre-defined color code followed by axial slicing perpendicular to the longitudinal axis of the descending duodenum, as previously described[42,43]. After a correct orientation of the specimen, which should follow in close collaboration with the surgical team (i.e., directly in the operating theater or, if not possible, by marking relevant anatomic landmarks with loose sutures), this method is rapid and easy to perform. Moreover, it offers technical advantages compared to other slicing methods, such as the opening of the specimen in a longitudinal manner by exposing the main pancreatic and the bile duct. In fact, the axial slice technique does not depend on the configuration of the pancreatic and the bile duct and it is not impaired by duct obstructions, which are a relative common finding in PDAC resections[44]. All relevant anatomic structures and their relationship to the tumor are easily displayed, the resection margins remain intact before slicing and their distance from the tumor can be easily measured after perpendicular sampling[42].
A striking characteristic of PDAC is its very dispersed and infiltrative type of growth, particularly evident at the tumor periphery, where tumor deposits may be found many mm away from the main tumor bulk (Figure 3). This simple morphological observation has been substantiated by a recent study where the minimum spanning tree analysis was used to calculate tumor cell dispersion in the tumor center and periphery[45]. The following logic consequence of this observation has been the introduction of the so-called 1-mm rule for the assessment of margin clearance in PDAC: in analogy to what has already been validated for the assessment of mesorectal clearance in rectal cancer, a pancreatic resection margin is defined as free (R0) if the tumor cells lies ≥ 1 mm from the margin itself[42,44].
Figure 3.
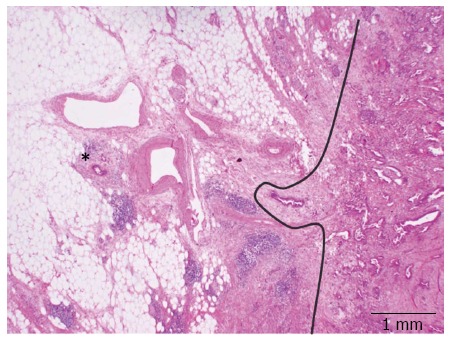
Dispersed growth of pancreatic ductal adenocarcinoma. Tumor deposits (star) of pancreatic ductal adenocarcinoma are found many mm away from the main tumor bulk. The curved line highlights the tumor front.
Relevance for the clinic
It has been shown that the rate of microscopic margin involvement (R-status) is related to the different grossing techniques. In particular, the use of the axial slicing technique and the application of the 1-mm rule lead to a significant higher rate of R1 resections compared to other techniques, with higher reproducibility and smaller deviation of this and other morphology-based parameters[42-44,46-50]. These results bear three important consequences. First of all, they highlight the fact that a high frequency of R1 resections for PDAC is not linked to the surgical techniques but mostly depend on high-quality pathology examination[42]. Second, a standardized pathology report based on a highly reproducible slicing technique allows a fair comparison between results from different institutions, which represents the basis for the execution of multicentric studies[51]. Third, the R-status assessed according to the axial slicing technique becomes a prognostic relevant factor at multivariate analysis that can be used to tailor post-operative treatments[50] according to the concepts of individualized medicine.
Future challenges
The 1-mm rule, although based on morphological evidence of a dispersed type of growth in PDAC, was introduced arbitrarily, mostly in analogy to mesorectal margin assessment in rectal cancer. Some studies have suggested that wider clearances are needed[50,52]. This aspect should be carefully evaluated in prospective studies, in order to define optimal margin clearance for PDAC.
MOLECULAR SUBTYPES OF PDAC: TOWARDS PERSONALIZED CANCER MANAGEMENT
Molecular genetics of PDAC
Recent advances in pancreatic cancer biology have led to the discovery of recurrent gene mutations in KRAS, SMAD4, TP53 and CDKN2A/p16, the identification of core signalling pathways and the definition of molecular subtypes with distinct prognosis and therapy response. In 2008, the first global genomic analysis of 24 advanced PDAC identified 12 core signalling pathways that are genetically altered in the great majority of PDAC. These include apoptosis, DNA damage control, Hedgehog and Wnt/Notch signalling pathways, among others[17]. Interestingly, the affected genes within these pathways highly differed between individual tumors. An average of 63 genetic alterations was found in the cancer cells, most commonly point mutations[17,53]. The new concept that mutations in PDAC are frequent and not restricted to single genes has important implications for treatment strategies: Firstly, the need to target key points and downstream targets of core signalling pathways rather that a single targetable oncogene/tumor suppressor gene. Secondly, the identification of less characterized signalling pathways in PDAC as additional drug targets (e.g., Hedgehog and integrin pathways)[17].
Relevance for the clinic
In 2011, Collisson et al[54] defined three distinct PDAC subtypes based on combined analysis of transcriptional profiles of primary tumor samples and human and mouse PDAC cell lines: classical, quasi-mesenchymal and exocrine-like subtypes. The authors found that stratification by subtype provided prognostic information reflected by the significantly better survival of individuals with classical subtype compared to individuals with quasi-mesenchymal subtype. Further analyses of human PDAC cell lines of known subtype suggested that drug responses differ by subtype: targeted therapies (erlotinib) were more effective in classical subtype, whereas cancer cell lines of the quasi-mesenchymal subtype were more sensitive to conventional chemotherapy (gemcitabine). In addition, targeting inactivated KRAS in classical and quasi-mesenchymal cell lines using RNA interference showed significantly more antiproliferative effect in the classical cell type compared to quasi-mesenchymal cell lines[54]. Likewise, the effect of concurrent inhibition of MEK, a downstream target of KRAS, and EGFR was demonstrated to be restricted to epithelial/classical subtype in a recent study[55]. The clinical relevance of the exocrine subtype, a subtype characterized by high expression of digestive enzymes, remains questionable. Although the exocrine subtype was identified in primary tumor samples, no representative tumor cell line could be identified among the investigated cell line collection, raising the possibility of an contamination artifact with normal pancreatic tissue[54].
The relationship between the four most common mutations of PDAC and the proposed molecular subtypes is largely unknown. However, it is increasingly clear that mutations in KRAS, SMAD4, TP53 and CDKN2A/p16 are “driver” mutations of PDAC, i.e., mutations that confer a selective growth advantage to the tumor cell[56], and they are key players within a complex, and not yet fully understood, network of core pathways[57].
The clinical significance of these driver mutations and intact core pathways has been highlighted by several publications. Among the four driver mutations, KRAS is most commonly mutated in PDAC and exhibits mutations rates up to 95%[58]. Although rare, PDAC patients with non-mutated KRAS tend to have a significantly better median survival than patients with mutated KRAS[59]. Yachida et al[60] investigated the mutation status of KRAS, TP53, CDKN2A/p16 and SMAD4 in an autopsy study of 79 patients and showed that the number of altered genes correlates with a significantly better overall and disease free survival and more indolent disease in patients with only 1 or 2 driver mutations.
Future challenges
These data suggest that the mutation status of KRAS, TP53, CDKN2A/p16 and SMAD4, also referred as the genetic fingerprint of PDAC, might be an integral part of a comprehensive pathology report in the future (Figure 4). Taken together, definition of altered major molecular pathways in PDAC and identification of molecular subtypes have allowed stratification of patients into groups with different biological behaviors.
Figure 4.
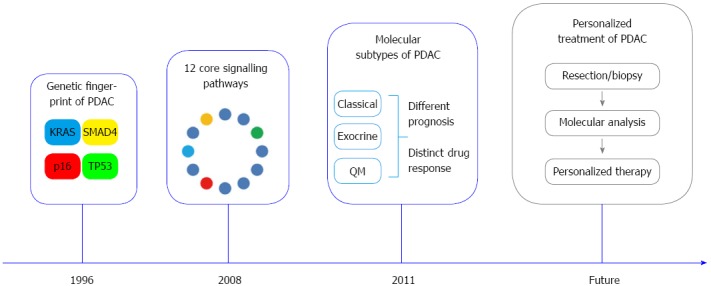
Milestones in pancreatic cancer biology. Mutations of KRAS, CDKN2A/p16, SMAD4 and TP53 represent the molecular fingerprint of pancreatic ductal adenocarcinoma (PDAC)[53]. Global genomic analysis of PDAC defined a set of 12 core signalling pathways commonly altered in the great majority of investigated tumors[17]. Three distinct molecular PDAC subtypes [“classical”, “quasi-mesenchymal” (QM) and “exocrine-like”] with prognostic relevance and distinct drug response were defined based on combined analysis of transcriptional profiles of primary tumor samples and human and mouse PDAC cell lines[54].
CONCLUSION
Despite enormous progresses during the last two decades, PDAC remains a disease with still high mortality rates. Pathology plays a fundamental role as interface discipline between basic research and clinic in improving our knowledge about the development of the disease and in providing tools to improve early diagnosis and personalized treatment. The morphological and molecular characterization of the precursor lesions may help in identifying biomarkers of early disease that could be first tested in high risk individuals and then validated in the general population. The introduction of a highly reproducible standardized pathological reporting method based on the axial slicing technique allows an exact definition of prognostic relevant parameters, such as the R-status, and represents the basis for the comparison of results obtained from multicentric clinical studies. Finally, the foreseeable identification of molecular subtypes of PDAC with different clinical behavior and response to therapy and the integration of these data into routine histopathological diagnosis will help to individualize the treatment of PDAC (Figure 4).
Footnotes
Supported by EU COST Action BM1204 EUPancreas "An integrated european platform for pancreas cancer research: from basic science to cinical and public health interventions for a rare disease"
P- Reviewer: Ijichi H, Li S S- Editor: Gou SX L- Editor: A E- Editor: Ma S
References
- 1.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 2.Hidalgo M. New insights into pancreatic cancer biology. Ann Oncol. 2012;23 Suppl 10:x135–x138. doi: 10.1093/annonc/mds313. [DOI] [PubMed] [Google Scholar]
- 3.Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra DS, Klöppel G, Longnecker DS, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eser S, Messer M, Eser P, von Werder A, Seidler B, Bajbouj M, Vogelmann R, Meining A, von Burstin J, Algül H, et al. In vivo diagnosis of murine pancreatic intraepithelial neoplasia and early-stage pancreatic cancer by molecular imaging. Proc Natl Acad Sci USA. 2011;108:9945–9950. doi: 10.1073/pnas.1100890108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43:128–133. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grünert A, Adler G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115:421–432. doi: 10.1016/s0016-5085(98)70209-4. [DOI] [PubMed] [Google Scholar]
- 8.Erkan M, Adler G, Apte MV, Bachem MG, Buchholz M, Detlefsen S, Esposito I, Friess H, Gress TM, Habisch HJ, et al. StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut. 2012;61:172–178. doi: 10.1136/gutjnl-2011-301220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erkan M, Reiser-Erkan C, Michalski CW, Deucker S, Sauliunaite D, Streit S, Esposito I, Friess H, Kleeff J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia. 2009;11:497–508. doi: 10.1593/neo.81618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paron I, Berchtold S, Vörös J, Shamarla M, Erkan M, Höfler H, Esposito I. Tenascin-C enhances pancreatic cancer cell growth and motility and affects cell adhesion through activation of the integrin pathway. PLoS One. 2011;6:e21684. doi: 10.1371/journal.pone.0021684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 12.Mazur PK, Siveke JT. Genetically engineered mouse models of pancreatic cancer: unravelling tumour biology and progressing translational oncology. Gut. 2012;61:1488–1500. doi: 10.1136/gutjnl-2011-300756. [DOI] [PubMed] [Google Scholar]
- 13.Trajkovic-Arsic M, Mohajerani P, Sarantopoulos A, Kalideris E, Steiger K, Esposito I, Ma X, Themelis G, Burton N, Michalski CW, et al. Multimodal molecular imaging of integrin αvβ3 for in vivo detection of pancreatic cancer. J Nucl Med. 2014;55:446–451. doi: 10.2967/jnumed.113.129619. [DOI] [PubMed] [Google Scholar]
- 14.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grüner BM, Hahne H, Mazur PK, Trajkovic-Arsic M, Maier S, Esposito I, Kalideris E, Michalski CW, Kleeff J, Rauser S, et al. MALDI imaging mass spectrometry for in situ proteomic analysis of preneoplastic lesions in pancreatic cancer. PLoS One. 2012;7:e39424. doi: 10.1371/journal.pone.0039424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigó R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 17.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 19.Sipos B, Frank S, Gress T, Hahn S, Klöppel G. Pancreatic intraepithelial neoplasia revisited and updated. Pancreatology. 2009;9:45–54. doi: 10.1159/000178874. [DOI] [PubMed] [Google Scholar]
- 20.Zamboni G, Hirabayashi K, Castelli P, Lennon AM. Precancerous lesions of the pancreas. Best Pract Res Clin Gastroenterol. 2013;27:299–322. doi: 10.1016/j.bpg.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730–733.e9. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlitter AM, Esposito I. [Pathology and classification of intraductal papillary mucinous neoplasms of the pancreas] Chirurg. 2012;83:110–115. doi: 10.1007/s00104-011-2181-x. [DOI] [PubMed] [Google Scholar]
- 23.Kosmahl M, Pauser U, Peters K, Sipos B, Lüttges J, Kremer B, Klöppel G. Cystic neoplasms of the pancreas and tumor-like lesions with cystic features: a review of 418 cases and a classification proposal. Virchows Arch. 2004;445:168–178. doi: 10.1007/s00428-004-1043-z. [DOI] [PubMed] [Google Scholar]
- 24.Dal Molin M, Matthaei H, Wu J, Blackford A, Debeljak M, Rezaee N, Wolfgang CL, Butturini G, Salvia R, Bassi C, et al. Clinicopathological correlates of activating GNAS mutations in intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann Surg Oncol. 2013;20:3802–3808. doi: 10.1245/s10434-013-3096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO Classification of Tumours of the Digestive System. IARC: Lyon; 2010. [Google Scholar]
- 26.Mohri D, Asaoka Y, Ijichi H, Miyabayashi K, Kudo Y, Seto M, Ohta M, Tada M, Tanaka Y, Ikenoue T, et al. Different subtypes of intraductal papillary mucinous neoplasm in the pancreas have distinct pathways to pancreatic cancer progression. J Gastroenterol. 2012;47:203–213. doi: 10.1007/s00535-011-0482-y. [DOI] [PubMed] [Google Scholar]
- 27.Ideno N, Ohtsuka T, Kono H, Fujiwara K, Oda Y, Aishima S, Ito T, Ishigami K, Tokunaga S, Ohuchida K, et al. Intraductal papillary mucinous neoplasms of the pancreas with distinct pancreatic ductal adenocarcinomas are frequently of gastric subtype. Ann Surg. 2013;258:141–151. doi: 10.1097/SLA.0b013e31828cd008. [DOI] [PubMed] [Google Scholar]
- 28.di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144:1220–1229. doi: 10.1053/j.gastro.2013.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, Grippo P, Stoffers DA, Silberg DG, Rustgi AK. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63:2005–2009. [PubMed] [Google Scholar]
- 30.Ray KC, Bell KM, Yan J, Gu G, Chung CH, Washington MK, Means AL. Epithelial tissues have varying degrees of susceptibility to Kras(G12D)-initiated tumorigenesis in a mouse model. PLoS One. 2011;6:e16786. doi: 10.1371/journal.pone.0016786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JP, Pan FC, Akiyama H, Wright CV, Jensen K, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:737–750. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maitra A, Leach SD. Disputed paternity: the uncertain ancestry of pancreatic ductal neoplasia. Cancer Cell. 2012;22:701–703. doi: 10.1016/j.ccr.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murtaugh LC, Leach SD. A case of mistaken identity? Nonductal origins of pancreatic “ductal” cancers. Cancer Cell. 2007;11:211–213. doi: 10.1016/j.ccr.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 34.Aichler M, Seiler C, Tost M, Siveke J, Mazur PK, Da Silva-Buttkus P, Bartsch DK, Langer P, Chiblak S, Dürr A, et al. Origin of pancreatic ductal adenocarcinoma from atypical flat lesions: a comparative study in transgenic mice and human tissues. J Pathol. 2012;226:723–734. doi: 10.1002/path.3017. [DOI] [PubMed] [Google Scholar]
- 35.Esposito I, Konukiewitz B, Schlitter AM, Klöppel G. [New insights into the origin of pancreatic cancer. Role of atypical flat lesions in pancreatic carcinogenesis] Pathologe. 2012;33 Suppl 2:189–193. doi: 10.1007/s00292-012-1673-x. [DOI] [PubMed] [Google Scholar]
- 36.Brune K, Abe T, Canto M, O’Malley L, Klein AP, Maitra A, Volkan Adsay N, Fishman EK, Cameron JL, Yeo CJ, et al. Multifocal neoplastic precursor lesions associated with lobular atrophy of the pancreas in patients having a strong family history of pancreatic cancer. Am J Surg Pathol. 2006;30:1067–1076. [PMC free article] [PubMed] [Google Scholar]
- 37.Meckler KA, Brentnall TA, Haggitt RC, Crispin D, Byrd DR, Kimmey MB, Bronner MP. Familial fibrocystic pancreatic atrophy with endocrine cell hyperplasia and pancreatic carcinoma. Am J Surg Pathol. 2001;25:1047–1053. doi: 10.1097/00000478-200108000-00009. [DOI] [PubMed] [Google Scholar]
- 38.Shi C, Hruban RH, Klein AP. Familial pancreatic cancer. Arch Pathol Lab Med. 2009;133:365–374. doi: 10.5858/133.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi C, Klein AP, Goggins M, Maitra A, Canto M, Ali S, Schulick R, Palmisano E, Hruban RH. Increased Prevalence of Precursor Lesions in Familial Pancreatic Cancer Patients. Clin Cancer Res. 2009;15:7737–7743. doi: 10.1158/1078-0432.CCR-09-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matthaei H, Norris AL, Tsiatis AC, Olino K, Hong SM, dal Molin M, Goggins MG, Canto M, Horton KM, Jackson KD, et al. Clinicopathological characteristics and molecular analyses of multifocal intraductal papillary mucinous neoplasms of the pancreas. Ann Surg. 2012;255:326–333. doi: 10.1097/SLA.0b013e3182378a18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esposito I, Seiler C, Bergmann F, Kleeff J, Friess H, Schirmacher P. Hypothetical progression model of pancreatic cancer with origin in the centroacinar-acinar compartment. Pancreas. 2007;35:212–217. doi: 10.1097/mpa.0b013e31805d0190. [DOI] [PubMed] [Google Scholar]
- 42.Esposito I, Kleeff J, Bergmann F, Reiser C, Herpel E, Friess H, Schirmacher P, Büchler MW. Most pancreatic cancer resections are R1 resections. Ann Surg Oncol. 2008;15:1651–1660. doi: 10.1245/s10434-008-9839-8. [DOI] [PubMed] [Google Scholar]
- 43.Verbeke CS, Leitch D, Menon KV, McMahon MJ, Guillou PJ, Anthoney A. Redefining the R1 resection in pancreatic cancer. Br J Surg. 2006;93:1232–1237. doi: 10.1002/bjs.5397. [DOI] [PubMed] [Google Scholar]
- 44.Verbeke CS. Resection margins in pancreatic cancer. Surg Clin North Am. 2013;93:647–662. doi: 10.1016/j.suc.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 45.Verbeke CS, Knapp J, Gladhaug IP. Tumour growth is more dispersed in pancreatic head cancers than in rectal cancer: implications for resection margin assessment. Histopathology. 2011;59:1111–1121. doi: 10.1111/j.1365-2559.2011.04056.x. [DOI] [PubMed] [Google Scholar]
- 46.Menon KV, Gomez D, Smith AM, Anthoney A, Verbeke CS. Impact of margin status on survival following pancreatoduodenectomy for cancer: the Leeds Pathology Protocol (LEEPP) HPB (Oxford) 2009;11:18–24. doi: 10.1111/j.1477-2574.2008.00013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campbell F, Smith RA, Whelan P, Sutton R, Raraty M, Neoptolemos JP, Ghaneh P. Classification of R1 resections for pancreatic cancer: the prognostic relevance of tumour involvement within 1 mm of a resection margin. Histopathology. 2009;55:277–283. doi: 10.1111/j.1365-2559.2009.03376.x. [DOI] [PubMed] [Google Scholar]
- 48.Jamieson NB, Foulis AK, Oien KA, Going JJ, Glen P, Dickson EJ, Imrie CW, McKay CJ, Carter R. Positive mobilization margins alone do not influence survival following pancreatico-duodenectomy for pancreatic ductal adenocarcinoma. Ann Surg. 2010;251:1003–1010. doi: 10.1097/SLA.0b013e3181d77369. [DOI] [PubMed] [Google Scholar]
- 49.Jamieson NB, Chan NI, Foulis AK, Dickson EJ, McKay CJ, Carter CR. The prognostic influence of resection margin clearance following pancreaticoduodenectomy for pancreatic ductal adenocarcinoma. J Gastrointest Surg. 2013;17:511–521. doi: 10.1007/s11605-012-2131-z. [DOI] [PubMed] [Google Scholar]
- 50.Chang DK, Johns AL, Merrett ND, Gill AJ, Colvin EK, Scarlett CJ, Nguyen NQ, Leong RW, Cosman PH, Kelly MI, et al. Margin clearance and outcome in resected pancreatic cancer. J Clin Oncol. 2009;27:2855–2862. doi: 10.1200/JCO.2008.20.5104. [DOI] [PubMed] [Google Scholar]
- 51.Katz MH, Merchant NB, Brower S, Branda M, Posner MC, William Traverso L, Abrams RA, Picozzi VJ, Pisters PW. Standardization of surgical and pathologic variables is needed in multicenter trials of adjuvant therapy for pancreatic cancer: results from the ACOSOG Z5031 trial. Ann Surg Oncol. 2011;18:337–344. doi: 10.1245/s10434-010-1282-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagakawa T, Nagamori M, Futakami F, Tsukioka Y, Kayahara M, Ohta T, Ueno K, Miyazaki I. Results of extensive surgery for pancreatic carcinoma. Cancer. 1996;77:640–645. [PubMed] [Google Scholar]
- 53.Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, Goodman SN, Sohn TA, Hruban RH, Yeo CJ, et al. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997;57:1731–1734. [PubMed] [Google Scholar]
- 54.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–503. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mirzoeva OK, Collisson EA, Schaefer PM, Hann B, Hom YK, Ko AH, Korn WM. Subtype-specific MEK-PI3 kinase feedback as a therapeutic target in pancreatic adenocarcinoma. Mol Cancer Ther. 2013;12:2213–2225. doi: 10.1158/1535-7163.MCT-13-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iacobuzio-Donahue CA. Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project. Gut. 2012;61:1085–1094. doi: 10.1136/gut.2010.236026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iacobuzio-Donahue CA, Velculescu VE, Wolfgang CL, Hruban RH. Genetic basis of pancreas cancer development and progression: insights from whole-exome and whole-genome sequencing. Clin Cancer Res. 2012;18:4257–4265. doi: 10.1158/1078-0432.CCR-12-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rachakonda PS, Bauer AS, Xie H, Campa D, Rizzato C, Canzian F, Beghelli S, Greenhalf W, Costello E, Schanne M, et al. Somatic mutations in exocrine pancreatic tumors: association with patient survival. PLoS One. 2013;8:e60870. doi: 10.1371/journal.pone.0060870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yachida S, White CM, Naito Y, Zhong Y, Brosnan JA, Macgregor-Das AM, Morgan RA, Saunders T, Laheru DA, Herman JM, et al. Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long-term survivors. Clin Cancer Res. 2012;18:6339–6347. doi: 10.1158/1078-0432.CCR-12-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]