

Abstract
Selenophosphate synthetase catalyzes the synthesis of selenophosphate which is a selenium donor for Sec biosynthesis. In Drosophila melanogaster, there are two types of selenophosphate synthetases designated dSPS1 and dSPS2, where dSPS2 is a selenoprotein. The mechanism of gene expression of dSPS2 as well as other selenoproteins in Drosophila has not been elucidated. Herein, we report an essential regulator system that regulates the transcription of the dSPS2 gene (dsps2). Through deletion/substitution mutagenesis, the downstream DNA replication-related element (DRE) located at +71 has been identified as an essential element for dsps2 promoter activity. Furthermore, double-stranded RNA interference (dsRNAi) experiments were performed to ablate transcription factors such as TBP, TRF1, TRF2 and DREF in Schneider cells. The dsRNAi experiments showed that dsps2 promoter activities in DREF- and TRF2-depleted cells were significantly decreased by 90% and 50%, respectively. However, the depletion of TBP or TRF1 did not affect the expression level of dsps2 even though there is a putative TATA box at –20. These results strongly suggest that the DRE/DREF system controls the basal level of transcription of dsps2 by interacting with TRF2.
INTRODUCTION
Selenium is a trace element that has been associated with cell proliferation, cancer, development and aging (1). This element occurs in animals primarily in selenium-containing proteins as the amino acid selenocysteine (Sec). Twenty-five selenoprotein genes have been identified in humans and swine and 24 in rodents (2). These genes code for selenoproteins such as glutathione peroxidase, selenoprotein P, selenoprotein W, thioredoxin reductase and thyroid hormone deiodinases. Sec is located at the active center in those selenoproteins whose functions have been characterized and it plays a major role in the enzyme’s function. The primary functions of selenoproteins are to play a role as antioxidants or in the regulation of cellular redox balance (3).
Selenoprotein biosynthesis in bacteria requires the participation of four gene products (4): Sec synthetase (SelA), a Sec-specific translation elongation factor (SelB), a Sec-tRNA (SelC) and selenophosphate synthetase (SelD). The mechanism of selenoprotein synthesis in eukaryotes, however, is only partially elucidated (5). Selenophosphate synthetase (SPS) catalyzes the synthesis of monoselenophosphate, AMP and orthophosphate (6). Selenophosphate is required for both the biosynthesis of Sec in selenium-dependent enzymes and for the conversion of 2-thiouridine residues to 2-selenouridine in tRNA (7). Drosophila has two types of SelD homologs named selenophosphate synthetase 1 and selenophosphate synthetase 2. In addition to the biochemical roles of SPS, the Drosophila selenophosphate synthetase 1 gene (dsps1) has been shown to be involved in imaginal disk morphogenesis, brain development and cell proliferation through the cis-control of the cellular redox balance (8,9). Drosophila selenophosphate synthetase 2 (dSPS2) is another SelD homolog found in eukaryotes. dSPS2 is a selenoprotein containing a Sec codon in the predicted active site of the enzyme instead of the arginine codon in dSPS1 (10). Although dSPS2 is anticipated to play similar biochemical roles to dSPS1, its functions in development, regulation mechanism of gene expression and signaling pathways have not been elucidated.
Promoters for mammalian selenoprotein genes such as the genes encoding glutathione peroxidase 1, glutathione peroxidase 2, phospholipid hydroperoxide glutathione peroxidase, thioredoxine reductase 1, selenoprotein W and selenoprotein P have been analyzed (11–17). These mammalian selenoprotein genes contain a TATA box as their core promoter and most mammalian selenoprotein genes require an auxiliary transcription factor such as SP1 (18). However, the mechanism of transcription of the genes involved in selenium metabolism in Drosophila has not been elucidated.
The DNA replication-related element (DRE) is a regulatory element frequently found in the promoters of Drosophila genes involved in cell proliferation (19) or cell cycle regulation (20). DRE sequences have been found within 1 kb upstream of the transcription initiation sites of many genes. A transcription factor DREF (DRE-binding factor) that specifically binds to the DRE sequence has also been isolated as an 80 kDa polypeptide homodimer (19). DREF can serve as an activator to regulate the transcription of the genes carrying DRE in their promoter region. Furthermore, this protein also associates with TRF2 (TBP-related factor 2), which directs promoter-selective expression of Drosophila genes such as PCNA (21). TRF2 has also been designated as either TLP, TLF or TRP (22). Double-stranded RNA interference (dsRNAi) has proven to be a powerful tool for disrupting gene expression in Drosophila both by making targeted gene knockdown flies (23) and by introducing dsRNA into cultured cells (24). In the present study, we identified the DRE/DREF system as an essential complex for the transcription of dsps2 by deletion/substitution mutation, electrophoretic mobility shift assays and dsRNAi knockdown of the expression of specifically targeted transcription factors (TFs). We introduced dsRNA directly into cultured Drosophila cells to knock down the expression of TFs and analyzed the promoter activity of dsps2 in TF-depleted cells. It was found that TRF2 was also required for the transcription of dsps2.
MATERIALS AND METHODS
Oligonucleotides
Oligonucleotides and their uses were as follows: (i) 5′-CGCGAATTTCTACGCACCGG-3′ was used as a primer for sequencing in RNase protection assays (RPA); (ii) 5′-GCTAGTGACGGCACTTCAACACTTATCG-3′ was used in primer extension experiments to determine the transcriptional initiation site; (iii) 5′-TACTAGGAATATCGATA AGTGTTGA-3′ (wtDRE), 5′-GCAGCTTCCGCGATCG CCTGTGGTC-3′ (non-specific competitor), 5′-TACTA TTCCTATCGATAAGTGTTGA-3′ (mNDRE 1), 5′-TACTA GGAAGCGAGATAAGTGTTGA-3′ (mDRE 1), 5′-TACTA GGAATATCTCGCAGTGTTGA-3′ (mDRE 2), 5′-TACTA GGAATATCGATACTGGTTGA-3′ (mNDRE 2) and 5′- TACTAGGAATCGATCTAAGTGTTGA-3′ (mut 3) were used to make double-stranded oligonucleotides used in electrophoretic mobility shift assays (EMSA) as probes or competitors; (iv) 5′-AATTAACCCTCACTAAAGGGAT GGACCAAATGCTAAGCCC –3′ (TBP cDNA forward), 5′-AATTAACCCTCACTAAAGGGTACTTTCTCGCTGCCAGTCT-3′ (TBP cDNA reverse), 5′-AATTAACCCTCACT AAAGGGGGTGGTTCAACAAGTCGTCT-3′ (TRF1 cDNA forward), 5′-AATTAACCCTCACTAAAGGGAATAGGT GATATCGCCTCCA-3′ (TRF1 cDNA reverse), 5′-AATT AACCCTCACTAAAGGGACAAGTCGTCGGTGGTTCAT-3′ (TRF2 cDNA forward), 5′-AATTAACCCTCACTAA AGGGGTGCCCAGTACGTTGACAATD-3′ (TRF2 cDNA reverse), 5′-AATTAACCCTCACTAAAGGGATGAGCG AAGGGGTACCAG-3′ (DREF cDNA forward) and 5′-AATTAACCCTCACTAAAGGGCCTCGTGCTTGATGACACG-3′ (DREF cDNA reverse) were used as primer sequences for making dsRNA.
Antibodies
Anti-TFIID (TBP) antibody was purchased from Santa Cruz. Anti-DREF antibodies Mab-#1 and Mab-#4 were obtained from M. Yamaguchi (25). Anti-actin antibody was purchased from Sigma.
Plasmid construction
An 841 bp of genomic DNA fragment containing the 5′-flanking region of dsps2 was produced by PCR amplification of Drosophila (Oregon R.) genomic DNA using 5′-ACT GACAGATCTCTTAGGAGCAGACATAAATG-3′ and 5′-GACTACAAGCTTGTAAACTTAGTAAGCTGAAA-3′ as primers, and subcloned into the BglII/HindIII restriction sites of the pGL3B vector (Promega). This plasmid was designated pGL3B-SPS2P. For RPA, the Drosophila genomic DNA insert in pGL3B-SPS2P was subcloned into the pBluscript II KS(+) vector (pBluescript-SPS2P). Internally deleted DNA fragments covering sequences from +21 to +82 were generated by ligating different combinations of 5′- and 3′- truncated fragments amplified by PCR and cloning into the BglII/HindIII restriction sites of pGL3B vector. Linker substitution mutants were generated by substituting six nucleotides at the target site with a BglII restriction sequence. DNAs encoding the upstream region and the downstream region from each target site were amplified by PCR. The forward primer for the upstream fragment has a XhoI restriction site and the reverse primer for the upstream fragment has a BglII restriction site which is the substitution of the six target nucleotides. The forward primer for the downstream fragment contains a BglII restriction site that is also the substitution of the same target sequence and the reverse primer for downstream fragment has a HindIII restriction site. The amplified DNAs were digested with BglII and ligated. The ligated fragment was redigested with XhoI and HindIII and cloned into pGL3B. Base substitution mutant plasmids were constructed from mutant DNA fragments generated in PCR-based mutagenesis reactions by using two sets of forward and reverse primers. The plasmid pT-luc (26) was used as a parental plasmid for constructing pTluc-sps2p(wt) and pTluc-sps2p(mDRE1). The 841 bp promoter regions of dsps2 containing the wild type DRE or mutant DRE1 was cloned into the BamHI site of pT-luc. TBP-luc and TFIIS-luc were described previously (27,28).
RNase protection assay
The antisense RNA probe was prepared by transcribing pBluescript II KS(+)-SPS2P using T7 RNA polymerase, after digesting the plasmid with HincII and then labeling with [α-32P]UTP (3000 Ci/mmol). Labeled RNA (5 × 105 c.p.m.) was hybridized to total RNA purified from adult Drosophila (Oregon R.). The hybridization reaction was carried out in a mixture containing 40 mM PIPES pH 6.4, 1 mM EDTA, 0.4 M NaCl and 80% formamide for 12–18 h at 50°C. Following hybridization the sample was digested with RNase digestion buffer containing 300 mM NaCl, 10 mM Tris–Cl pH 7.4, 5 mM EDTA pH 7.5 and 10 µl of RNase A/T1 mix (2 mg/ml RNase A, 5000 U/ml RNase T1) for 1 h at 30°C. This reaction was terminated by adding 0.6% SDS and 50 µg of proteinase K. The reaction was then extracted with phenol–chloroform and precipitated with ethanol, and the products were analyzed on a 4% polyacrylamide gel containing 7 M urea.
Primer extension
A synthetic oligonucleotide complementary to nucleotide positions +74 to +101 of the dsps2 was 5′-end-labeled with [γ-32P]ATP using T4 polynucleotide kinase. Total RNA isolated from adult Drosophila was mixed with 2 × 106 c.p.m. of labeled primer in 30 µl of hybridization buffer containing 40 mM PIPES pH 6.4, 1 mM EDTA, 0.4 M NaCl and 80% formamide. After denaturation, the RNA–primer mixture was hybridized for 12 h at 30°C and precipitated with ethanol. The extension process was carried out at 42°C for 1 h by adding Moloney murine leukemia virus (M-MLV) reverse transcriptase. The extended product was then ethanol precipitated and analyzed on a sequencing gel consisting of 6% polyacrylamide/7 M urea and a sequencing standard using the same primer was also loaded on the same gel.
DNA transfection and luciferase assay
Drosophila Schneider cells (SL2) were cultured at 25°C in HyQ SFX-INSECT media (HyClone). Schneider cells were plated at 2 × 106 cells per well in a six-well plate and cultured for 24 h before DNA transfection using Lipofectin reagent (Invitrogen). Each transfection includes 3 µg of wild-type or mutant plasmids. For normalization, all of the constructs were cotransfected with 0.6 µg of pG5E16-CAT and MT-GVP, each of which expresses chloramphenicol acetyl transferase (CAT) and its activator. Transfection was performed for 24 h and then the media was replenished. Cells were harvested 72 h after transfection and luciferase assay was carried out by means of a Luciferase Reporter Assay System (Promega). All transient transfection data represented relative activities that were compared with the wild-type activity and each transfection was performed in triplicate.
Preparation of nuclear extracts and EMSA
Nuclear extracts were prepared from Drosophila Schneider cells (SL2) as previously described (29). The GST-DREF (1–125) (30) fusion protein was expressed from a pGEX-DREF(1–125) clone in Escherichia coli BL21, and purified as previously described (30). Thirty femtomoles of labeled probe was incubated in a 20 µl reaction mixture containing 1 µg of BSA, 5 mM DTT, 0.5 µg of salmon sperm DNA, 1 mM EDTA, 105 mM KCl and 10 µg of SL2 nuclear extract or 100 ng of purified GST-DREF (1–125) fusion protein at 25°C for 15 min. In competition experiments, unlabeled competitors were added to the reaction in amounts 20–50× molar excess of the probes.
Double-stranded RNA interference
To generate double-stranded RNAs of the transcription factors, cDNA fragments ∼600–700 bp in length were amplified by PCR with forward and reverse primers containing T3 promoter sequence tag (AATTAACCCTC ACTAAAGGG) (see Oligonucleotides). The amplified cDNA fragments were employed as templates with T3 Megascript kit (Ambion) to transcribe in both directions. Both sense and antisense strand RNA were annealed as follows: The RNA products were resuspended in 2× annealing buffer (40 mM HEPES–KOH pH 7.6, 10 mM EDTA pH 8.0) and annealed by incubation at 95°C for 5 min followed by slow cooling to room temperature. The annealed products were then extracted with acid phenol–chloroform–IAA followed by ethanol precipitation. The RNA pellets were resuspended in sterile water and analyzed by 1% agarose gel electrophoresis to confirm that the dsRNA products were produced.
To introduce dsRNAs into cells, 1 × 106 SL2 cells were plated on a 24-well plate in 400 µl of HyQ SFX-INSECT media (HyClone). dsRNAs were added directly to the media to a final concentration of 37 nM and incubated for 8 h (24). The cells were transfected with CAT- and luciferase-reporter-containing vectors, harvested in 40 h and the promoter activity assayed by measuring luciferase activity. Luciferase activity was normalized with CAT activity.
Northern blotting
Total RNA was isolated from SL2 cells using TRIzol Reagent (Invitrogen) according to the manufacturer’s instructions. For northern blot analysis, 25 µg of total RNA was loaded on a 1% agarose–formaldehyde gel and transferred onto a HybondN+ membrane (Amersham). The cDNAs of TBP, TRF1, TRF2 and DREF were used as probes and these sequences corresponded to the C-terminal 500 bp of each gene, which did not include the sequences for dsRNA production. Ribosomal protein 49 (Rp-49) cDNA was used as an internal control. The filters were hybridized overnight, washed and autoradiogrammed.
Western blot analysis
Cells were lysed in 1× reporter lysis buffer (Invitrogen). Proteins were separated by SDS–polyacrylamide gel electrophoresis (8%) and transferred to a Hybond-ECL nitrocellulose membrane (Amersham). The membrane was blocked with TBST (50 mM Tris–Cl pH 8.5, 150 mM NaCl, 0.1% Tween 20) containing 5% low-fat dried milk (Sigma) for 1 h at room temperature, and then incubated with an appropriate antibody for 1 h at room temperature. After washing with TBST, the membrane was incubated with goat anti-mouse IgG (Sigma) or goat anti-rabbit IgG (Sigma) for 1 h at room temperature. The signal was detected using an ECL kit (Amersham).
RESULTS
Cloning and sequencing of 5′-flanking region of dsps2
The 5′-flanking region of dsps2, which was 854 bp in length, was PCR amplified from genomic DNA of Drosophila (Oregon R.), and subcloned into the pGL3B vector. Ten independently cloned plasmids were then sequenced to exclude the possibility of mutagenesis which might be produced during PCR. By analyzing the sequence, we found four nucleotides located upstream and downstream from the translation initiation point that were different from the genomic sequence published previously (31) (Fig. 1). One plasmid containing a typical sequence was desiganted pGL3B-SPS2P. Two putative TATA box sequences (5′-TATAAA) were found at nucleotide positions between –77 and –72 and between –23 and –18.
Figure 1.

Nucleotide sequence of the 5′-flanking region of dsps2. The transcription start site is italicized and described as +1. The underlined nucleotide sequences indicate putative TATA boxes (TATAAA). Nucleotides in the open boxes indicate the nucleotide sequences different from those previously reported. The DRE consensus sequence is italicized and underlined at +71 and +78. The translational initiation codon is located at positions +165 to +167 and the partial N-terminal amino acid sequence is shown as single amino acid letters.
Determination of transcriptional initiation site of dsps2
RPA was performed prior to the primer extension experiment to measure the length of 5′-UTR of dsps2 mRNA. The antisense RNA probe corresponding to 300 bp of the 5′-flanking sequence was generated by in vitro transcription and hybridized with Drosophila total RNA (Fig. 2A). After hybridization, single-stranded RNA was digested with RNases. A single protected fragment 161 nt long was observed (Fig. 2B, lane 2). The size of protected RNA was estimated by comparison with the DNA sequencing ladder generated on a pBluescript-SPS2P plasmid by RPA primer that contains the same 5′-end as that of the antisense RNA probe used in this experiment (Fig. 2B, left four lanes).
Figure 2.

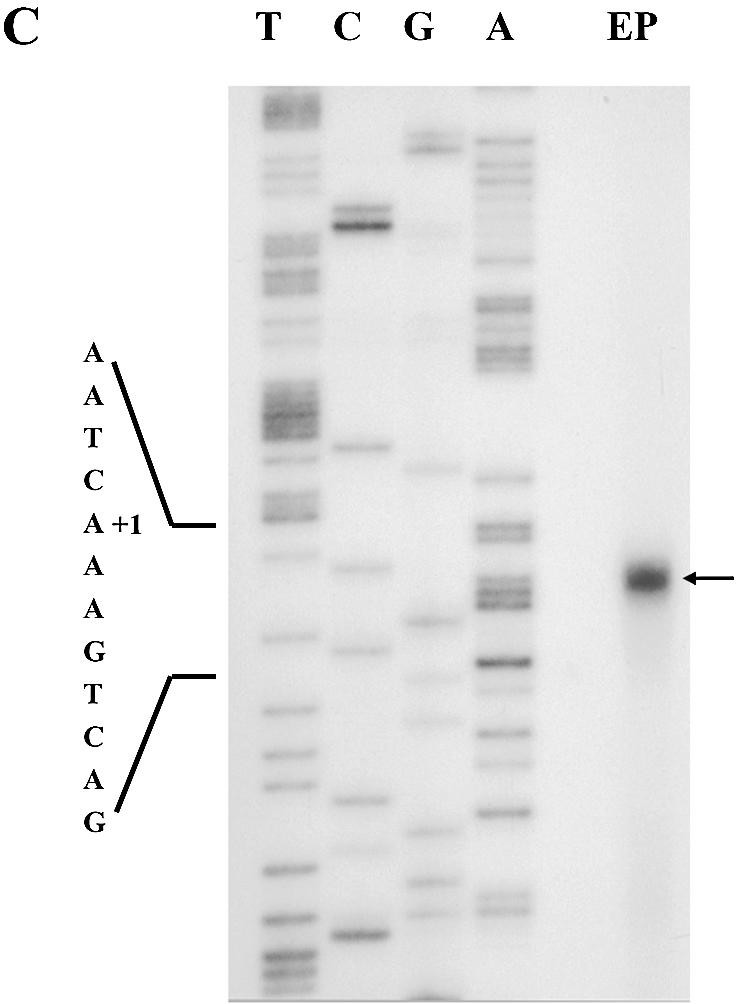
Identification of the transcriptional start site. (A) Schematic representation of the 5′-flanking region of dsps2, the open reading frame (ORF), the 300 bp probe, the 161 bp protected fragment and the primer used for sequencing. (B) Autoradiogram of RPA: lane 1, the probe used for RPA; lane 2, the protected probe from RNaseA/T1 by Drosophila RNA; lane 3, the negative control using yeast RNA. The assay was performed as described in Materials and Methods. The sequence of the 5′-flanking region of dsps2 that was generated by using the RPA primer is shown on the left. (C) Results of primer extension. The arrow designates the extended band in the lane marked as EP (Extended Product). The sequencing reaction surrounding the transcription start site is shown on the left.
In addition to the RPA, primer extension was performed by using a 5′-labeled 28mer antisense primer (corresponding to nucleotide positions +74 to +101) (see Materials and Methods for the sequence) with total RNAs isolated from Drosophila. As shown in Figure 2C, a single 101 bp extended product was detected. By comparing this product with the DNA sequencing ladder generated from the pGL3B-SPS2P plasmid using the same primer, the transcriptional initiation site of dsps2 was determined to be located 164 bp upstream from the initiation site of translation (see Fig. 1). The position of the transcription initiation site as determined by primer extension coincided with that obtained by RPA.
Identification of the dsps2 promoter region
Several deletion mutants were constructed to determine the core promoter of dsps2. By employing exonuclease serial deletion mutagenesis, it was found that the region downstream of the transcriptional initiation point (+21 to +82) was more essential than the upstream region (data not shown). Therefore we examined the promoter activities of two downstream deletion mutants and the putative downstream TATA box mutant (located at –23 to –18) relative to those observed with the wild type (pGL3B-SPS2P) and negative control (pGL3-Basic) sequences. As shown in Figure 3A, DPD2 contained a deletion from +40 to +78 and its promoter activity was <5% of that of wild type. DPD1 contained a deletion from +17 to +41, but its promoter activity appeared to be stimulated compared with that of wild type. Surprisingly, the mutated putative TATA box upstream of dsps2 showed only 40% decreased promoter activity compared with wild type. Similar results were obtained in DPD1-TM and DPD2-TM, which are double mutants at both the TATA box and downstream regions. These results suggest that the downstream region spanning +40 to +80 contains an important element for the transcription of dsps2. Therefore we focused in greater detail by using linker substitution and site-directed mutagenesis. As shown in Figure 3B, linker mutants were generated over the +49 to +88 region at 10-bp intervals and the middle sequences of each interval were substituted with a BglII restriction sequence. These four linker mutant plasmids were then transfected transiently into SL2 cells and their relative promoter activities measured. Promoter activities of linkers 2, 3 and 4 relative to wild type were decreased by 81%, 98% and 68%, respectively. These results suggest that the essential element in dsps2 is located within the region spanning +62 to +91 and presumably the core promoter would be in the linker 3 region, since the latter region exhibited the lowest activity.
Figure 3.


Effect of deletion and base substitution mutagenesis on the promoter activity of dsps2. Each mutant construct was transfected into SL2 cells and their relative promoter activities measured by luciferase assay. (A) Deletion mutant analysis. A series of deletion constructs are shown on the left; their designations are described in the middle and their corresponding luciferase activities on the right. The uppermost construct designated pGL3-Basic is the negative control, the construct designated pGL3B-SPS2P is the wild-type control, wherein +1 shows the transcription start site, and the upstream TM in the box designates the TATA box mutant. (B) Linker mutant analysis. Sequences of linker mutants are shown in the upper portion, wherein the BglII sequence is underlined, and the graph in the lower portion represents their luciferase activities relative to that of the wild-type activity. The standard deviations from the mean are indicated by error bars.
DRE is the essential element of dsps2
Linker 3 (+72 to +81) contains an 8 bp palindromic sequence (+71 to +78; TATCGATA), which is the consensus sequence of DRE. To confirm whether the DRE consensus sequence regulates the transcription of dsps2, a set of base substitution mutations in and around the DRE sequence was constructed. The effects of these mutations on the promoter activity were examined with the luciferase assay system. As shown in Figure 4, plasmid mNDRE1 carrying four base substitutions immediately upstream of the DRE sequence did not affect the transcriptional activity of dsps2, while plasmid mNDRE2 carrying three base substitutions immediately downstream had ∼50% reduced activity compared with wild type. In contrast, plasmids mDRE1 and mDRE2 that contained mutations within the 8 bp sequence motif manifested severe reduction, and to a level of the negative control. These data strongly suggest that DRE is the essential element of dsps2.
Figure 4.

Effect of mutagenesis in and around DRE on promoter activity. Base substitution mutations in and around DRE are represented in the upper portion of the figure, and their relative luciferase activities in the lower portion. SPS2P designates the wild-type sequence and the others designate the mutant sequences. Mutations are shown in italics and bold in the DRE region within the box. Arrows designate the palindromic sequence of DRE. The standard deviations from the mean are indicated by error bars.
Identification of specific transcription factor that binds to DRE of dsps2
EMSA experiments were used to determine if there is protein binding specificity to the DRE sequence of the dsps2 promoter. Extracts of SL2 cells were mixed with 32P-labeled probe, with or without an excess of unlabeled potential competitor, and the complex electrophoresed as shown in Figure 5. A single DNA–protein complex was detected as a shifted band using the synthetic wtDRE oligonucleotide as probe. This shifted band was diminished by adding excessive amounts of unlabeled wtDRE and mNDRE1. However, the addition of mDRE1, mDRE2 and Mut3, which has five nucleotide substitution mutations in the center of DRE, did not affect formation of the retarded band (Fig. 5A). It should be noted that the addition of cold mNDRE2 in which the 3′flank of DRE was mutated resulted in a weak reduction in band retardation. This result is consistent with that obtained from the promoter activity assay mentioned above.
Figure 5.


Identification of DREF as a protein factor that binds to dsps2 promoter. (A) EMSA of DRE-containing sequence in SL2 cell extracts. Sequences of probe or competitor DNAs are shown in the upper portion of the figure, the competitor DNA fragments in increasing amounts in the center portion and the results of the competition experiments in the lower portion. The DRE consensus sequence is shown in the box. In the mutant competitors, only mutated sequences are indicated and the sequences that are the same as wild type are marked as dots. NC designates non-specific competitor. (B) Supershift of retarded band by anti-DREF antibody. 32P-labeled probes were incubated with SL2 nuclear extracts in the absence of antibody (lanes 2, 5 and 6) or in the presence of anti-DREF monoclonal antibody (lanes 3 and 4). The extracts were also incubated with an excess amount of unlabeled wild-type oligonucleotide as specific competitor (lane 5) or mutant3 oligonucleotide (see Materials and Methods) as non-specific competitor (lane 6). The probe was also incubated without extract, antibody and competitor (lane 1). (C) EMSA with GST-DREF fusion protein. SL2 nuclear extracts were incubated with GST protein (lane 1) or with GST-DREF (1–125) in the absence (lane 2) or presence of excess amounts of specific competitor (wtDRE, lanes 3–5) or non-specific competitor (NC, lanes 6–8) as shown at the top of the panel. Concentrations of competitors used are given in Materials and Methods.
To determine that the protein bound to DRE is DREF, a supershift experiment was performed. When the anti-DREF antibody, designated Mab-#4, was added to the reaction, a supershifted band was generated (Fig. 5B, lane 3). However, the addition of Mab-#1 caused inhibition of protein binding to the probe (Fig. 5B, lane 4). Mab-#1 specifically binds to the DNA binding domain of DREF (25). These results indicate that the protein bound to DRE is DREF. Using a recombinant fusion protein of the N-terminal fragment (1–125 amino acid residues) of DREF, designated GST-DREF (1–125), the electrophoretic mobility shift assay was performed. GST-DREF (1–125) was observed to bind specifically to a labeled wtDRE probe in vitro (Fig. 5C). In considering the EMSA and supershift assay, we can conclude that DREF binds to the DRE sequence which is located at bases +71 to +78 downstream of the dsps2 initiation site.
Activation of the dsps2 promoter by DREF overexpression
To determine the promoter-selective properties of DREF, we examined whether DREF can stimulate the dsps2 promoter activity by cotransfection with the DREF expression plasmid. The plasmids pTluc-sps2p(wt) or pTluc-sps2p(mDRE1), which contain the luciferase reporter gene under control of the wild type or the mDRE1 mutated dsps2 promoter, respectively, were cotransfected into SL2 cells with DREF expression plasmid. The transient expression of luciferase was then measured. As a positive and a negative control, Drosophila TBP and TFIIS gene promoter–luciferase fusion plasmids were used. The Drosophila TBP gene promoter contains DREs and was previously known to be regulated by the DRE/DREF system (26). Cotransfection with the DREF expression plasmid resulted in ∼2.5- and 2-fold stimulation of the wild-type dsps2 and TBP promoter activities, respectively (Fig. 6). However, the activity of TFIIS promoter, which does not contain DRE, was not stimulated. The cells transfected with pTluc-sps2p(mDRE1) with or without DREF expression plasmid show almost no luciferase activity (data not shown). These data suggest that DREF activates the dsps2 promoter in a DRE-dependent manner and is required for transcription of dsps2.
Figure 6.

The dsps2 promoter is activated by DREF overexpression in SL2 cells. The luciferase activities of the cotransfected cells by the promoter– luciferase fusion plasmids with or without DREF expression plasmid were compared with that of the transfected cells by pTluc-sps2p(wt) without DREF expression plasmid. The TBP and TFIIS promoters were used as positive and negative controls, respectively. The standard deviations from the mean are indicated by error bars.
Identification of DREF as an essential transcription factor by dsRNAi
We employed dsRNAi strategy to ablate general transcription factors (GTFs) such as dTBP, dTRF1 and dTRF2 and DREF. Double-stranded RNAs, ∼700 bp long, were prepared and introduced into Schneider cells by adding them directly into the culture medium. The endogenous target RNAs (DREFi, TBPi, TRF1i and TRF2i) were confirmed to be depleted specifically by northern blot analysis within 8 h after treatment (Fig. 7A). Eight hours after double-stranded RNA treatment, the plasmid pGL3B-SPS2P, which contains the luciferase reporter under control of the dsps2 promoter, or the plasmid pGL3-Basic, which lacks the dsps2 promoter, was transfected and promoter activity examined after 40 h incubation. The levels of TBP and DREF were also monitored by western blot analysis before the luciferase assay and it was revealed that the amounts of both proteins were specifically and significantly reduced by dsRNA treatment (Fig. 7B, inset). As shown in Figure 7B, the dsps2 promoter activity in DREF-depleted cells was strongly diminished (10-fold) compared with that of non-depleted cells (Fig. 7B). Deletion of TRF2 mRNA resulted in a 2-fold reduction of dsps2 promoter activity. However, ablation of TBP and TRF1 did not affect the promoter activity of dsps2. The fact that the ablation of TRF2 decreased the dsps2 promoter activity suggests that it is required for the transcription of dsps2. It should be noted that ablation of TBP did not affect the promoter activity even though the dsps2 flanking sequence contains a putative TATA box at –20. On the contrary, the TBP-dependent attacin promoter activity was reduced ∼3.3-fold in TBP-ablated cells. The cells treated with other dsRNAs, however, did not show any significant effect on the activity of the attacin promoter (Fig. 7B, open bar). These results were consistent with the previous data obtained from DRE mutant experiments and strongly suggest that DREF is an essential transcription factor for dsps2 transcription. The fact that the depletion of TRF2 decreased the dsps2 promoter activity also suggests that DERF forms an initiation complex, presumably by interacting with TRF2 (see below).
Figure 7.


Effect of ablating transcription factors on the dsps2 promoter activity by dsRNAi. (A) Confirmation of the depletion of target mRNAs after dsRNA treatment. After adding dsRNA to Drosophila cells, total RNA was isolated and the level of target mRNA examined by northern blot analysis. The film was exposed for 24 h after hybridization. dsRNAs used in treating cells are shown on the top and probes used for hybridization on the left of each panel. Control designates the cells that were not treated by dsRNA. Rp49 was used as a loading control. (B) Effect of depleting transcription factors on the activity of dsps2 promoter. After introducing dsRNA into cells, the reporter plasmids were transfected and the level of luciferase activity was measured. The relative luciferase activity designates the amount of luciferase activity in dsRNA-treated cells relative to that of the non-treated control cells. SPS2P (closed bars) and pAttacin (open bars) designate plasmids containing the luciferase gene fused to the dsps2 and attacin promoters, respectively. No promoter designates basic plasmids. The standard deviations from the mean are indicated by error bars. The inset shows monitoring of the ablation of target protein by western blot analysis. Forty-eight hours after dsRNA treatment, cell extracts were prepared and western blot analysis was carried out using anti-DREF or anti-TBP antibody. Actin was used as a loading control. The experiments were performed in triplicate.
DISCUSSION
Although selenium is an essential element for many life forms and selenoproteins are key selenium-containing molecules, the regulation of selenoprotein gene expression in Drosophila has not been elucidated. In this study, the regulation of selenoprotein gene expression in dsps2 was examined as a model selenoprotein gene to elucidate the essential element region(s) used in transcription. By deletion or substitution mutagenesis and dsRNAi analysis, we identified the essential element region and at least one of the essential protein factors required for transcription of dsps2. Initially, we cloned the 5′-flanking region of dsps2. Sequence analysis showed that there are four nucleotides that are different from the sequence published previously (31) and this discrepancy is assumed to be the result of different polymorphic forms. In silico promoter analysis showed that the 5′-flanking region contains >20 putative promoter elements including a putative TATA box upstream of the transcription start site at –20. Promoter analysis through deletion and base substitution mutagenesis demonstrated that the DRE positioned at +71 from transcriptional initiation point is the major regulatory element of dsps2. In all DRE-containing genes with the exception of the DREF gene, which has three DREs both upstream and downstream of the transcription start site at –554 to –543, –81 to –80 and +225 to +234, respectively (32), this element was found only upstream of the transcription initiation point in dsps2. The downstream DRE in the DREF gene is involved in the autoregulation of DREF, but not in the activation of transcription (32). Therefore dsps2 is the first example in which DRE is located only downstream from the transcription initiation site and plays a role as the essential element. The unique location and function of DRE in dsps2 implicates that the DRE/DREF complex might exploit interactions with different components of transcription factors to recruit RNA polymerase II from other DRE-containing genes whose DREs are located upstream of the transcription start site. It should be also noted that the promoter activity of the mutated 3′-neighboring sequences of DRE was also decreased by 50% in the luciferase assay, and the sequences also showed weak competition in the EMSA experiment. These results indicate that the 3′-neighboring sequences (3 nt) of DRE are also required, perhaps as an auxiliary element in forming the DRE/DREF complex.
DREF overexpression in SL2 cells resulted in a stimulatory effect on the dsps2 promoter activity (wild type), but no effect on the DRE mutant promoter. This indicated that the DRE/DREF system is responsible for the transcription of dsps2. The increase in the promoter activity by DREF overexpression was only ∼2-fold, presumably due to a high level of endogenous DREF in SL2 cells.
In the dsRNA interference experiment, TBP did not affect the promoter activity even though the putative TATA box was found at –20. The significant decrease in promoter activity observed in DREF-depleted cells corresponded to that obtained with the DRE mutant analysis, and these results provided direct evidence that DREF is an essential transcription factor of dsps2 transcription. The promoter activity that decreased by 50% in TRF2-depleted cells supported the idea that TRF2 associates with DREF in a DRE-dependent manner (21). Since the ablation of TRF2 caused a reduction in dsps2 promoter activity, and since TFR2 is known to interact with DREF (21), it is tempting to speculate that TRF2 is recruited to interact with DREF in the pre-initiation complex. However, the precise mechanism by which TRF2 acts on the dsps2 promoter has not yet been determined. As noted above, the DRE/DREF system was originally shown to activate transcription of several DNA replication and cell-cycle-related genes (19,20). This indicates that Drosophila SPS2 may be required during DNA replication or for cell cycle regulation. However, the specific biological function of dsps2 remains to be clarified.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr M. Yamaguchi for pGEX-DREF(1–125) and anti-DREF antibody. We also thank Dr Y. J. Kim for pAttacin. This work was supported by a Korea Research Foundation Grant (KRF-2001-015-DS0066) to B.J.L. J.S.J., S.H.B., H.S.L., M.Y.O. and M.S.S. were supported by BK21 Program (Ministry of Education and Human Resources, Korea) and K.B. was supported by KOSEF Grant (R01-2001-000-00173-0).
REFERENCES
- 1.Lee B.J., Park,S.I., Park,J.M., Chittum,H.S. and Hatfield,D.L. (1996) Molecular biology of selenium and its role in human health. Mol. Cells, 6, 509–520. [Google Scholar]
- 2.Kryukov G.V., Castellano,S., Novoselov,S.V., Lobanov,A.V., Zehtab,O., Guigo,R. and Gladyshev,V.N. (2003) Characterization of mammalian selenoproteomes. Science, 300, 1439–1443. [DOI] [PubMed] [Google Scholar]
- 3.Stadtman T.C. (1996) Selenocystein. Annu. Rev. Biochem., 65, 83–100. [DOI] [PubMed] [Google Scholar]
- 4.Böck A., Forchhammer,K., Heider,J. and Baron,C. (1991) Selenocysteine: the 21st amino acid. Trends Biochem. Sci., 5, 515–520. [DOI] [PubMed] [Google Scholar]
- 5.Driscoll D.M. and Copeland,P.R. (2003) Mechanism and regulation of selenoprotein synthesis. Annu. Rev. Nutr., 23, 17–40. [DOI] [PubMed] [Google Scholar]
- 6.Veres Z., Kim,I.Y., Scholz,T.D. and Stadtman,T.C. (1994) Selenophosphate synthetase. J. Biol. Chem., 269, 10597–10603. [PubMed] [Google Scholar]
- 7.Leinfelder W., Forchhammer,K., Verprek,B., Zehelein,E. and Bock,A. (1990) In vitro synthesis of selenocysteinyl-tRNAUCA from seryl-tRNAUCA: involvement and characterization of the selD gene product. Proc. Natl Acad. Sci. USA, 87, 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alsina B., Corominas,M., Berry,M.J., Baguna,J. and Serras,F. (1999) Disruption of selenoprotein biosynthesis affects cell proliferation in the imaginal discs and brain of Drosophila melanogaster. J. Cell Sci., 112, 2875–2884. [DOI] [PubMed] [Google Scholar]
- 9.Morey M., Corominas,M. and Serras,F. (2003) DIAP1 suppresses ROS-induced apoptosis caused by impairment of the selD/sps1 homolog in Drosophila. J. Cell Sci., 116, 4597–604. [DOI] [PubMed] [Google Scholar]
- 10.Hirosawa-Takamori M., Jackle,H. and Vorbruggen,G. (2000) The class selenophosphate synthetase gene of Drosophila contains a functional of mammalian-type SECIS. EMBO Rep., 1, 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelner M.J., Bagnell,R.D., Montoya,M.A. and Lanham,K.A. (2000) Structural organization of the human gastrointestinal glutathione peroxidase (GPX2) promoter and 3′-nontranscribed region: transcriptional response to exogenous redox agents. Gene, 248, 109–116. [DOI] [PubMed] [Google Scholar]
- 12.Rundlöf A-K.,,, Carlsten,M. and Arnér,E.S.J. (2001) The core promoter of human thioredoxine reductase 1. J. Biol. Chem., 276, 30542–30551. [DOI] [PubMed] [Google Scholar]
- 13.Rundlöf A-K., Janaro,M., Miranda-Vizuete,A. and Arnér,E.S.J. (2004) Evidence for intriguingly complex transcription of human thioredoxine reductase 1. Free Radic. Biol. Med., 36, 641–656. [DOI] [PubMed] [Google Scholar]
- 14.Dreher I., Jakobs,T.C. and Köhrle,J. (1997) Cloning and characterization of the human selenoprotein P promoter. J. Biol. Chem., 272, 29364–29371. [DOI] [PubMed] [Google Scholar]
- 15.Maiorino M., Scapin,M., Ursini,F., Biasolo,M., Bosello,V. and Flohe,L. (2003) Distinct promoters determine alternative transcription of gpx-4 into phospholipid-hydroperoxide glutathione peroxidase variants. J. Biol. Chem., 278, 34286–34290. [DOI] [PubMed] [Google Scholar]
- 16.Amantana A., Vorachek,W.R., Butler,J.A., Costa,N.D. and Whanger,P.D. (2002) Effect of copper, zinc and cadmium on the promoter of selenoprotein W in glial and myoblast cells. J. Inorg. Biochem., 91, 356–362. [DOI] [PubMed] [Google Scholar]
- 17.Merante F., Altamentova,S.M., Mickle,D.A., Weisel,R.D., Thatcher,B.J., Martin,B.M., Marshall,J.G., Tumiati,L.C., Cowan,D.B. and Li,R.K. (2002) The characterization and purification of a human transcription factor modulating the glutathione peroxidase gene in response to oxygen tension. Mol. Cell. Biochem., 229, 73–83. [DOI] [PubMed] [Google Scholar]
- 18.Ufer C., Borchert,A. and Kuhn,H. (2003) Functional characterization of cis- and trans-regulatory elements involved in expression of phospholipid hydroperoxide glutathione peroxidase. Nucleic Acids Res., 31, 4293–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirose F., Yamaguchi,M., Handa,H., Inomata,Y. and Matsukage,A. (1993) Novel 8-base pair sequence (Drosophila DNA Replication-related Element) and specific binding factor involved in the expression of Drosophila genes for DNA polymerase α and proliferating cell nuclear antigen. J. Biol. Chem., 268, 2092–2099. [PubMed] [Google Scholar]
- 20.Ohno K., Hirose,F., Sakaguchi,K., Nishida,Y. and Matsukage,A. (1996) Transcriptional regulation of the Drosophila CycA gene by the DNA replication-related element (DRE) and DRE binding factor (DREF). Nucleic Acids Res., 24, 3942–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hochheimer A., Zhou,S., Zheng,S., Holmes,M.C. and Tjian,R. (2002) TRF2 associates with DREF and directs promoter-selective gene expression in Drosophila. Nature, 420, 439–445. [DOI] [PubMed] [Google Scholar]
- 22.Sugiura S., Kashiwabara,S., Iwase,S. and Baba T. (2003) Expression of a testis-specific form of TBP-related factor (TRF2) mRNA during mouse spermatogenesis. J. Reprod. Dev., 49, 107–111. [DOI] [PubMed] [Google Scholar]
- 23.Kwon S.Y., Badenhorst,P., Martin-Romero,F.J., Carlson,B., Paterson,B.M., Gladyshev,V.N., Lee,B.J. and Hatfield,D. (2003) The Drosophila selenoprotein BthD is required for survival and has a role in salivary gland development. Mol. Cell. Biol., 23, 8495–8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clemens J.C., Worby,C.A., Simonson-Leff,N., Muda,M., Maehama,T., Hemmings,B.A. and Dixon,J.E. (2000) Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc. Natl Acad. Sci. USA, 97, 6499–6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirose F., Yamaguchi,M., Kuroda,K., Omori,A., Hachiya,T., Ikeda,M., Nishimoto,Y. and Matsukage,A. (1996) Isolation and characterization of cDNA for DREF, a promoter-activating factor for Drosophila DNA replication-related genes. J. Biol. Chem., 271, 3930–3937. [DOI] [PubMed] [Google Scholar]
- 26.Lee K.S., Oh,Y., Baek,G., Yoon,J., Han,K., Cho,N.Y. and Baek,K. (1999) Analysis of the structure and expression for the TFIIB gene in Drosophila melanogaster. Mol. Cells, 7, 374–379. [PubMed] [Google Scholar]
- 27.Choi T.Y., Cho,N.Y., Oh,Y., Yoo,M.A., Matsukage,A., Ryu,Y., Han,K., Yoon,J. and Baek,K. (2000) The DNA replication-related element (DRE) -DRE binding factor (DREF) system may be involved in the expression of the Drosophila melanogaster TBP gene. FEBS Lett., 483, 71–77. [DOI] [PubMed] [Google Scholar]
- 28.Oh Y., Lee,S., Yoon,J., Han,K. and Baek,K. (2001) Promoter analysis of the Drosophila melanogaster gene encoding transcription elongation factor TFIIS. Biochim. Biophys. Acta, 1518, 276–281. [DOI] [PubMed] [Google Scholar]
- 29.Andrews N. and Faller,D. (1991) A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res., 19, 2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sawado T., Hirose,F., Takahashi,Y., Sasaki,T., Shinomiya,T., Sakaguchi,K., Matsukage,A. and Yamaguchi,M. (1998) The DNA replication-related element (DRE)/DRE-binding factor system is a transcriptional regulator of the Drosophila E2F gene. J. Biol. Chem., 273, 26042–26051. [DOI] [PubMed] [Google Scholar]
- 31.Adams M.D. and Celinker,S.E. (2000) The genome sequence of Drosophila melanogaster. Science, 287, 2185–2195. [DOI] [PubMed] [Google Scholar]
- 32.Kwon E., Seto,H., Hirose,F., Ohshima,N., Takahashi,Y., Nishida,Y. and Yamaguchi,M. (2003) Transcription of a gene for Drosophila transcription factor, DREF by DRE and cis-elements conserved between Drosophila melanogaster and virilis. Gene, 309, 101–116. [DOI] [PubMed] [Google Scholar]