

Abstract
The leukocyte integrin αMβ2/Mac-1 appears to support the inflammatory response through multiple ligands, but local engagement of fibrin(ogen) may be particularly important for leukocyte function. To define the biological significance of fibrin(ogen)-αMβ2 interaction in vivo, gene-targeted mice were generated in which the αMβ2-binding motif within the fibrinogen γ chain (N390RLSIGE396) was converted to a series of alanine residues. Mice carrying the Fibγ390–396A allele maintained normal levels of fibrinogen, retained normal clotting function, supported platelet aggregation, and never developed spontaneous hemorrhagic events. However, the mutant fibrinogen failed to support αMβ2-mediated adhesion of primary neutrophils, macrophages, and αMβ2-expressing cell lines. The elimination of the αMβ2-binding motif on fibrin(ogen) severely compromised the inflammatory response in vivo as evidenced by a dramatic impediment in leukocyte clearance of Staphylococcus aureus inoculated into the peritoneal cavity. This defect in bacterial clearance was due not to diminished leukocyte trafficking but rather to a failure to fully implement antimicrobial functions. These studies definitively demonstrate that fibrin(ogen) is a physiologically relevant ligand for αMβ2, integrin engagement of fibrin(ogen) is critical to leukocyte function and innate immunity in vivo, and the biological importance of fibrinogen in regulating the inflammatory response can be appreciated outside of any alteration in clotting function.
Introduction
The β2 (CD18) subfamily of integrins serves a vital role in leukocyte function and the development of an effective inflammatory response in vivo (1). Genetic deficiency in β2 results in the severe immunological disorder, leukocyte adhesion deficiency type I (LAD I), that is characterized by a profound reduction in neutrophil emigration at sites of inflammation and chronic infections (2). Four members of the integrin β2 subfamily have been recognized: αMβ2 (Mac-1, CD11b/CD18, CR3), αLβ2 (LFA-1, CD11a/CD18), αXβ2 (p150,95, CD11c/CD18), and αDβ2 (CD11d/CD18) (3, 4). Based in part on the prominent expression of αMβ2 and αLβ2 on the surface of key inflammatory cells, including neutrophils, monocytes, macrophages, and mast cells, these integrins are generally thought to play a dominant role in inflammatory cell function. The critical importance of αMβ2 and αLβ2 in leukocyte function in vivo has been affirmed and clarified through detailed studies of mice with specific genetic deficits in αM, αL, and β2 (5–8). Working in concert, αLβ2 and αMβ2 appear to be instrumental in (a) firm adhesion of leukocytes to the vessel wall that follows selectin-mediated rolling events (b) transendothelial cell migration and leukocyte trafficking (c) leukocyte activation, and (d) cell survival/apoptosis (1, 9–11).
Central to unraveling the precise biological roles of αMβ2 and αLβ2 is defining the biologically relevant ligands for each of these integrins. While common ligands are known, these integrins are clearly designed to engage a distinct repertoire of cell-surface and ECM proteins (12). αLβ2 specifically binds intercellular adhesion molecule (ICAM) family members, whereas αMβ2 is capable of binding a remarkable assortment of seemingly unrelated ligands, including ICAM-1, the complement C3 derivative, iC3b, the urokinase-type plasminogen activator receptor (uPAR), platelet membrane glycoprotein GP1bα, and immobilized fibrin(ogen) (12). The physiological and/or pathological significance of these and other possible ligands that might regulate leukocyte function and innate immunity remains to be fully defined.
Given the increasingly persuasive evidence suggesting an important interplay between hemostatic factors and inflammatory systems, fibrin(ogen) has gained increasing attention as a possible biologically significant ligand for αMβ2. As a symmetrical dimer that is recognized by a variety of integrin and nonintegrin receptors on multiple cell types, fibrinogen could serve as a bridging molecule between leukocytes and other cells, including platelets. Furthermore, as a protein that is deposited in the form of a provisional fibrin matrix at virtually any site of overt tissue damage, fibrin could serve as a significant nondiffusible cue in regulating leukocyte targeting. Consistent with this general theory, multiple in vitro studies have shown that leukocyte engagement of fibrin(ogen) can profoundly alter leukocyte function, leading to changes in cell migration, phagocytosis, NF-κB–mediated transcription, production of chemokines and cytokines, degranulation, and other processes (13–18).
These findings have driven efforts to better define the molecular details of the interaction between fibrinogen and αMβ2. One notable facet of the binding interaction is that both fibrin and immobilized fibrinogen are bound with high-affinity/avidity by αMβ2, whereas soluble fibrinogen is a relatively poor ligand (19, 20). This conformation-dependent binding implies that αMβ2 would generally not be occupied when circulating leukocytes passively encounter plasma fibrinogen. The integrin, however, would mediate avid cellular engagement of immobilized fibrin(ogen) at sites of tissue damage, locations where copious fibrin deposition would be universally observed. Multiple sequences in fibrinogen can interact with αMβ2, but the most compelling data point to a critical role of the γ chain in the region of γ377–395 (P2 sequence; ref. 21, 22) and particularly its carboxy-terminal region (P2-C). To definitively establish whether the P2-C region of the fibrinogen γ chain constitutes the critical motif for αMβ2 binding in the context of the whole fibrinogen molecule, and to provide the means to evaluate the biological importance of fibrin(ogen)-αMβ2 interactions in inflammatory processes in vivo, we employed a gene-targeting approach to convert the mouse fibrinogen γ chain sequence N390RLSIGE396 to a series of seven alanine residues. This mutant form of fibrinogen (which we term fibrinogen γ390–396A) maintained normal hemostatic properties. Fibrinogen γ390–396A was unable to support αMβ2-mediated cellular adhesion in vitro, however, and mice constitutively expressing this mutant form of fibrinogen exhibited a major impediment in inflammatory cell clearance of Staphylococcus aureus in the context of acute peritonitis.
Methods
Generation of gene-targeted mice.
The fibrinogen γ chain targeting vector was constructed using a portion of the mouse gene cloned from a 129-strain genomic DNA library. PCR-based mutagenesis was used to convert the exon 9 sequence encoding amino acid residues 390–396 of the mature protein (N390RLSIGE396) to a series of alanines. The selected nucleotide substitutions also resulted in the introduction of a PvuII site that served to flag the mutant allele in establishing animal genotypes. All nucleic acid substitutions were confirmed by DNA sequence analysis. HPRT and HSV-tk minigenes were introduced into the γ chain targeting vector as described (23). E14TG2a (24) ES cells that had incorporated the targeting vector by homologous recombination were initially identified by PCR analysis using primers complementary to the HPRT minigene (primer 5: 5′-CCTGAAGAACGAGATCAGCAGCCTCTGTTC-3′) and the γ chain gene (primer 6: 5′-ATACATGGATATTAGCCAGGCAGTAGTGAC-3′) that generated a PCR product of 754 bp. Positive clones were confirmed using a second primer set complementary to the HPRT minigene (primer 4: 5′-AAATGCTCCAGACTGCCTTG-3′) and the γ chain gene (primer 3: 5′-ATTGACATGATCACCAAAATTGCTTATTG-3′), which yielded a 5,735-bp PCR product. Homologous recombination of the targeting vector was further confirmed by Southern blot analysis of PvuII-digested genomic DNA. A hybridization probe was prepared from a 320-bp NcoI/PvuII restriction enzyme fragment located downstream of the fibrinogen γ chain gene. Mice were routinely genotyped by PCR analysis using primers immediately upstream (primer 1: 5′-ATTGACATGATCACCAAAATTGCTTATTG-3′) and downstream (primer 2: 5′-CCATTTAAGGCTAGGTATCATCTTAAGAAAG-3′) of exon 9, which yielded a PCR product of 527 bp. The PCR product from the mutant allele was recognized by the generation of 304-bp and 223-bp fragments following PvuII digestion. Founder mice were bred to NIH Black Swiss females (Taconic Farms, Germantown, New York, USA) to generate heterozygous mice that were then interbred to produce homozygous mutant animals. Animals carrying the mutant allele were subsequently backcrossed six generations to C57Bl/6 mice, and these C57Bl/6-inbred mice were used in all of the in vivo studies presented.
Hematological analyses.
Hematological analyses of blood cells were done using a Cell-Dyn 4000 analyzer, and plasma thrombin times were established as described previously (25). Fibrin polymerization was evaluated by standard turbidity assays using both plasma and purified fibrinogen. Briefly, citrate plasma (diluted tenfold in 20 mM HEPES, pH 7.4, containing 0.15 M NaCl and 5 mM ε-amino caproic acid) or 0.15 mg/ml purified fibrinogen in the same buffer was combined with bovine thrombin (final concentration 0.2 U/ml; Enzyme Research Laboratories, South Bend, Indiana, USA), and Ca2+ (10 mM), and OD350 measurements were taken every 30 seconds. Western blot analysis of plasma fibrinogen was performed using rabbit anti-mouse fibrinogen in conjunction with an ECL detection system (Amersham Pharmacia Biotech, Piscataway, New Jersey, USA) (25). Covalent cross-linking of the fibrinogen γ chain by factor XIIIa was analyzed using purified fibrinogen. Briefly, 0.25 mg/ml fibrinogen was combined with 2 U/ml bovine thrombin, 0.15 mg/ml human factor XIII (Enzyme Research Laboratories), and 25 mM Ca2+ and incubated for up to 10 minutes at 37°C. Reactions were terminated by addition of 2% SDS, 10% β-mercaptoethanol, and 0.1 M EDTA, and the fibrin chains were analyzed by SDS-PAGE.
Platelet aggregation and flow cytometry.
Platelet aggregation was performed at 37°C using a ChronoLog 560 aggregometer as described (25). Briefly, platelet suspensions in autologous plasma were prepared from WT, Fibγ390–396A, and FibγØ5 mice (23) and adjusted to approximately 300,000 platelets per microliter. Aggregation was initiated with ADP (final concentration of 10 ∝M). Flow-cytometric analyses of fibrinogen binding to ADP-activated platelets by αIIbβ3 were done using a FITC-conjugated rabbit anti-human fibrinogen Ab (DAKO A/S, Glostrup, Denmark), as previously described (23).
Purification of plasma fibrinogen.
Fibrinogen was isolated from citrated mouse plasma using Gly-Pro-Arg affinity chromatography (26). The affinity matrix was prepared using a Gly-Pro-Arg-Pro-Cys peptide (Invitrogen Corp. Carlsbad, California, USA) conjugated to SulfoLink coupling gel (Pierce Biotechnology Inc., Rockford, Illinois, USA). Purified fibrinogen was then dialyzed exhaustively against 20 mM HEPES, pH 7.4, containing 0.15 M NaCl and 5 mM ε-amino caproic acid. Fibrinogen concentration was determined spectrophotometrically and its quality established by SDS-PAGE.
Thrombus formation following FeCl3 injury in vivo.
Thrombosis was induced in the carotid artery of anesthetized mice using FeCl3 as described in detail elsewhere (27). Thrombus formation was recorded for up to 30 minutes using a miniature video camera (ProVideo CVC-514; CSI/SPECO, Amityville, New York, USA). Blood flow through the carotid artery was monitored using a Doppler flow probe (0.5VB307; Transonic Systems Inc., Ithaca, New York, USA) connected to a flow meter (T106; Transonic Systems Inc.). After the 30-minute recording period, anesthetized animals were perfused with 4% paraformaldehyde in PBS, and the injured carotid arteries were collected for electron microscopy and histology.
Cell adhesion assays using cell lines and primary phagocytes.
Human embryonic kidney 293 (HEK293) cell lines expressing β2 integrins were described previously (21, 28). THP-1 monocytoid cells were obtained from American Type Culture Collection (Manassas, Virginia, USA). Human neutrophils were isolated from heparinized peripheral blood from consenting volunteers by centrifugation though leukocyte separation media (Histopaque-1119 and Histopague-1077; Sigma-Aldrich, St. Louis, Missouri, USA) according to the manufacturer’s recommendations. Human blood monocytes were isolated from EDTA anticoagulated peripheral blood of consenting volunteers according to an established protocol (29, 30). Mouse resident peritoneal macrophages were isolated from lavage fluid by placing cell suspensions in serum containing RPMI-1640 on uncoated Cellstar microtiter plates. After incubation for 4 hours at 37°C, unbound cells were aspirated, and the adherent macrophages were subsequently removed using ice-cold PBS containing 2 mg/ml glucose and 0.5 mM EDTA. These preparations were shown to be primarily macrophages by staining cytospin preparations with Diff-Quik (Dade Behring Inc., Deerfield, Illinois, USA). Prior to use in adhesion assays, cell preparations were suspended in either HBSS (HEK293 cells and monocytes) containing 5 mM Ca2+ and 5 mM Mg2+ or serum-free RPMI-1640 (THP-1, neutrophils, and macrophages) at a concentration of 0.5 ∞ 105/ml to 1 ∞ 106/ml.
Cell adhesion assays were performed in 48-well or 96-well polystyrene plates (Costar; Corning Inc., Corning, New York, USA) that were coated with purified fibrinogen. Nonspecific binding was blocked using either 1% polyvinylpyrrolidone (HEK293 cells, primary monocytes, and primary macrophages) or 1% nonfat dry milk (THP-1 cells and primary neutrophils). HEK293 cells were harvested using cell-dissociation buffer (Invitrogen Corp.). Aliquots of cells (200 ∝l) were added to wells and incubated at 37°C for 25 minutes and subsequently washed with buffered saline to remove nonadherent cells. Inhibition experiments were performed using the following Ab’s: M1/70 rat anti-mouse αM (known to block both mouse and human αMβ2; eBioscience, San Diego, California, USA), IB4 anti-β2 (American Type Culture Collection), or control rat anti-mouse IgG (Pierce Biotechnology Inc.). Ab’s were incubated with cells at 20 μg/ml for 15 minutes at room temperature before transfer to coated wells. The number of adherent cells was determined by either counting multiple high-powered (∞100) fields or using a CyQuant (Molecular Probes Inc., Eugene, Oregon, USA) fluorescence detection system. All analyses were performed in triplicate.
Differential analysis of resident peritoneal cells.
Peritoneal lavage fluid was collected by injection of 5 ml of PBS into the abdominal cavity of anesthetized mice. The total number of cells present within the lavage fluid was determined manually using a hemocytometer. Differential cell counts were made using cytospin preparations stained with Diff-Quik (Dade Behring Inc.). The percentage of neutrophils was confirmed by Leder stain. Flow cytometry was performed using fluorochrome-labeled rat anti-mouse CD3 (BD Biosciences, San Jose, California, USA), rat anti-mouse CD19 (BD Biosciences), and rat anti-mouse F4/80 (CalTag Laboratories Inc., Burlingame, California, USA) to confirm the percentage of T cells, B cells, and macrophage/monocytes, respectively.
Clearance of intraperitoneal S. aureus.
A WT Newman strain of S. aureus and a clumping factor A–deficient (ClfA-deficient) derivative (DU5852) (kindly provided by T.J. Foster, Trinity College, Dublin, Ireland) were employed. Overnight cultures of bacteria were harvested by centrifugation, washed, and then resuspended in cold sterile saline at 4 ∞ 108/ml to 10 ∞ 108/ml. In experiments using heat-killed bacteria, suspensions were boiled for 5 minutes prior to injection. Bacterial kill was confirmed by plating dilutions of the boiled suspensions on tryptic soy agar. Mice were given an intraperitoneal injection with 1 ml of bacterial suspension, and the peritoneal lavage fluids were later collected using ice-cold buffered saline as described above. Serial dilutions were plated on tryptic soy agar to establish the bacterial CFUs.
Results
Site-directed mutagenesis of the endogenous fibrinogen γ chain gene.
To determine the physiological and pathological importance of fibrin(ogen) as a ligand for αMβ2, we generated a replacement-type gene-targeting vector to selectively eliminate the αMβ2-binding site previously identified within the carboxy-terminal portion of the fibrinogen γ chain (21). The specific residues selected for mutation, N390RLSIGE396, were chosen based on four findings and/or criteria. First, this sequence was a consistent element within P2 peptides, known to block αMβ2 binding to immobilized fibrinogen. Second, these amino acids were conserved between species (note that the sequence is N390RLTIGE396 in the human molecule). Third, these residues were largely exposed to solvent based on the crystal structure of fibrinogen derivatives. Finally, this sequence was spatially far removed from the γ chain “hole” known to support fibrin polymerization (31). Thus, an alteration within residues 390–396 was not expected to alter clotting function. Using portions of the cloned mouse γ chain gene and a PCR-based mutagenesis strategy, a targeting vector was generated in which 12 nucleotide substitutions were introduced within the exon 9 sequence (Figure 1, A and B). These substitutions simultaneously converted the codons encoding γ chain residues 390–396 to a series of seven alanine residues and introduced a diagnostic PvuII site. Embryonic stem cell transfectants that had incorporated the targeting vector by homologous recombination based on PCR analyses (see Figure 1A and Methods) were used to generate transgenic founder mice. The introduction of the Fibγ390–396A targeting vector into the mouse genome by homologous recombination was confirmed by both PCR (see representative data in Figure 1C) and Southern blot analysis of genomic DNA purified from tail biopsies. Crosses between hemizygous mice established that the Fibγ390–396A mutation was compatible with Mendelian inheritance; of the first 235 pups generated from hemizygous breeding pairs, 62 (26%) carried only the WT allele (WT/WT), 107 (46%) were hemizygous (WT/γ390–396A), and 66 (28%) carried only the mutant allele (γ390–396A/γ390–396A, hereafter simply referred to as Fibγ390–396A mice). Unlike fibrinogen-null mice (25), Fibγ390–396A mice never developed overt spontaneous bleeding events, and the survival profile of unchallenged Fibγ390–396A mice was indistinguishable from WT littermates. Furthermore, unlike fibrinogen-null females, which uniformly develop fatal intrauterine bleeding during pregnancy, Fibγ390–396A females exhibited excellent reproductive success and were capable of supporting multiple pregnancies to term (data not shown). Finally, Fibγ390–396A offspring could be consistently raised from homozygous mutant parents, indicating that neither reproduction nor development was compromised by the mutant fibrinogen, regardless of its maternal and/or embryonic source.
Figure 1.

Modification of the fibrinogen γ chain gene. (A) Overall structure of the Fibγ390–396A gene-targeting vector, the WT fibrinogen γ chain gene, and the targeted Fibγ390–396A allele. Exons are depicted as solid areas, and the introduced nucleotide substitutions and PvuII site are indicated in text boxes as 390–396A and PvuII, respectively. The PvuII fragments that were diagnostic for the WT and targeted alleles by Southern blot assays are indicated by thin lines. Arrowheads indicate the position of diagnostic PCR primers used for detecting homologous recombination events in ES cells. Routine animal genotyping was done using primers 1 and 2 followed by PvuII digestion. (B) Partial nucleotide sequence of exons 9 and exon 10 of the WT and γ390–396A genes. Asterisks indicate nucleotides that were mutated in the Fibγ390–396A allele. A vertical arrow indicates the position of the exon 9 to 10 splice junction. Amino acids altered by the nucleotide substitutions are in italics. Underlined amino acids indicate the glutamine and lysine that participate in transglutaminase-mediated cross-linking. Amino acids that are known to be critical for platelet integrin receptor αIIbβ3 binding (23) are bracketed. (C) Representative PCR analyses to establish animal genotypes using DNA template from ear biopsies of WT, hemizygous, and homozygous mutant Fibγ390–396A mice. Primers 1 and 2 were used to amplify a 527-bp fragment, which was subsequently digested with PvuII to yield the diagnostic fragments of 304 bp and 223 bp. ddH2O, double distilled water.
Fib γ 390–396A mice carry normal levels of fibrinogen and exhibit a normal hematological profile.
The seven amino acid substitutions introduced into the γ chain did not alter the steady-state concentration or size of the component chains within circulating fibrinogen based on Western blot analyses (see Figure 2). The overall structural integrity of the mutant plasma fibrinogen was further established by comparative analysis of purified fibrinogen (Figure 2). The component fibrinogen chains were indistinguishable in both ratio and size in WT and Fibγ390–396A mice. A more general hematological analysis of blood cells showed that the complete blood cell counts, including red cells, white cells, and platelets, were virtually identical in control (n = 6) and Fibγ390–396A (n = 6) mice (Table 1). The fact that the mutant fibrinogen did not alter blood platelet count is consistent with the view that fibrinogen γ390–396A presents no hemorrhagic consequences that might lead to inordinate platelet consumption.
Figure 2.

Characterization of Fibγ390–396A fibrinogen. (A) Western blot of fibrinogen in plasma from WT and mutant mice. (B) Coomassie blue–stained SDS polyacrylamide gel (reducing conditions) showing affinity-purified fibrinogen preparations from WT and Fibγ390–396A mice. (C) Comparative analysis of thrombin-induced fibrin polymerization in plasma from WT and homozygous Fibγ390–396A mice. (D) Analysis of fXIIIa-mediated fibrin cross-linking in reaction mixtures containing either purified WT or γ390–396A fibrinogen. The electrophoretic positions of Aα, Bβ, γ chains are indicated at left along with γ-γ dimer and α polymer cross-linking products.
Table 1.
Hematological analysis of WT and Fibγ390–396A mice

Fibrinogen γ390–396A maintains normal coagulation properties.
One objective in generating the Fibγ390–396A mutation was to explore the potential role(s) of fibrinogen in innate immunity in a context where clotting function was in no way compromised. To confirm that the Fibγ390–396A mutation did not impede fibrin polymer formation, multiple coagulation analyses were performed. Thrombin clotting time is sensitive to both fibrinogen concentration and structure. As shown in Table 1, plasma samples from WT and Fibγ390–396A mice exhibited indistinguishable clotting times following addition of bovine thrombin and Ca2+. As a second comparative analysis of fibrin polymer formation, standard turbidity measurements were made as a function of time following addition of thrombin and Ca2+ to diluted plasma (see Figure 2C for representative polymerization profiles) or purified fibrinogen (data not shown) prepared from control and Fibγ390–396A mice. No significant difference in polymerization profile was observed using samples prepared from multiple WT and Fibγ390–396A mice. Both the lag phase (indicative of the time required for fibrinopeptide release) and the subsequent fibrin monomer assembly rates were indistinguishable using multiple preparations of WT and mutant fibrinogen. Furthermore, detailed electrophoretic analyses of denatured and reduced fibrin clots prepared using purified fibrinogen from control and mutant mice showed that there was no apparent difference in factor XIIIa-catalyzed (transglutaminase-catalyzed) cross-linking of the γ chain to form γ-γ dimers (Figure 2D). The γ-γ dimers were also readily detected within plasma clots from both WT and Fibγ390–396A mice (data not shown). These data infer that the overall structural integrity of fibrinogen is maintained in the γ390–396A mutant and the most dramatic property of fibrinogen, the ability to polymerize into an insoluble matrix, was preserved in fibrinogen γ390–396A.
Fibrinogen γ390–396A supports platelet aggregation and normal thrombus formation.
By engaging the platelet integrin αIIbβ3, fibrinogen supports platelet aggregation and promotes formation of stable platelet thrombi at sites of vascular damage. To determine if the γ390–396A mutation altered the ability of fibrinogen to engage αIIbβ3, mutant and WT fibrinogen were compared for (a) their binding activity to ADP-activated platelets (b) their ability support ADP-induced platelet aggregation in vitro, and (c) their ability to support thrombus formation in vivo. Using an established flow-cytometry approach, we found no quantitative difference in WT and γ390–396A fibrinogen binding to αIIbβ3 on ADP-activated platelets (see supplemental material; available at http://www.jci.org/cgi/content/full/113/11/1596/DC1). Furthermore, ADP-induced platelet aggregation in vitro followed a similar pattern using platelet-rich plasma from WT and Fibγ390–396A animals (Figure 3). To control for the fibrinogen dependence of platelet aggregation in these experiments, parallel studies were done using FibγØ5 mice (23), a mutant line expressing a form of fibrinogen lacking the αIIbβ3-binding motif. As shown previously, no ADP-induced platelet aggregation was observed (Figure 3). As a final test of the ability of fibrinogen γ390–396A to support platelet/fibrin deposition, thrombus formation within FeCl3-injured carotid arteries was compared in WT and Fibγ390–396A mice by both real-time intravital videomicroscopy and scanning electron microscopy of fixed tissues. No appreciable difference was observed in the time to vessel occlusion (10.2 ± 0.9 minutes, n = 4 for WT and 10 ± 2 minutes, n = 3 for Fibγ390–396A animals) or thrombus stability/appearance (see supplemental material). Therefore, in this model, thrombus formation in vivo appears to be unimpaired in mice expressing fibrinogen γ390–396A.
Figure 3.

Fibrinogen γ390–396A supports normal platelet aggregation. Comparative analysis of ADP-induced platelet aggregation in vitro using platelet-rich plasma prepared from WT and Fibγ390–396A mice. The fibrinogen dependence of aggregation in vitro was demonstrated with FibγØ5 mice that express a mutant form of fibrinogen lacking the αIIbβ3-binding motif. The decline in transmitted light that immediately follows ADP addition is a consequence of platelet shape change.
Fibrinogenγ390–396A does not supportαM β2-mediated cell adhesion in vitro.
To test the central hypothesis that the γ390–396A mutation would specifically eliminate αMβ2 engagement of fibrinogen, affinity-purified fibrinogen preparations from WT and Fibγ390–396A mice were immobilized on microtiter plates and compared for their ability to support αMβ2-dependent cell adhesion. Our initial studies focused on a cell type that plays a critical role in the inflammatory response in vivo and that is known to engage immobilized fibrin(ogen) almost exclusively through αMβ2 in vitro — primary neutrophils (7, 32). Consistent with earlier reports, isolated human peripheral blood neutrophils were found to readily adhere to immobilized WT fibrinogen (Figure 4A), and this adhesion was largely αMβ2-dependent based on inhibitor studies with the αM-specific mAb, M1/70 (Figure 4D). More significantly, purified fibrinogen γ390–396A failed to support the adhesion of primary human neutrophils (Figure 4, B and D). This profound difference in cell adhesion to WT and γ390–396A fibrinogen was not a trivial reflection of a difference in the amount of fibrinogen immobilized; quantitative immunological analyses indicated that WT and mutant fibrinogen was absorbed onto the surface of plastic wells with equal efficiency (data not shown). As a final specificity control in these cell adhesion studies, we showed that another fibrinogen derivative, fibrinogen γØ5, lacking the carboxy-terminal γ chain residues 407–411, supported αMβ2-dependent neutrophil adhesion to the same extent as WT fibrinogen (Figure 4, C and D).
Figure 4.

Fibrinogen γ390–396A does not support αMβ2-dependent adhesion of primary neutrophils. Primary human neutrophils (106 cells/ml) were transferred to either uncoated wells, wells coated with 10 ∝g/ml WT mouse fibrinogen (A), wells coated with 10 ∝g/ml fibrinogen γ390–396A (B), or wells coated with 10 ∝g/ml fibrinogen γØ5 (C). Shown are representative views of bound cells after 25 minutes. (D) Quantitative comparison of neutrophil adhesion to WT, fibrinogen γ390–396A, and fibrinogen γØ5. The specificity of neutrophil engagement was established by preincubating neutrophils with 20 ∝g/ml of the rat anti-mouse αM I-domain Ab, M1/70, or control rat anti-mouse IgG for 15 minutes at room temperature before addition to the fibrinogen-coated wells. The data shown are means ± SD. HPF, high power field; Con, control.
These comparative studies of primary leukocyte adhesion to WT and γ390–396A fibrinogen were also extended to isolated peripheral blood human monocytes (Figure 5A) and mouse resident peritoneal macrophages (Figure 5B) with a similar outcome. As expected, monocytes and macrophages exhibited excellent adhesion to WT fibrinogen, and this adhesion was effectively blocked by αMβ2-blocking Ab’s to either the αM subunit (e.g., 44a or M1/70) or β2 subunit (e.g., IB4), but not control Ab’s. Neither primary monocytes nor macrophages were adherent to fibrinogen γ390–396A, however (Figure 5, A and B). A similar pattern was also seen with THP-1 cells, a human monocytic cell line known to express high levels of αMβ2 and to adhere to fibrinogen through αMβ2 (33). Robust, concentration-dependent adhesion of THP-1 cells was observed for WT, but not γ390–396A, fibrinogen (Figure 5, C and D). In agreement with previous reports that αMβ2 engagement of fibrinogen is conformation dependent (i.e., strong when fibrinogen is surface immobilized or converted to fibrin, but weak with soluble fibrinogen), THP-1 cell adhesion to immobilized WT fibrinogen was not inhibited by either soluble WT or γ390–396A fibrinogen, even at concentrations that were orders of magnitude above the Kd of αMβ2 for other established high-affinity ligands (data not shown). Taken together, these data suggest that fibrinogen γ390–396A neither supports αMβ2-dependent cell adhesion in vitro nor results in a gain-of-function property whereby the soluble molecule is bound with high affinity by αMβ2.
Figure 5.

Fibrinogen γ390–396A fails to support αMβ2-dependent adhesion of monocytes, macrophages, or the monocytoid cell line, THP-1. (A) Comparative analysis of primary human blood monocyte adhesion to immobilized WT and γ390–396A fibrinogen (coating concentration 2 ∝g/ml). Specificity of integrin-mediated binding was established by preincubation of cells with blocking αM- or β2-specific mAb’s (44a and IB4, respectively). The nonblocking αM mAb, OKM1, was used as a control. (B) Comparative analysis of mouse peritoneal macrophage adhesion to WT and γ390–396A fibrinogen. The blocking Ab M1/70 was used to demonstrate αMβ2-specificity. (C) THP-1 cell adhesion as a function of fibrinogen coating concentration using either WT or γ390–396A fibrinogen. (D) Adhesion of THP-1 cells to WT or γ390–396A fibrinogen in the presence of either EDTA or αMβ2 Ab, M1/70. The data shown are means ± SD. fl, fluorescence.
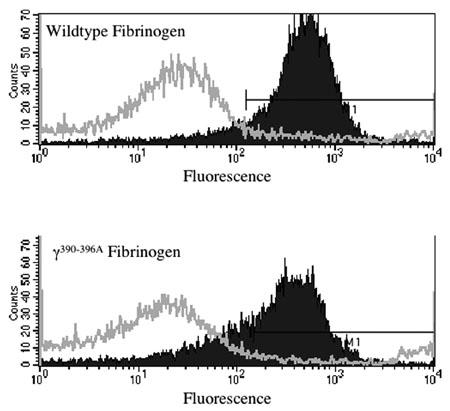
To more rigorously define the specific impact of the fibrinogen γ390–396A mutation on interaction with β2 integrin receptors, we evaluated the binding potential of HEK293 transfectants expressing αMβ2 or the related integrin, αLβ2 (21, 28, 34). Consistent with the requirement for a specific β2 integrin to support cell adhesion to fibrinogen, mock transfectants (empty expression vector) or transfectants expressing αLβ2 (an integrin known not to bind fibrinogen) failed to adhere to any immobilized fibrinogen preparation (Figure 6A), whereas HEK293 transfectants expressing αMβ2 displayed excellent adhesion to WT fibrinogen (Figure 6, A and B). More importantly, immobilized fibrinogen γ390–396A failed to support HEK293 cell adhesion regardless of the presence or absence of αMβ2 or αLβ2 (Figure 6, A and B). Similar results were observed in multiple independent experiments.
Figure 6.

Cell adhesion to fibrinogen is supported by integrin αMβ2, but not the related integrin, αLβ2. (A) Adhesion of HEK293 transfectants expressing either αMβ2, αLβ2, or no β2 integrin (mock transfectants) to microtiter wells coated with either 10 ∝g/ml WT or γ390–396A fibrinogen. (B) Adhesion of αMβ2-expressing HEK293 cells to WT or γ390–396A fibrinogen in the absence or presence of Ab’s against the αM I-domain (M1/70), β2 (IB4), and irrelevant rat anti-mouse IgG. The data shown are means ± SD.
Leukocyte clearance of peritoneal bacteria is strongly impeded in mice expressing fibrinogen γ390–396A.
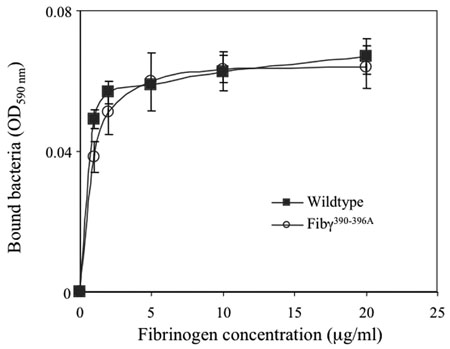
One important incentive for using a gene-targeting approach in mice was that it provided an opportunity to readily define the biological significance, if any, of αMβ2-mediated leukocyte engagement of fibrin(ogen) in vivo. To begin to explore the in vivo consequences of the fibrinogen γ390–396A mutation on the innate immune system, we compared the ability of inflammatory cells to clear the common gram-positive pathogen, S. aureus, following intraperitoneal inoculation. Quantitative analysis of viable bacteria within the peritoneal lavage fluid from cohorts of WT and mutant mice 1 hour after infection revealed that Fibγ390–396A mice exhibited a profound impediment in bacterial clearance relative to control animals in this acute peritonitis model (Figure 7A). This same pattern of inefficient bacterial clearance in Fibγ390–396A mice was observed when mice were challenged with a mutant strain of S. aureus lacking the bacterial fibrinogen receptor, termed ClfA (Figure 7B). One inference of these results is that host leukocyte engagement of fibrinogen through αMβ2, rather than bacterial engagement of fibrinogen through ClfA, is the key determinant of bacterial clearance in the context of acute peritonitis. In this regard, it should be noted that control studies showed that fibrinogen γ390–396A, like WT fibrinogen, retained the ability to support ClfA-dependent S. aureus adhesion in vitro (see supplemental material).
Figure 7.

Fib γ390–396Amice exhibit a severe impediment in innate immunity resulting in inefficient clearance of S. aureus in an acute peritonitis model. (A) WT S. aureus was introduced by intraperitoneal injection into WT or Fibγ390–396A mice, and peritoneal lavage fluid was collected from each animal 1 hour after inoculation to determine the total number of CFUs. (B) Comparative analysis of bacterial clearance in WT and Fibγ390–396A mice challenged with a S. aureus strain lacking the bacterial fibrinogen-binding protein ClfA. Note that bacterial clearance is inefficient in Fibγ390–396A mice relative to WT animals regardless of the presence or absence of ClfA. The data shown are means ± SDs. P < 0.035 and P < 0.021 for WT and ClfA S. aureus, respectively (Mann-Whitney U test).
The genotype-dependent difference in bacterial clearance within the abdominal cavity was evident even as early as 30 minutes after infection, but was more pronounced at 3 hours as a consequence of the expansion of viable microbes (Figure 8A). Consistent with a defect in the activation of leukocyte antimicrobial functions in Fibγ390–396A mice, microscopic analysis of lavage fluid collected 3 hours after S. aureus inoculation revealed few appreciable bacteria on cytospin spreads from WT mice, whereas both free and leukocyte-associated bacteria were prevalent in the mutant mice (Figure 8B). Many phagocytes appeared to be overwhelmed by bacteria and seemed physically disrupted. A similar failure to effectively clear peritoneal S. aureus has also been documented in fibrinogen-null mice (Du et al., unpublished observations), further underscoring the fibrinogen dependence of this clearance process. The difference in early bacterial clearance was not due to any genotype-related difference in either the number or composition of resident peritoneal leukocytes; quantitative analysis of peritoneal lavage fluids collected from unchallenged WT and Fibγ390–396A mice indicated that the number of peritoneal macrophages, neutrophils, and mast cells (all of which are known to express αMβ2) was indistinguishable in WT and Fibγ390–396A mice (Figure 8C). Similarly, whole blood analyses showed that the total number and differential counts of circulating white cells was not different in WT and Fibγ390–396A mice (data not shown). Furthermore, the impediment in bacterial clearance in Fibγ390–396A was not due to any obvious impediment in leukocyte trafficking. A significant increase in the neutrophil infiltrate was observed within the peritoneal cavity of both control and Fibγ390–396A mice within 3 hours after infection (compare Figure 8, C and D). Although the neutrophil infiltrate tended to be modestly diminished in Fibγ390–396A mice in these studies, the difference did not reach statistical significance (P = 0.08 in the Mann-Whitney U test). Given that phagocytes were clearly failing in Fibγ390–396A mice, and often appeared to be themselves disrupted by engorged bacteria in cytospin spreads (see Figure 8B), we further explored leukocyte trafficking in WT and Fibγ390–396A mice in response to heat-killed S. aureus. Analyses of lavage fluids collected from control and Fibγ390–396A mice 5 hours after introduction of nonviable bacteria showed that there was a pronounced increase in total peritoneal neutrophils in challenged mice of both genotypes (Figure 8E) relative to unchallenged animals (Figure 8C). More notably, Fibγ390–396A mice exhibited the same robust neutrophil efflux that was observed in WT controls (Figure 8E). A similar increase in the number of leukocytes present within the peritoneal cavity was observed in control and mutant mice at 24 hours, with macrophages being a predictably higher fraction of total leukocytes (Figure 8F). Thus, neither neutrophil nor macrophage trafficking appears to be appreciably impeded by the loss of the αMβ2-binding motif on fibrinogen, a finding that is consistent with the fact that leukocyte trafficking is generally maintained in αMβ2-deficient mice. Rather, the loss of the fibrin(ogen)-αMβ2 interactions appear to compromise leukocyte activation pathways whereby host inflammatory cells kill microbial pathogens.
Figure 8.

Time course analysis of S. aureus clearance and leukocyte trafficking within the peritoneal cavity of WT and Fibγ390–396A mice following intraperitoneal infection. (A) Higher levels of S. aureus in Fibγ390–396A mice relative to WT animals were appreciable 30 minutes after inoculation, and obvious differential expansion was apparent after 3 hours. P < 0.02, Mann-Whitney U test (n = 5 pairs). (B) Representative microscopic views of peritoneal lavage cytospins prepared at the 3-hour time point from WT (left) and Fibγ390–396A mice (right). Bacterial phagocytosis (arrows) was found in both genotypes, but few residual bacteria were found within WT mice. In contrast, leukocytes from mutant animals appeared overwhelmed with bacteria, which often led to phagocyte disruption (double arrowhead). (C–F) Total cell counts and leukocyte differentials within peritoneal lavage fluid from WT and Fibγ390–396A mice. Unchallenged mice (C) displayed no significant genotype-related difference in resident cells (P – 0.5, Mann-Whitney U test using four pairs). Three hours after infection (D) a major increase in neutrophil levels was observed in mice of both genotypes compared with naive mice (compare panels C and D), with a trend toward fewer total cells and fewer neutrophils in mutant mice (P ♠ 0.1, Mann-Whitney U test using five pairs). However, comparison of WT and Fibγ390–396A mice challenged with heat-killed bacteria for 5 hours (E) or 24 hours (F) indicated that there was, if anything, an increase in total leukocyte and neutrophil accumulation in mutant mice (P > 0.2 for all comparisons between genotypes using a Mann-Whitney U test using five pairs). The data shown are means ± SD. Neut, neutrophil; Mac, macrophage.
Discussion
The leukocyte integrins αMβ2 and αLβ2 are central mediators of leukocyte adhesion, transmigration, activation, and the expression of specialized functions. Despite the fact that these two integrins share an ability to bind certain ligands (e.g., ICAM-1), it is increasingly clear that these receptors are not functionally equivalent in their binding properties, both in terms of their repertoire of ligands and in terms of their differential binding to shared ligands (8, 10, 35, 36). These integrins seem to play complementary, albeit partially overlapping, roles in regulating leukocyte function (7, 8, 10, 11, 37–39). One obvious distinction between αMβ2 and αLβ2 is that the former integrin can engage immobilized fibrin(ogen) and other ligands that are not recognized by the later receptor (36). To better understand the interplay between hemostatic factors and the inflammatory response in vivo and to specifically define the biological significance, if any, of αMβ2 engagement of fibrin(ogen), we genetically modified the endogenous fibrinogen γ chain gene in mice to selectively eliminate the αMβ2-binding motif in the carboxy-terminal portion of the molecule. As an experimental approach to specifically discern the functional importance of the fibrinogen-αMβ2 interaction in vivo, the generation of this fibrinogen variant offered the distinct advantage of neither imposing any alteration in αMβ2 itself nor precluding αMβ2 interaction with other potential ligands or crosstalk with other receptors. Here we show that homozygous mice carrying the mutant fibrinogen γ390–396A allele are viable to adulthood, never experience spontaneous bleeding events, carry normal levels of circulating fibrinogen, maintain normal clotting function, retain normal fibrinogen engagement by other integrin receptors (e.g., αIIbβ3), retain normal platelet aggregation, and exhibit normal thrombus formation in vivo. Unlike WT fibrinogen, however, immobilized fibrinogen γ390–396A failed to support αMβ2-mediated adhesion of a variety of cell types, including primary neutrophils and macrophages. Most importantly, the disruption in αMβ2 engagement of fibrin(ogen) was found to have dramatic consequences on the inflammatory response in vivo. Fibγ390–396A mice exhibited a remarkable impediment in the elimination of the microbial pathogen, S. aureus, in an acute peritonitis model. Four major conclusions are drawn from these studies. First, fibrin(ogen) is an important regulator of inflammatory cell function and innate immunity. Second, fibrin(ogen) constitutes a physiologically relevant ligand for the leukocyte integrin αMβ2. Third, the biological consequences of a loss in the αMβ2-fibrin(ogen) interaction is not (fully) compensated by the continued availability of all other potential ligands (e.g., ICAM-1, iC3b, GPIbα, uPAR, etc). Finally, the biological importance of fibrinogen in regulating the inflammatory response can be appreciated outside of any alteration in either clotting function or platelet thrombus formation.
The specific inflammatory processes that are impeded in mice expressing fibrinogen γ390–396A remains to be fully defined, but a few inferences can be drawn based on the present and previous studies. Consistent with the observation that αMβ2 is not strictly required for efficient leukocyte trafficking in many inflammatory settings in vivo (7, 8), the loss of the αMβ2-binding motif on fibrinogen did not restrict leukocyte trafficking in response to intraperitoneal bacteria, live or heat killed. A prevailing hypothesis consistent with the available data is that αMβ2 primarily controls leukocyte function upon arrival at sites of inflammatory challenge (7, 8, 11). A simple extension of this theory is that in the context of soluble inflammatory mediators, leukocyte engagement of immobilized fibrin(ogen) within inflamed and/or damaged tissues may be an important cue in leukocyte target recognition, ultimately regulating the expression of specialized functions. Consistent with this view, neutrophil engagement of fibrin(ogen) through αMβ2 results in dramatic cellular changes in vitro (15–18, 40–42), including calcium mobilization, activation of NF-κB, increased phosphorylation events, degranulation, upregulation of cell surface adhesion molecules, increased migration, and decreased apoptosis. The concept that leukocyte interaction with immobilized fibrin(ogen) is an important event in target recognition has two attractive features. First, fibrin could provide a unique, nondiffusible or spatially defined signal modulating inflammatory cell function. Second, fibrin would be found within the ECM at virtually any site of tissue damage, regardless of the underlying insult, but would be distinctly absent within normal tissues. Thus, fibrin could provide a universal cue flagging the precise site of any challenge and provide another means to locally regulate leukocyte function. Of course, this theory does not preclude the seminal contribution of soluble inflammatory mediators (e.g., cytokines and chemokines) or a significant contribution of other αMβ2 ligands (e.g., iC3b). Nevertheless, the present studies show that even when the engagement of all other ligands remains intact, the loss of αMβ2 interaction with fibrin(ogen) compromises leukocyte function, including the ability to efficiently clear an infectious agent in vivo.
The present data show that in the context of intact fibrinogen the preeminent αMβ2-binding motif is located in the carboxy-terminal portion of the γ chain. This region may not constitute the sole αMβ2-binding element. Other regions, including the AαE splice variant found in just 2% of plasma fibrinogen, might also contribute in vivo to αMβ2 binding (43). Presuming, however, that the fraction of fibrinogen carrying AαE remains a constant feature of both WT and Fibγ390–396A mice, any αMβ2-binding potential conferred by the AαE domain would appear to be comparatively poor relative to the contribution of the far more abundant standard γ chain.
Given that the αMβ2-binding motif on fibrinogen is tied to the inflammatory response in vivo, then a question still unresolved is the relative importance of the many distinct forms of fibrin(ogen) in regulating leukocyte function, including (a) fibrin matrices (b) fibrinogen immobilized on cell surfaces by other specific receptors (e.g., αvβ3, α5β1, αIIbβ3, ICAM-1), and (c) soluble fibrinogen. The fact that circulating leukocytes would be constantly exposed to high concentrations of fibrinogen would seem to preclude any obvious utility of the soluble molecule in defining inflammatory processes. Furthermore, soluble fibrinogen is a relatively poor ligand for αMβ2 (20). The fact that neutrophils avidly engage surface-immobilized fibrinogen or fibrin by αMβ2 implies that these forms of fibrin(ogen) are likely to be the most instructive to inflammatory cells in vivo. Regardless of the relative importance of fibrin polymer formation, the present findings show that there are functional elements within fibrinogen that are relevant to the inflammatory response distinct from those critical for generating a provisional fibrin matrix.
While bacterial clearance is clearly inefficient in Fibγ390–396A mice relative to control animals, there appears to be a significant residual capacity to eliminate microbes in the mutant animals. This residual capacity to control S. aureus may be a reflection of either αMβ2-independent leukocyte activation pathways or αMβ2-dependent signaling events mediated by alternative sites on fibrinogen and/or alternative ligands (e.g., bacterial opsonization with iC3b) (16, 44). The residual capacity to contain overt microbial challenges in Fibγ390–396A mice is not surprising based on two previous observations. First, spontaneous infections are not commonplace in fibrinogen-null mice, at least when adaptive immunity remains intact (25). Second, while spontaneous infections are frequently observed in the absence of all β2 integrins, these are not commonplace in the absence of just αMβ2 (5, 7, 45). Nevertheless, like Fibγ390–396A mice, αMβ2-deficient mice have been shown to exhibit a diminished ability to clear gram-positive pathogens (i.e., Streptococcus pneumoniae), and as a result αMβ2-deficient mice exhibit increased mortality relative to control mice (39). Interestingly, this increased sensitivity to infection in αMβ2-deficient mice was apparent despite the fact that these mice exhibited no diminution in leukocyte emigration in response to intraperitoneal inoculation of bacteria. Thus, neither αMβ2 nor its ligand, fibrin(ogen), are strictly required for leukocyte efflux, but both are important for efficient bacterial clearance.
Despite the fact that leukocyte emigration is generally maintained in αMβ2-deficient mice following some inflammatory challenges, both αMβ2 and fibrin(ogen) may contribute to leukocyte trafficking in certain contexts, particularly when there is direct endothelial damage (46). In the absence of endothelial damage, αLβ2-mediated engagement of endothelial cell ICAM-1 is likely to be dominant in supporting firm leukocyte adhesion and trafficking (10, 11). On the other hand, αMβ2 engagement of fibrin(ogen) may supercede αLβ2 engagement of ICAM-1 in supporting leukocyte trafficking in contexts involving frank endothelial denudation and secondary platelet/fibrin(ogen) deposition on the vessel wall (e.g., following balloon angioplasty) (46). Consistent with this view, αMβ2 is known to support the firm adhesion of neutrophils to surface-bound platelets under flow conditions in vitro, and this adhesion is fibrinogen dependent (47). Furthermore, adherent neutrophils have been shown to efficiently migrate through platelet monolayers in vitro (47). It seems likely that fibrinogen γ390–396A bound to platelet αIIbβ3 will not support neutrophil adhesion to surface-bound platelets. If so, then local leukocyte emigration in vivo may be significantly diminished in Fibγ390–396A mice in the context of direct vascular injury.
One interesting facet of the finding that αMβ2 engagement of fibrin(ogen) supports host clearance of microbes is that bacterial pathogens have also evolved a variety of secretory and cell surface proteins that are designed to disrupt integrin function, engage fibrin(ogen), and/or promote fibrinolysis within vertebrate hosts. These bacterial factors presumably serve a variety of purposes, but undoubtedly all are designed to increase the success of the microbe at the expense of the host, and many may have evolved as countermeasures to innate immune surveillance mechanisms. In addition to expressing the bacterial fibrinolytic agent, streptokinase, group A Streptococcus secrete a factor that is structurally similar to the αM subunit of αMβ2, termed GAS-Mac, which increases bacterial virulence by inhibiting host phagocyte function (48). Similarly, S. aureus expresses an extraordinary array of factors designed to subvert the hemostatic system, including fibrinogen-binding proteins (e.g., ClfA), coagulase, and the fibrinolytic agent, staphlyokinase (49, 50). Indeed, the expression of bacterial plasminogen activators is a particularly common theme among both gram-positive and gram-negative bacteria. Taken together, it appears that leukocyte engagement of fibrin(ogen) may constitute an important nexus of inflammatory cell action that bacterial pathogens seek to subvert through multiple mechanisms.
The finding that fibrin(ogen) can serve as a powerful inflammatory mediator has potential clinical implications. First, the fibrinogen-integrin axis can now be viewed as a potentially useful target in the development of new therapeutic strategies for the treatment or prevention of inflammatory diseases such as sepsis and inflammatory lung, bowel, and joint disease. Given that other hemostatic factors, including activated protein C (51), are also known to be promising focal points for controlling inflammatory processes, fibrinogen may be one of many coagulation system components that stand at the interface between the hemostatic and inflammatory systems. In fact, the potential utility of fibrinogen as a target in inflammatory disease has already been underscored in studies showing that the pharmacological depletion of fibrinogen in mice can diminish the progression of arthritis (52). Of course, an important second implication of the present study is that effective anti- or proinflammatory strategies focusing on fibrin(ogen)-leukocyte interactions potentially could be devised that would not necessarily compromise hemostatic function. Thus, in principle, inflammatory responses could be controlled at the level of hemostatic factors without increasing the risk of bleeding or thrombotic events.
Supplementary Material
Acknowledgments
We thank Kathryn Talmage, Christine La Jeunesse, and Keith Kombrinck for their excellent technical assistance. We thank Joseph Palumbo for his helpful advice. We also thank Ronald E. Gordon and Norman Katz, Department of Pathology, Mount Sinai School of Medicine, New York, USA, for their help with scanning electron microscopy. Finally, we gratefully acknowledge the assistance of Heikki Vaananen, Department of Physiology and Biophysics, Mount Sinai School of Medicine. This work was supported by grants from the NIH to J.L. Degen (HL-63194), E.F. Plow (HL-66197), and M.J. Flick (T32HL-00742).
Footnotes
Matthew J. Flick and XinLi Du contributed equally to this work.
Nonstandard abbreviations used: clumping factor A (ClfA); human embryonic kidney (HEK); intercellular adhesion molecule (ICAM); leukocyte adhesion deficiency type I (LAD I); urokinase-type plasminogen activator receptor (uPAR).
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, D.C., Kishimoto, T.K., and Smith, C.W. 1995. Leukocyte adhesion deficiency and other disorders of leukocyte adherence and motility. In The metabolic and molecular bases of inherited disease. W.S. Sly, C.R. Scriver, A.L. Beaudet, and D. Valle, editors. McGraw-Hill. New York, New York, USA. 3955–3994.
- 3.Larson RS, Springer TA. Structure and function of leukocyte integrins. Immunol. Rev. 1990;114:181–217. doi: 10.1111/j.1600-065x.1990.tb00565.x. [DOI] [PubMed] [Google Scholar]
- 4.Van der Vieren M, et al. A novel leukointegrin, αdβ2, binds preferentially to ICAM-3. Immunity. 1995;3:683–690. doi: 10.1016/1074-7613(95)90058-6. [DOI] [PubMed] [Google Scholar]
- 5.Coxon A, et al. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 6.Mizgerd JP, et al. Neutrophil emigration in the skin, lungs, and peritoneum: different requirements for CD11/CD18 revealed by CD18-deficient mice. J. Exp. Med. 1997;186:1357–1364. doi: 10.1084/jem.186.8.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu H, et al. LFA-1 is sufficient in mediating neutrophil emigration in Mac-1-deficient mice. J. Clin. Invest. 1997;99:1340–1350. doi: 10.1172/JCI119293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding ZM, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J. Immunol. 1999;163:5029–5038. [PubMed] [Google Scholar]
- 9.Walzog B, Jeblonski F, Zakrzewicz A, Gaehtgens P. β2 integrins (CD11/CD18) promote apoptosis of human neutrophils. FASEB J. 1997;11:1177–1186. doi: 10.1096/fasebj.11.13.9367353. [DOI] [PubMed] [Google Scholar]
- 10.Dunne JL, Ballantyne CM, Beaudet AL, Ley K. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood. 2002;99:336–341. doi: 10.1182/blood.v99.1.336. [DOI] [PubMed] [Google Scholar]
- 11.Henderson RB, et al. The use of lymphocyte function-associated antigen (LFA)-1-deficient mice to determine the role of LFA-1, Mac-1, and α4 integrin in the inflammatory response of neutrophils. J. Exp. Med. 2001;194:219–226. doi: 10.1084/jem.194.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. Ligand binding to integrins. J. Biol. Chem. 2000;275:21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- 13.Forsyth CB, Solovjov DA, Ugarova TP, Plow EF. Integrin αMβ2-mediated cell migration to fibrinogen and its recognition peptides. J. Exp. Med. 2001;193:1123–1133. doi: 10.1084/jem.193.10.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Languino LR, et al. Fibrinogen mediates leukocyte adhesion to vascular endothelium through an ICAM-1-dependent pathway. Cell. 1993;73:1423–1434. doi: 10.1016/0092-8674(93)90367-y. [DOI] [PubMed] [Google Scholar]
- 15.Shi C, Zhang X, Chen Z, Robinson MK, Simon DI. Leukocyte integrin Mac-1 recruits toll/interleukin-1 receptor superfamily signaling intermediates to modulate NF-κB activity. Circ. Res. 2001;89:859–865. doi: 10.1161/hh2201.099166. [DOI] [PubMed] [Google Scholar]
- 16.Rubel C, et al. Fibrinogen promotes neutrophil activation and delays apoptosis. J. Immunol. 2001;166:2002–2010. doi: 10.4049/jimmunol.166.3.2002. [DOI] [PubMed] [Google Scholar]
- 17.Rubel C, et al. Soluble fibrinogen modulates neutrophil functionality through the activation of an extracellular signal-regulated kinase-dependent pathway. J. Immunol. 2002;168:3527–3535. doi: 10.4049/jimmunol.168.7.3527. [DOI] [PubMed] [Google Scholar]
- 18.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 2001;167:2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 19.Loike JD, et al. The role of protected extracellular compartments in interactions between leukocytes, and platelets, and fibrin/fibrinogen matrices. Ann. N. Y. Acad. Sci. 1992;667:163–172. doi: 10.1111/j.1749-6632.1992.tb51608.x. [DOI] [PubMed] [Google Scholar]
- 20.Lishko VK, Kudryk B, Yakubenko VP, Yee VC, Ugarova TP. Regulated unmasking of the cryptic binding site for integrin αMβ2 in the γ C-domain of fibrinogen. Biochemistry. 2002;41:12942–12951. doi: 10.1021/bi026324c. [DOI] [PubMed] [Google Scholar]
- 21.Ugarova TP, et al. Identification of a novel recognition sequence for integrin αMβ2 within the γ-chain of fibrinogen. J. Biol. Chem. 1998;273:22519–22527. doi: 10.1074/jbc.273.35.22519. [DOI] [PubMed] [Google Scholar]
- 22.Ugarova TP, et al. Sequence γ 377-395(P2), but not γ 190-202(P1), is the binding site for the αM I-domain of integrin αMβ2 in the γ C-domain of fibrinogen. Biochemistry. 2003;42:9365–9373. doi: 10.1021/bi034057k. [DOI] [PubMed] [Google Scholar]
- 23.Holmback K, Danton MJ, Suh TT, Daugherty CC, Degen JL. Impaired platelet aggregation and sustained bleeding in mice lacking the fibrinogen motif bound by integrin αIIbβ3. EMBO J. 1996;15:5760–5771. [PMC free article] [PubMed] [Google Scholar]
- 24.Hooper M, Hardy K, Handyside A, Hunter S, Monk M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 1987;326:292–295. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- 25.Suh TT, et al. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995;9:2020–2033. doi: 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]
- 26.Farrell DH, Thiagarajan P. Binding of recombinant fibrinogen mutants to platelets. J. Biol. Chem. 1994;269:226–231. [PubMed] [Google Scholar]
- 27.Jirousková M, Chereshnev I, Väänänen H, Degen JL, Coller BS. Antibody blockade or mutation of the fibrinogen γ chain C-terminus are more effective in inhibiting murine arterial thrombus formation than complete absence of fibrinogen. Blood. 2004;103:1995–2002. doi: 10.1182/blood-2003-10-3401. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Plow EF. Overlapping, but not identical, sites are involved in the recognition of C3bi, neutrophil inhibitory factor, and adhesive ligands by the αMβ2 integrin. J. Biol. Chem. 1996;271:18211–18216. doi: 10.1074/jbc.271.30.18211. [DOI] [PubMed] [Google Scholar]
- 29.Kumagai K, Itoh K, Hinuma S, Tada M. Pretreatment of plastic Petri dishes with fetal calf serum. A simple method for macrophage isolation. J. Immunol. Methods. 1979;29:17–25. doi: 10.1016/0022-1759(79)90121-2. [DOI] [PubMed] [Google Scholar]
- 30.Adams, D.O., Edelson, P.J., and Koren, H.S. 1981. Methods for studying mononuclear phagocytes. Academic Press. New York, New York, USA. xxiv, 1023 pp.
- 31.Doolittle RF, Yang Z, Mochalkin I. Crystal structure studies on fibrinogen and fibrin. Ann. N. Y. Acad. Sci. 2001;936:31–43. doi: 10.1111/j.1749-6632.2001.tb03492.x. [DOI] [PubMed] [Google Scholar]
- 32.Wright SD, et al. Complement receptor type three (CD11b/CD18) of human polymorphonuclear leukocytes recognizes fibrinogen. Proc. Natl. Acad. Sci. U. S. A. 1988;85:7734–7738. doi: 10.1073/pnas.85.20.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altieri DC, Plescia J, Plow EF. The structural motif glycine 190-valine 202 of the fibrinogen γ chain interacts with CD11b/CD18 integrin (αMβ2, Mac-1) and promotes leukocyte adhesion. J. Biol. Chem. 1993;268:1847–1853. [PubMed] [Google Scholar]
- 34.Zhang L, Plow EF. Identification and reconstruction of the binding site within αMβ2 for a specific and high affinity ligand, NIF. J. Biol. Chem. 1997;272:17558–17564. doi: 10.1074/jbc.272.28.17558. [DOI] [PubMed] [Google Scholar]
- 35.Diamond MS, et al. ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18) J. Cell Biol. 1990;111:3129–3139. doi: 10.1083/jcb.111.6.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yakubenko VP, Lishko VK, Lam SC, Ugarova TP. A molecular basis for integrin αMβ2 ligand binding promiscuity. J. Biol. Chem. 2002;277:48635–48642. doi: 10.1074/jbc.M208877200. [DOI] [PubMed] [Google Scholar]
- 37.Gregory SH, et al. Complementary adhesion molecules promote neutrophil-Kupffer cell interaction and the elimination of bacteria taken up by the liver. J. Immunol. 2002;168:308–315. doi: 10.4049/jimmunol.168.1.308. [DOI] [PubMed] [Google Scholar]
- 38.Miyamoto M, et al. Neutrophilia in LFA-1-deficient mice confers resistance to listeriosis: possible contribution of granulocyte-colony-stimulating factor and IL-17. J. Immunol. 2003;170:5228–5234. doi: 10.4049/jimmunol.170.10.5228. [DOI] [PubMed] [Google Scholar]
- 39.Prince JE, et al. The differential roles of LFA-1 and Mac-1 in host defense against systemic infection with Streptococcus pneumoniae. J. Immunol. 2001;166:7362–7369. doi: 10.4049/jimmunol.166.12.7362. [DOI] [PubMed] [Google Scholar]
- 40.Sitrin RG, Pan PM, Srikanth S, Todd RF., 3rd Fibrinogen activates NF-κB transcription factors in mononuclear phagocytes. J. Immunol. 1998;161:1462–1470. [PubMed] [Google Scholar]
- 41.Szaba FM, Smiley ST. Roles for thrombin and fibrin(ogen) in cytokine/chemokine production and macrophage adhesion in vivo. Blood. 2002;99:1053–1059. doi: 10.1182/blood.v99.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takami M, Terry V, Petruzzelli L. Signaling pathways involved in IL-8-dependent activation of adhesion through Mac-1. J. Immunol. 2002;168:4559–4566. doi: 10.4049/jimmunol.168.9.4559. [DOI] [PubMed] [Google Scholar]
- 43.Lishko VK, Yakubenko VP, Hertzberg KM, Grieninger G, Ugarova TP. The alternatively spliced αE C domain of human fibrinogen-420 is a novel ligand for leukocyte integrins αMβ2 and αXβ2. Blood. 2001;98:2448–2455. doi: 10.1182/blood.v98.8.2448. [DOI] [PubMed] [Google Scholar]
- 44.Gordon DL, Rice J, Finlay-Jones JJ, McDonald PJ, Hostetter MK. Analysis of C3 deposition and degradation on bacterial surfaces after opsonization. J. Infect. Dis. 1988;157:697–704. doi: 10.1093/infdis/157.4.697. [DOI] [PubMed] [Google Scholar]
- 45.Scharffetter-Kochanek K, et al. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J. Exp. Med. 1998;188:119–131. doi: 10.1084/jem.188.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simon DI, et al. Decreased neointimal formation in Mac-1(–/–) mice reveals a role for inflammation in vascular repair after angioplasty. J. Clin. Invest. 2000;105:293–300. doi: 10.1172/JCI7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer TA. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the β2-integrin CD11b/CD18. Blood. 1996;88:146–157. [PubMed] [Google Scholar]
- 48.Lei B, et al. Evasion of human innate and acquired immunity by a bacterial homolog of CD11b that inhibits opsonophagocytosis. Nat. Med. 2001;7:1298–1305. doi: 10.1038/nm1201-1298. [DOI] [PubMed] [Google Scholar]
- 49.Moreillon P, et al. Role of Staphylococcus aureus coagulase and clumping factor in pathogenesis of experimental endocarditis. Infect. Immun. 1995;63:4738–4743. doi: 10.1128/iai.63.12.4738-4743.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McDevitt D, et al. Characterization of the interaction between the Staphylococcus aureus clumping factor (ClfA) and fibrinogen. Eur. J. Biochem. 1997;247:416–424. doi: 10.1111/j.1432-1033.1997.00416.x. [DOI] [PubMed] [Google Scholar]
- 51.Esmon CT. Protein C pathway in sepsis. Ann. Med. 2002;34:598–605. doi: 10.1080/078538902321117823. [DOI] [PubMed] [Google Scholar]
- 52.Busso N, et al. Exacerbation of antigen-induced arthritis in urokinase-deficient mice. J. Clin. Invest. 1998;102:41–50. doi: 10.1172/JCI2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}