

Abstract
Pansclerotic morphea is a rare subtype of localised scleroderma. Very few cases have been reported in the literature. This case discusses a young man with a 15-year history of pansclerotic morphea, illustrating the clinical course and complications of this disease. Notably, recurrent squamous cell carcinomas with several high-risk features and frequent cellulitis were the basis of many admissions. As often seen with this disease, autoimmune markers were negative. However, marked eosinophilia was noted late in the course. Furthermore, this disease is quite resistant to treatment. Immunosuppressive therapy thus far has perhaps slowed the progression, although not cured the disease in this patient.
Background
Pansclerotic morphea is a subtype of localised scleroderma.1 It is a rare yet extremely disabling disease,1 with significant morbidity and mortality. Few cases have been reported since the disease was first described in 1923 by Roudinesco and Vallery-Radot.2 In 1980, Diaz-Perez et al2 completed the first series analysing 14 children. Female to male ratio is reported to be approximately 4:1.3 Disease onset is usually before age 14,2 but may present after adolescence4 and follows a rapidly progressive and disabling course.2 It is characterised by pansclerosis, affecting the dermis and subcutaneous fat, and can extend to fascia, muscle and bone.2 Polymorphous plaques develop and rapidly progress on the extremities, trunk, face and scalp, usually with sparing of fingers and toes.3 Other clinical features include chronic ulcers resistant to treatment, frequent skin infections, contracture deformities, musculoskeletal atrophy, immobility, squamous cell carcinoma (SCC), ectropion and alopecia secondary to sclerosis.5 6 Lab findings are often non-specific,6 but may reveal eosinophilia and hypergammaglobuminaemia.2 The disease is resistant to immunosuppressive treatment and prognosis is grim.7
Case presentation
A 25-year-old Hispanic man with a 15-year history of pansclerotic morphea presented with rapidly recurrent large SCCs at two sites on his left foot, along with a right peroneal venous thrombosis and right forearm inflammatory tenosynovitis. He had a large SCC excised with clear margins from the same foot a few weeks prior to this presentation, as well as 1 year earlier.
He first developed features of pansclerotic morphea at the age of 10. Initially, erythematous pruritic plaques developed bilaterally on his feet, which then spread to his legs and rapidly became sclerotic. Shortly afterwards, he developed sclerotic plaques on his trunk, neck, face and upper extremities. Over the years, these polymorphous plaques expanded in size and became confluent, until almost his entire body surface became involved. His skin continues to become progressively more sclerotic, which he finds quite painful and which makes movement difficult. There is no internal organ involvement. There is no significant personal medical or family history.
Over the course of the disease, he has developed frequent lower extremity skin infections, osteomyelitis, recurrent SCCs, chronic ulcers, bilateral ankle joint contractures, reduced mobility, bilateral upper and lower ectropion, deep venous thrombosis, anaemia of chronic disease, eosinophilia and adrenal insufficiency. Most notable has been the frequent recurrent lower extremity infections and ulcers. Over the past 7 years, he has been admitted approximately 20 times for lower extremity pain and infection.
On examination, the patient was a cachectic and distressed young Hispanic man. There was severe pansclerosis involving almost the entire surface of his body, sparing only the palms, fingers, soles, toes, axillae, scalp and ears. There were two 5 cm×5 cm SCCs on the dorsum of the left foot, overlying the areas of chronic ulcers. There was marked muscle atrophy, particularly of the lower limbs. There were large areas of patchy hyperpigmentation of the lower extremity. There was a hypopigmented patch on his anterior right shoulder. There were bilateral ankle joint contractures. There was no range of motion (ROM) at both ankles, and severely reduced ROM of both wrists. Knee and ankle jerk reflexes were absent. Sensation was intact throughout. There was bilateral upper and lower lid ectropion (figures 1 and 2).
Figure 1.
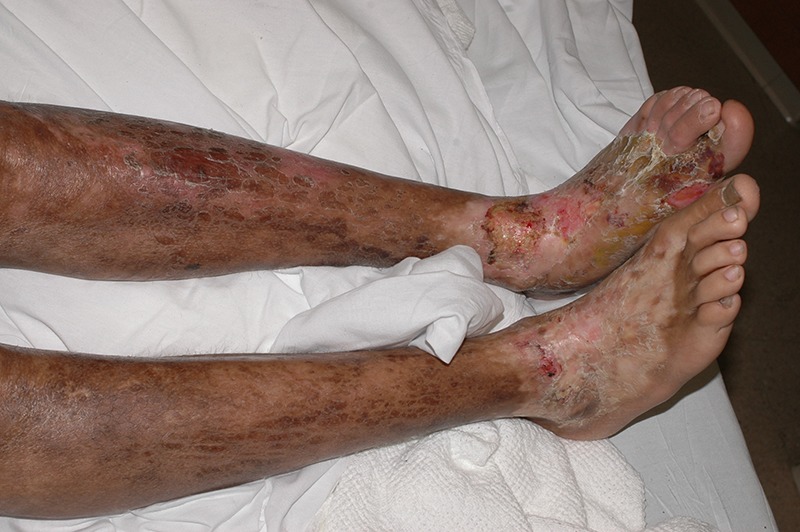
(2006) Pansclerotic morphea of the lower limbs at age 18, with ulcers and hair loss. Relative sparing of toes and plantar surfaces.
Figure 2.
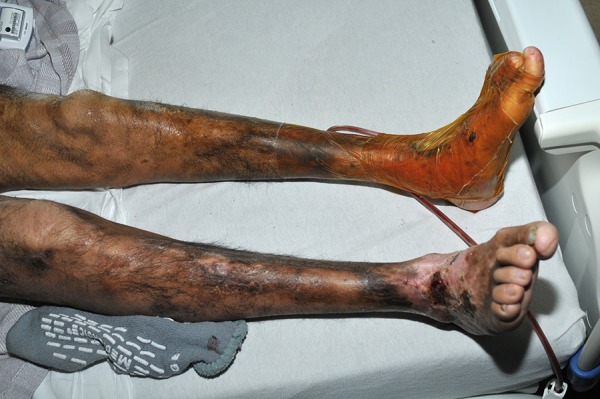
(2013) Post-excision of SCCs of the left foot at age 25. Compared to 2006 (Figure 1), there is considerable muscle wasting and hyperpigmentation, with patchy hair distribution. Toes and plantar surfaces are still relatively spared.
Investigations
Laboratory investigations revealed marked eosinophilia, indicating at one point a relative eosinophil count of 70% (normal<8%) and an absolute eosinophil count of 16 800 µ/L (normal 0–500 µ/L). Also noted were adrenal insufficiency (AM cortisol 1.5 µg/dL; normal 4.46) and anaemia (haemoglobin 7.8 g/dL). Urinalysis and renal function were within normal range. The eosinophilia had been present over the previous 12-month period, and had gradually increased during this time. Bone marrow biopsy showed increased eosinophils along with an atypical lymphoid aggregate. PDGFRA and PDGFRB genes were normal. Blood cultures for bacteria, fungus and parasites were all negative. Immunoglobulins were also within normal range.
Antinuclear antibody, single-stranded A antibody, antidouble stranded DNA antibody, anticentromere antibody, antineutrophil cytoplasmic antibodies and rheumatoid factor were all negative.
Ultrasound identified a right peroneal venous thrombosis. Right forearm MRI revealed extensive subcutaneous oedema of the anterior forearm, and inflammatory tenosynovitis of the flexor tendons and extensor carpi ulnaris.
Treatment
During the course of this disease, he has been treated with prednisone, methotrexate, bosentan, etanercept and mycophenolate, with minimal improvement noted. Pregabalin and morphine provide him with adequate pain relief. Current medications include citalopram, hydroxyurea, morphine, oxycodone, prednisone, pregabalin, iron supplementation and zinc sulfate.
Outcome and follow-up
This patient eventually required a below-knee amputation of his left leg due to recurrent high-risk SCCs. The eosinophilia was successfully treated with hydroxyurea and prednisone.
Discussion
Pansclerotic morphea has a rapid and progressively disabling course, with significant morbidity and mortality.3 This is a unique case with a 15-year follow-up period, illustrating the clinical course and long-term complications of this disease.
Eosinophilia has been reported in other cases of pansclerotic morphea.2 The aetiology of this patient's eosinophilia may have been multifactorial. Initially, typical causes such as infection and drug reaction were ruled out. A high eosinophil count may have been due to or have been compounded by his recurrent SCCs. This may be due to a paraneoplastic effect causing secondary eosinophilia due to increased interleukins and granulocyte-macrophage colony-stimulating factor.8 Additionally, he had inflammatory tenosynovitis with subcutaneous oedema in his right forearm, which was intensely pruritic. Furthermore, he may have had reactive eosinophilia in response to his adrenal insufficiency. Glucocorticoids inhibit proliferation of eosinophils.9 Low glucocorticoid levels in adrenal insufficiency may lead to the proliferation of eosinophils.9
Patients with pansclerotic morphea appear to be at a higher risk of developing SCCs.5 In the general population, SCCs tend to occur in sun-exposed areas, such as the head, neck and upper extremities, with lighter skin tones being at greater risk. This patient developed multiple recurrent SCCs of his left foot at 14 and 15 years after disease onset. He had Fitzpatrick skin type IV with very limited sun-exposure. His SCCs had several high-risk features, including rapid recurrence, large diameter, location in a chronic wound site, perineural invasion histologically, associated neurological symptoms and comorbid immunosuppression.10 Relevant risk factors for SCC development in the pansclerotic morphea population include immunosuppression, chronic ulcers, frequent infections, chronic inflammation, scar tissue and previous non-melanoma skin cancer.7 He also developed a right peroneal venous thrombosis, with predisposing factors of malignancy, recent surgery and reduced mobility.
This rare case of pansclerotic morphea illustrates the clinical course and complications of a severely debilitating disease. This patient developed pansclerotic morphea at 10 years of age. Early clinical features included development of sclerotic plaques in the lower extremities, which later rapidly spread to the rest of the body. Acral sparing is still present, particularly of the fingers, toes, palms and soles. Chronic ulcers and frequent skin infections have been present throughout the course of the disease. Later clinical features included recurrent high-risk SCCs, muscular atrophy especially of the lower extremities, joint contractures, reduced mobility, hyperpigmentation of the lower extremity, anaemia of chronic disease and deep vein thrombosis. He had no internal organ involvement. Autoimmune markers were negative, but laboratory investigations late in the course of the disease revealed eosinophilia, adrenal insufficiency and anaemia of chronic disease. Treatment thus far has not cured the pansclerotic morphea, but may have slowed the progression of this rare disease.
Patient's perspective.
The psychosocial impact of pansclerotic morphea has been quite significant on this patient. He has a history of depression with suicide attempts related to this chronic disease. He has aspirations to become a lawyer, however, he is hindered by the frequent hospitalisations and restricted mobility. Additionally, he reports that he is often the subject of ridicule by the general public due to his physical appearance. He is often mistaken for a burns patient, and subsequently mocked for it. Despite all of this, he tries to maintain a positive outlook on life and remains determined to overcome these obstacles to achieve his goals.
Learning points.
Pansclerotic morphea is a rare subtype of localised scleroderma.
Clinical features include: polymorphous plaques, which usually begin in the extremities and progress rapidly to involve almost all other areas, with acral sparing often present, chronic ulcers, frequent cellulitis, muscular atrophy, contracture deformities, reduced mobility, squamous cell carcinoma, ectropion and alopaecia secondary to sclerosis.
Patients with pansclerotic morphea have many risk factors predisposing them to squamous cell carcinoma including: immunosuppression, chronic ulcers, frequent skin infections, chronic inflammation, scar tissue and previous skin cancers.
Laboratory investigations are usually non-specific, but may reveal eosinophilia and hypergammaglobuminaemia.
Footnotes
Contributors: IG was involved in conception, acquisition of data, drafting of manuscript, final approval and accountability. OK and WD were involved in acquisition of data, critical revision of manuscript, final approval and accountability.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Roldan R, Morote G, Castro M, et al. Efficacy of bosentan in treatment of unresponsive cutaneous ulceration in disabling pansclerotic morphea in children. J Rheumatol 2006;33:2538–40 [PubMed] [Google Scholar]
- 2.Diaz-Perez JL, Connolly SM, Winkelmann RK. Disabling pansclerotic morphea of children. Arch Dermatol 1980;116:169–73 [PubMed] [Google Scholar]
- 3.Banks TA, Steen V, Katona I, et al. Successful treatment of pansclerotic morphea with imatinib mesylate in a pediatric patient. Ann Paediatr Rheumatol 2013;2:43–9 [Google Scholar]
- 4.Maragh SH, Davis MDP, Bruce AJ, et al. Disabling pansclerotic morphea: clinical presentation in two adults. J Am Acad Dermatol 2005;53(2 Suppl 1):S115–19 [DOI] [PubMed] [Google Scholar]
- 5.Wollina U, Buslau M, Heinig B, et al. Disabling pansclerotic morphea of childhood poses a high risk of chronic ulceration of the skin and squamous cell carcinoma. Int J Low Extrem Wounds 2007;6:291–8 [DOI] [PubMed] [Google Scholar]
- 6.Wollina U, Buslau M, Petrov I, et al. Disabling panslerotic morphea of childhood. Expert Rev Dermatol 2007;2:775–84 [Google Scholar]
- 7.Singh DS, Kumar L, Radotra BD, et al. Disabling pansclerotic morphea of childhood and hypogammaglobulinemia: a curious association. Rheumatol Int 2002;21:158–60 [DOI] [PubMed] [Google Scholar]
- 8.Sonoda Y, Arai N, Ogawa M. Humoral regulation of eosinophilopoiesis in vitro: analysis of the targets of interleukin-3, granulocyte/macrophage colony-stimulating factor (GM-CSF), and interleukin-5. Leukemia 1989;3:14–18 [PubMed] [Google Scholar]
- 9.Montgomery ND, Dunphy CH, Mooberry M, et al. Diagnostic complexities of eosinophilia. Arch Pathol Lab Med 2013;137:259–69 [DOI] [PubMed] [Google Scholar]
- 10.Hardin J. Chapter 13: cutaneous conditions. In: Knoop KJ, Stack LB, Storrow AB, et al., eds The atlas of emergency medicine. 3rd edn McGraw-Hill, 2010 [Google Scholar]