

Abstract
The past decade has witnessed huge advances in our understanding of the genetics underlying Parkinson’s disease. Identifying commonalities in the biological function of genes linked to Parkinson’s provides an opportunity to elucidate pathways that lead to neuronal degeneration and eventually to disease. We propose that the genetic forms of Parkinson’s disease largely associated with α-synuclein-positive neuropathology (SNCA, LRRK2, and GBA) are brought together by involvement in the autophagy/lysosomal pathway and that this represents a unifying pathway to disease in these cases.
Keywords: Parkinson’s disease, LRRK2, GBA
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by the preferential death of dopaminergic neurons. Historically, PD has been defined histopathologically by two key features: the loss of neurons in the substantia nigra pars compacta and the presence of intracellular proteinaceous inclusion bodies called Lewy bodies (LBs) (1). The major advances in the field of medical genetics during the 1980s and 1990s opened a new chapter in the study of PD by facilitating the identification of genes linked to mendelian forms of this disease, starting with SNCA, coding for α-synuclein, in 1997 (2). Both point mutations and gene multiplications in α-synuclein are causative for familial PD (3). The association of α-synuclein with inherited parkinsonism led, in turn, to an important breakthrough in the classification of the pathology of PD with the realization that the neuropathological hallmark of this disease, the LB, was made up of aggregates of α-synuclein (4).
FOLLOWING THE PATHOLOGY
A number of other genes have subsequently been identified in familial forms of PD (Table 1), the most common of which are mutations in the LRRK2 gene on chromosome 12, coding for leucine-rich repeat kinase 2 (LRRK2) (5). Dominant mutations in LRRK2 cause a parkinsonian syndrome clinically similar to sporadic PD. Although the majority of cases (>90%) present with LB pathology, a significant minority of cases manifest with Tau, Aβ, TDP-43, or ubiquitin-positive inclusions in the absence of LBs (6).
Table 1. Genetic synucleinopathies.
| Gene | Protein | Phenotype | Pathology | Inheritance | Mutations |
|---|---|---|---|---|---|
| SNCA | α-Synuclein | PD, DLB | LB | Dominant | Point mutations (A53T, A30P, E46K, H50Q, and G51D), gene multiplications |
| GBA | GBA | GD, PD | LB | Dominant | Point mutations (numerous described, most common L444P and N370S) |
| LRRK2 | LRRK2 | PD | Pleiomorphic, mostly LB | Dominant | Point mutations (N1437H, R1441G/C, Y1699C, G2019S, and I2020T) |
| ATP13a2 | ATP13a2 | KRS, NCL | Unknown | Recessive | Truncations |
| VPS35 | VPS35 | PD | Unknown | Dominant | Point mutation (D620N) |
• ATP13a2, ATPase 13a2; DLB, dementia with Lewy bodies; GBA, glucocerebrosidase; GD, Gaucher’s disease; KRS, Kufor-Rakeb syndrome; LRRK2, leucine-rich repeat kinase 2; NCL, neuronal ceroid lipofuscinosis; SNCA, synuclein α; VPS35, vacuolar protein sorting 35.
Homozygous mutations in GBA, encoding glucocerebrosidase (GBA), cause Gaucher’s disease (GD), a lysosomal storage disorder. It has long been noted that a subset of patients with GD present with parkinsonism, and this clue has led recently to the realization that individuals with a mutation in the heterozygous state have an increased risk of developing PD, with abundant LB pathology (7).
In addition to SNCA, LRRK2, and GBA, a number of other inherited parkinsonian syndromes have been identified, most notably those linked to mutations in PARK2 (coding for parkin), PINK1, and PARK7 (coding for DJ-1). These disorders are inherited in an autosomal recessive manner, with the protein products of PARK2 and PINK1 localized to a common pathway involved in mitochondrial quality control (8). PARK7 has been implicated in a number of cellular processes, including the response to oxidative stress, RNA regulation, and protein folding, and may act in a parallel pathway to the parkin/PINK1 pathway (9, 10). The relationship of these recessive disorders to the synucleinopathies is unclear. For PINK1 and PARK7, the paucity of neuropathological reports makes it difficult to judge whether these are indeed LB disorders (11). A number of PARK2 cases have come to autopsy, and although a minority of cases present with LBs, it has been suggested that these are secondary to the disease process (12).
Finally, several genes have been recently associated with parkinsonism. Mutations in vacuolar protein sorting 35 (VPS35) cause a parkinsonian syndrome clinically similar to sporadic PD (13, 14). Mutations in the P5-type ATPase 13a2 (ATP13a2) are associated with a clinical syndrome called Kufor-Rakeb. Although this syndrome is clinically distinct from PD (it is characterized by pyramidal degeneration, widespread neurodegeneration, and dementia), one of the clinical features is early-onset parkinsonism (15). There are no published neuropathological reports for individuals harboring mutations in either of these genes, so it is not known whether they are associated with α-synuclein aggregation and LB pathology. Notably, however, the functions of both ATP13a2 and VPS35 have been the subject of a number of studies that aid us in interpreting how their dysfunction might lead to disease.
LOOKING FOR PATTERNS
One of the major benefits of identifying inherited forms of disease is that this allows a systematic analysis of pathways that are involved in a particular disorder. An example of this is the realization that the 3 genes linked to mendelian forms of Alzheimer’s disease (AD), APP, PSEN1, and PSEN2, are all involved in the production of the Aβ peptide (16). The identification of this pathway has led to a 20-yr effort to target the proteolytic events leading to the production of Aβ, resulting in the identification of the secretases that cleave the Aβ precursor protein (APP), and to the development of compounds to manipulate these proteolytic events. This example highlights the utility of understanding the links between different genes connected to the same pathology.
In contrast to AD, grouping the inherited forms of PD and parkinsonism has shown only limited success. As noted above, PARK2 and PINK1 have been robustly demonstrated to map to the same cellular pathway, but how the other genes implicated in PD group together into a pathogenic pathway or pathways is unclear. A recent review suggested lysosomal dysfunction as a unifying feature of PD (17). The hypothesis that we propose here is that the genes associated with LB-positive PD (SNCA, LRRK2, and GBA) are linked by a common role in the autophagy/lysosomal pathway. Based on phenotypic and cellular studies, we propose that parkinsonism caused by mutations in VPS35 and ATP13a2 also result from disruption of the autophagy/lysosomal system.
EXAMINING THE SUSPECTS
The central player in genetic LB disorders is α-synuclein. As such, this protein is the logical starting point when examining the evidence supporting a common leitmotif linking the inherited synucleinopathies.
α-Synuclein
α-Synuclein is a 140-aa protein of unknown function (3). Accumulating evidence shows that dysfunction of α-synuclein affects the autophagic pathway. Modulation of synuclein protein levels inhibits autophagy via Rab1a, and dominant mutations in α-synuclein associated with PD can decrease lysosomal functionality, thus reducing the flux through the autophagy pathway (18, 19). α-Synuclein can also be degraded via chaperone-mediated autophagy (CMA), a process disrupted by mutations, which suggests that synuclein can take several routes to degradation by the lysosomes (19, 20). Abnormal accumulation of monomeric α-synuclein within the cell is likely to be the first step to misfolding and aggregation, leading to deposition in LBs, and dysfunction of degradation pathways will have a profound effect on this process (3). In the context of the hypothesis under discussion here, a potentially important aspect of synuclein biology is its link to the regulation of vesicle trafficking. Much of the data relating to α-synuclein function focuses on a role in the regulation of SNARE proteins, which form multicomponent complexes that are essential for vesicle fusion events. In particular, α-synuclein has been implicated in synaptic vesicle fusion, highlighted by a study investigating a mouse lacking α-, β-, and γ-synucleins (21). It is noteworthy that SNARE complexes are also required for the correct function and fusion of autophagic vesicles, which provides a potential link between the physiological role of α-synuclein and autophagy (22). Whether the physiological function of α-synuclein and its dysfunction in disease are both directly linked to the autophagy/lysosomal pathway is unclear; a recent report suggests an explicit divergence between function and toxicity (23). However, it is abundantly clear that this protein is closely linked to autophagy in disease.
GBA
GBA is a lysosomal enzyme that hydrolyzes glucocerebroside, releasing ceramide. Loss-of-function mutations in GBA cause the lysosomal storage disorder GD in the homozygous state, which leads to an accumulation of glucocerebrosides in the lysosomes. In the heterozygous state, the same mutations greatly increase the risk of developing PD (7). What is indisputable is that mutations in GBA that increase the risk of PD disrupt lysosomal function, which provides a concrete link between the synucleinopathy that results from these mutations and the autophagy/lysosomal pathway. Altered ceramide levels due to GBA dysfunction can also affect the Akt and mTORC1 signaling pathways, altering the autophagy/lysosomal pathway from upstream as well as at the point of lysosomal function (24).
LRRK2
LRRK2 is a multidomain enzyme that has been implicated in a large number of cellular processes. An interaction with membranes and a putative role in the regulation of autophagy are reoccurring themes in the LRRK2 literature (5). A number of studies have reported mutations in LRRK2 affecting autophagy via an as yet unidentified mechanism (25, 26). Also, several reports suggest that LRRK2 may have a physiological role in regulating autophagy, with both acute knockdown and knockout of LRRK2 affecting autophagy. Mice lacking LRRK2 display alterations in markers of autophagy and in the lysosomal pathway (27), and siRNA knockdown of LRRK2 in human cells results in an increase in the lipid-associated form of the autophagy marker LC3 (25). A recent study reported an interaction between LRRK2 and CMA, and given the previously documented link between α-synuclein and CMA, this finding may be of relevance to the accumulation of this protein in the synucleinopathies (28). These data in toto suggest a close link between LRRK2 and the autophagy/lysosomal pathway.
VPS35 and ATP13a2
Although the neuropathology associated with mutations in VPS35 and ATP13a2 has not been described, the physiological roles of these proteins place them in the autophagy/lysosomal pathway.
VPS35 is part of the retromer complex that is involved in the transport of endosomes back to the trans-Golgi and in the sorting of receptors for hydrolases, thus affecting vacuole/lysosomal function and biogenesis. Loss of function in VPS35 has been shown to produce vacuolar/lysosomal abnormalities (29).
ATP13a2 is a lysosomal protein, a P5-type ATPase thought to be responsible for cation transport and the regulation of manganese levels. Loss of ATP13a2 in cellular and animal models results in lysosomal dysfunction, with a reduction in proteolytic activity and accumulation of lysosomes and autophagosomes (30, 31). The phenotypic spectrum for mutations at the ATP13a2 locus also includes neuronal ceroid lipofuscinosis, a lysosomal disorder (32).
JOINING THE DOTS
As described above, each of the genes conclusively linked to inherited synucleinopathy has also been linked to either lysosomal function or autophagy. Two further genes linked to parkinsonian syndromes of undetermined neuropathological status display clear links to these pathways. What evidence suggests that these genes are linked and that mutations in them may have a common effect on the autophagy/lysosomal pathway?
A number of recent studies have highlighted a reciprocal relationship between α-synuclein and GBA. A study by Cullen et al. (33) examined the effect of mutations in GBA on α-synuclein, observing a marked increase in synuclein levels in the presence of disease associated mutants. Mazzulli et al. (34) demonstrated that a decrease in GBA activity results in a preferential increase in α-synuclein levels due to reduced lysosomal proteolysis. This study also reported that increased α-synuclein levels had an effect on the trafficking of GBA to the lysosomes, creating a positive feedback loop. The exact mechanics of the relationship between α-synuclein and GBA are not clear; however, some evidence supports a direct interaction between these two proteins (35).
The nature of the interaction between α-synuclein and LRRK2 is a matter of greater conjecture. Several lines of evidence suggest crosstalk between these proteins, leading to neurodegeneration and PD. A study by Lin et al. (36) using a combination of mice transgenic for LRRK2 and α-synuclein, as well as LRRK2-knockout mice, demonstrated that LRRK2 has a major effect on synuclein neuropathology. Several studies of LRRK2-knockout mice have observed accumulation of α-synuclein, albeit in the kidneys of these mice rather than their brains (27). A connection between LRRK2, CMA, and α-synuclein has also been reported (28). Whether these data reflect a direct interaction between synuclein and LRRK2, mediated by a LRRK2-dependent phosphorylation event or a more distant relationship with LRRK2 acting upstream of α-synuclein, remains an area of intense investigation (5).
Several studies link loss of ATP13a2 function to the accumulation of α-synuclein. Both knockout and knockdown of ATP13a2 result in an accumulation of insoluble α-synuclein, as well as neuronal toxicity (37,-,39). Very little has been reported regarding the effect of mutations in VPS35. Macleod et al. (40) report a link between dysfunction of LRRK2, VPS35, and a locus recently identified by the genome wide association study (GWAS) for PD: the PARK16 locus. The researchers report that manipulating LRRK2 and a candidate gene for the PARK16 locus, RAB7L1, results in changes in endosomal trafficking, linking to the previously reported function of VPS35 (40).
Somewhat distinct from the familial and genetic forms of disease under examination here, two novel mouse models of neurodegeneration have been reported where disruption of a key protein in the autophagy pathway (Atg7) results in accumulation of α-synuclein and LRRK2 (41, 42). These data suggest that, in addition to an effect on autophagy resulting from alteration in the function of LRRK2 and α-synuclein, a reciprocal relationship exists between the physiological process of autophagy and these pathologically linked proteins. A summary of these relationships is displayed in Fig. 1.
Figure 1. Links between the genetic synucleinopathies.

Venn diagram illustrating the overlaps between the key genes discussed in this review and their physiological roles. Solid arrows indicate reported biological interactions, dotted arrow represents untested interaction.
CONCLUSIONS AND TESTS
The data considered in the present work support a central role for the autophagy/lysosomal pathway in the pathogenesis of the genetic synucleinopathies, a possibility that has important ramifications for our understanding of the pathways that lead to Parkinson’s. Understanding these pathways is the key to the development of new strategies to target a disease that is of growing importance to health care systems across the globe. We propose that disorders connected to alterations in SNCA, LRRK2, and GBA, as well as disease caused by mutations in ATP13a2 and VPS35, can all be mechanistically linked to the autophagy/lysosomal pathway (Fig. 2). Key experimental tests of our hypothesis are summarized in Table 2. Our prediction is that a common theme in the nature of the disruption of these pathways for each of the mutant genes under consideration here will emerge, in all cases resulting in an alteration in the biology of α-synuclein. As with any hypothetical model, it is important to highlight data that are not consistent with these predictions. Why, for example, are there LRRK2 mutation carriers who present with PD in the absence of LBs? Where do the rare cases of PARK2 with LBs fit into this scheme? Another important consideration is whether this model can (or should) be applied to the sporadic synucleinopathies; the evidence in the literature that this may be the case is unclear (43, 44). In common with many neurodegenerative disorders, substantial gaps remain in our understanding of PD, both inherited and idiopathic. Research over the coming years will provide answers to some of the questions posed above and, it is to be hoped, allow us to judge whether the autophagy/lysosomal pathway does indeed occupy a central role in the inherited synucleinopathies.
Figure 2. Autophagy/lysosomal pathways to PD: hypothetical framework for the interaction of the genes implicated in the genetic synucleopathies at the convergence of the autophagy and lysosomal pathways showing location of the genes discussed in the text.
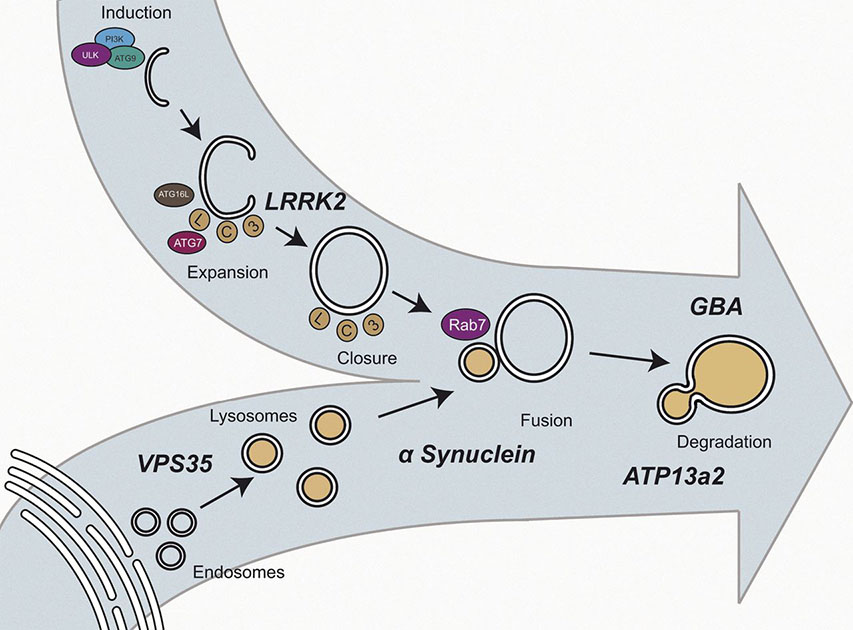
Table 2. Tests of this hypothesis.
| Experimental test | Expected outcome |
|---|---|
| Neuropathological examination of ATP13a2 and VPS35 mutation carriers | Cases manifesting with extensive LB pathology |
| Parallel analysis of autophagic flux and lysosomal function in cellular models for mutated genes | A consistent effect of mutations, pushing these pathways in a similar direction |
| Investigating autophagy/lysosomal markers in the brains of sporadic cases of Parkinson’s disease | Alterations in markers congruent with those observed in familial forms of disease |
| Examination of models for disorders where LBs are a secondary lesion, such as Alzheimer’s disease | Disruption of autophagic/lysosomal pathways as a secondary effect of the primary pathology |
Acknowledgments
The authors acknowledge generous research support from the Michael J. Fox Foundation and Parkinson’s UK. P.A.L. is a Parkinson’s UK research fellow (grant F1002). C.M. is funded by the Rosetrees Trust.
This work was supported in part by the Wellcome Trust/Medical Research Council (MRC) Joint Call in Neurodegeneration award (WT089698) to the UK Parkinson’s Disease Consortium (UKPDC), whose members are from the University College London Institute of Neurology (London, UK), the University of Sheffield (Sheffield, UK), and the MRC Protein Phosphorylation Unit at the University of Dundee (Dundee, UK).
Glossary
- AD
Alzheimer’s disease
- ATP13a2
ATPase 13a2
- CMA
chaperone-mediated autophagy
- GBA
glucocerebrosidase
- GD
Gaucher’s disease
- LB
Lewy body
- LRRK2
leucine-rich repeat kinase 2
- PD
Parkinson’s disease
- VPS35
vacuolar protein sorting 35
REFERENCES
- 1.Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 2.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 3.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 5.Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010;11:791–797. doi: 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 7.Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012;11:986–998. doi: 10.1016/S1474-4422(12)70190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deas E, Wood NW, Plun-Favreau H. Mitophagy and Parkinson’s disease: the PINK1-parkin link. Biochim. Biophys. Acta. 2011;1813:623–633. doi: 10.1016/j.bbamcr.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kahle PJ, Waak J, Gasser T. DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Rad. Biol. Med. 2009;47:1354–1361. doi: 10.1016/j.freeradbiomed.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A, Miller D, Maric D, Cedazo-Minguez A, Cookson MR. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2010;20:40–50. doi: 10.1093/hmg/ddq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Houlden H, Singleton AB. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 2012;124:325–338. doi: 10.1007/s00401-012-1013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doherty KM, Silveira-Moriyama L, Parkkinen L, Healy DG, Farrell M, Mencacci NE, Ahmed Z, Brett FM, Hardy J, Quinn N, Counihan TJ, Lynch T, Fox ZV, Revesz T, Lees AJ, Holton JL. Parkin disease: a clinicopathologic entity? JAMA Neurol. 2013;70:571–579. doi: 10.1001/jamaneurol.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, Behrouz B, Melrose HL, Hentati E, Puschmann A, Evans DM, Conibear E, Wasserman WW, Aasly JO, Burkhard PR, Djaldetti R, Ghika J, Hentati F, Krygowska-Wajs A, Lynch T, Melamed E, Rajput A, Rajput AH, Solida A, Wu RM, Uitti RJ, Wszolek ZK, Vingerhoets F, Farrer MJ. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011;89:162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC, Klopp N, Wolf E, Seppi K, Pirker W, Presslauer S, Mollenhauer B, Katzenschlager R, Foki T, Hotzy C, Reinthaler E, Harutyunyan A, Kralovics R, Peters A, Zimprich F, Brucke T, Poewe W, Auff E, Trenkwalder C, Rost B, Ransmayr G, Winkelmann J, Meitinger T, Strom TM. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011;89:168–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 16.Lichtenthaler SF, Haass C, Steiner H. Regulated intramembrane proteolysis-lessons from amyloid precursor protein processing. J. Neurochem. 2011;117:779–796. doi: 10.1111/j.1471-4159.2011.07248.x. [DOI] [PubMed] [Google Scholar]
- 17.Tofaris GK. Lysosome-dependent pathways as a unifying theme in Parkinson’s disease. Mov. Disord. 2012;27:1364–1369. doi: 10.1002/mds.25136. [DOI] [PubMed] [Google Scholar]
- 18.Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O’Kane CJ, Rubinsztein DC. alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J. Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 20.Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, Baba M, McNew JA, Jiang X, Reggiori F, Melia TJ, Klionsky DJ. SNARE proteins are required for macroautophagy. Cell. 2011;146:290–302. doi: 10.1016/j.cell.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burre J, Sharma M, Sudhof TC. Systematic mutagenesis of alpha-synuclein reveals distinct sequence requirements for physiological and pathological activities. J. Neurosci. 2012;32:15227–15242. doi: 10.1523/JNEUROSCI.3545-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Young MM, Kester M, Wang HG. Sphingolipids: regulators of crosstalk between apoptosis and autophagy. J. Lipid Res. 2013;54:5–19. doi: 10.1194/jlr.R031278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alegre-Abarrategui J, Christian H, Lufino MM, Mutihac R, Venda LL, Ansorge O, Wade-Martins R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009;18:4022–4034. doi: 10.1093/hmg/ddp346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomez-Suaga P, Luzon-Toro B, Churamani D, Zhang L, Bloor-Young D, Patel S, Woodman PG, Churchill GC, Hilfiker S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012;21:511–525. doi: 10.1093/hmg/ddr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tong Y, Giaime E, Yamaguchi H, Ichimura T, Liu Y, Si H, Cai H, Bonventre JV, Shen J. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations ofthe autophagy pathway. Mol. Neurodegener. 2012;7:2. doi: 10.1186/1750-1326-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013;16:394–406. doi: 10.1038/nn.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arighi CN, Hartnell LM, Aguilar RC, Haft CR, Bonifacino JS. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol. 2004;165:123–133. doi: 10.1083/jcb.200312055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park JS, Mehta P, Cooper AA, Veivers D, Heimbach A, Stiller B, Kubisch C, Fung VS, Krainc D, Mackay-Sim A, Sue CM. Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing Kufor-Rakeb syndrome, a form of early-onset parkinsonism. Hum. Mutat. 2011;32:956–964. doi: 10.1002/humu.21527. [DOI] [PubMed] [Google Scholar]
- 31.Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. U. S. A. 2012;109:9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum. Mol. Genet. 2012;21:2646–2650. doi: 10.1093/hmg/dds089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ, Kolodziej P, Kahn I, Saftig P, Woulfe J, Rochet JC, Glicksman MA, Cheng SH, Grabowski GA, Shihabuddin LS, Schlossmacher MG. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann. Neurol. 2011;69:940–953. doi: 10.1002/ana.22400. [DOI] [PubMed] [Google Scholar]
- 34.Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yap TL, Gruschus JM, Velayati A, Westbroek W, Goldin E, Moaven N, Sidransky E, Lee JC. Alphasynuclein interacts with Glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J. Biol. Chem. 2011;286:28080–28088. doi: 10.1074/jbc.M111.237859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-diseaserelated mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeger-Lotem E, Riva L, Su LJ, Gitler AD, Cashikar AG, King OD, Auluck PK, Geddie ML, Valastyan JS, Karger DR, Lindquist S, Fraenkel E. Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat. Genet. 2009;41:316–323. doi: 10.1038/ng.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012;32:4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, Dickson DW, Mazzulli JR, Bardgett ME, Haik KL, Ekhator O, Chava AK, Howard J, Gannon M, Hoffman E, Chen Y, Prasad V, Linn SC, Tamargo RJ, Westbroek W, Sidransky E, Krainc D, Shull GE. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum. Mol. Genet. 2013;22:2067–2082. doi: 10.1093/hmg/ddt057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Macleod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, Maccabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A. RAB7L1 Interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahmed I, Liang Y, Schools S, Dawson VL, Dawson TM, Savitt JM. Development and characterization of a new Parkinson’s disease model resulting from impaired autophagy. J. Neurosci. 2012;32:16503–16509. doi: 10.1523/JNEUROSCI.0209-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, Holstein GR, Yue Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of alphasynuclein and LRRK2 in the brain. J. Neurosci. 2012;32:7585–7593. doi: 10.1523/JNEUROSCI.5809-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009;35:385–398. doi: 10.1016/j.nbd.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 44.Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AH. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010;67:1464–1472. doi: 10.1001/archneurol.2010.198. [DOI] [PubMed] [Google Scholar]