

Abstract
Excess glucocorticoid levels cause Cushing's syndrome (CS) and may be due to pituitary, adrenal or ectopic tumours. Adrenocorticotropic hormone (ACTH) levels are useful in identifying adrenal tumours. In rare cases, ACTH-producing phaeochromocytomas are the cause of CS. We present two cases of ACTH-secreting phaeochromocytoma as the underlying cause of CS. In both cases, female patients presented with the classical clinical signs of CS and an adrenal mass. High ACTH levels raised the suspicion of an ACTH-secreting phaeochromocytoma. The diagnosis was confirmed by urinary catecholamine levels and positive fluorine-18-L-dihydroxyphenylalanine (18F-DOPA) positron emission tomography (PET) CT (Case 1) and fluorodeoxyglucose PET-CT (Case 2). Both patients were treated with an α-blocker prior to surgical intervention. The two cases underline the importance of thorough diagnostic workup in patients with CS. An ACTH-secreting phaeochromocytoma should be checked for in patients with an adrenal mass and elevated ACTH levels.
Background
Cushing's syndrome (CS) is caused by excess glucocorticoid levels,1 and may be due to corticosteroid treatment, pituitary tumour (Cushing's disease (CD)), cortisol-secreting adrenal tumour or increased production of an adrenocorticotropic hormone (ACTH) by an extrapituitary (ectopic) tumour.1 2
While CD accounts for 70% of CS cases, about 10% of patients with CS have an ectopic ACTH-secreting tumour.3 The incidence of CD in Denmark is 1.2–1.7/million/year, and that of CS caused by adrenal tumours is 0.6/million/year.4
Patients with ectopic ACTH-producing tumours are most often diagnosed by pulmonary carcinoids, neuroendocrine carcinoids or occult cancers. In the rare cases where CS is caused by an ACTH-producing phaeochromocytoma,5 the patients may present with classical CS and an adrenal mass. High levels of ACTH and catecholamines should give the suspicion of an ACTH-secreting phaeochromocytoma. The treatment success of phaeochromocytoma depends on the patient's presurgical condition.6 Surgery on a phaeochromocytoma can have severe consequences for the patient.7 In a case series of 54 autopsies with verified phaeochromocytoma, 27% of the patients had died either due to a hypertensive or hypotensive crisis during surgery for non-adrenal pathologies.8 9 As up to 25% of phaeochromocytomas are caused by genetic mutations, genetic screening is recommended.10
We present two cases of patients with CS due to an ACTH-producing phaeochromocytoma.
Case presentation
Case 1: A 70-year-old woman was admitted to the emergency ward at a regional hospital because of vertigo. The patient had a history of cardiovascular events and was treated with platelet inhibitors, statins and β-blockers. The patient had smoked for about 54 pack-years and there was no family history of endocrine disease. At presentation, the patient was clinically dehydrated and had clinical signs of CS with a moon face, a buffalo hump, subcutaneous haematomas, truncal obesity and hirsutism, but no abdominal striae. She had neutrophilia and hypokalaemia. Intravenous rehydration with a potassium supplement was initiated. The patient needed 90 mmol of potassium daily to maintain electrolyte homeostasis. Owing to suspected CS, she was transferred to the Department of Endocrinology at Odense University Hospital for further diagnostic workup.
Case 2: A 67-year-old woman was referred to our endocrinology outpatient clinic due to uncontrolled high blood pressure. The patient was taking four antihypertensive drugs and 20 mmol potassium supplements daily. She had osteoporosis with compression fractures of Th12, L3 and L4 and there was no family history of endocrine diseases. At presentation, she had clinical signs of CS including a reddish moon face, violet/purple abdominal striae and hirsutism. She had a normal neutrophil count and normal potassium levels.
Investigations
Laboratory findings are summarised in table 1.
Table 1.
Summary of laboratory results
| Laboratory test | Reference range | Case 1 |
Case 2 |
||
|---|---|---|---|---|---|
| Preoperative | Postoperative | Preoperative | Postoperative | ||
| Cortisol | |||||
| 24 h urinary cortisol | 8–125 mol/day | 2651 | – | 305 | – |
| P-cortisol | 200–700 nmol/L | 1711 | – | 1060 | 430 |
| P-ACTH | 2–14 pmol/L | 96 | 2 | 16 | 3 |
| Catecholamines | |||||
| Night-urinary norepinephrine | 2.2–13.4 nmol/h | 32.2 | – | 40.9 | – |
| Night-urinary epinephrine | <0.8 nmol/h | 31.6 | – | 15.7 | – |
| Night-urinary dopamine | 23–141 nmol/h | 169 | – | 190 | – |
| P-metanephrine (free) | <90 ng/L | 213 | N.A. | N.A. | N.A. |
| P-normetanephrine (free) | <180 ng/L | 99 | N.A. | N.A. | N.A. |
| Metabolism | |||||
| HbA1c | 31–44 mmol/mol | 101 | – | 55 | – |
| Electrolytes | |||||
| P-potassium | 3.5–4.4 mmol/L | 2.8 | – | 3.1 | – |
| Tumour markers | |||||
| Chromogranin A | <85 µg/L | 990 | – | N.A | – |
Selected preoperative and postoperative laboratory results are shown for the two cases.
ACTH, adrenocorticotropic hormone; HbA1c, glycated haemoglobin; N.A., not available.
Case 1: Twenty-four hour urinary cortisol levels were elevated and a 1 mg dexamethasone suppression test showed incomplete suppression of cortisol. ACTH levels were elevated. MRI of the pituitary gland was normal.
The patient reported abdominal pain and a CT scan of the abdomen showed bilateral adrenal adenomas. The largest adenoma was located in the right adrenal gland and measured 30×18 mm. The adenoma in the left adrenal gland measured 17×12 mm. Both adenomas had high HU-scores of 100. Night-urinary catecholamines and plasma metanephrines were elevated. A fluorine-18-L-dihydroxyphenylalanine (18F-DOPA) positron emission tomography (PET) CT showed a significant pathological uptake of 18F-DOPA in the larger adrenal adenoma (figure 1). The 18F-DOPA PET-CT also revealed a pulmonary infiltration without 18F-DOPA uptake, but it was highly positive on a subsequent fluorodeoxyglucose (FDG) PET-CT. Bronchoalveolar lavage and endoscopic bronchial ultrasound identified the pulmonary lesion as an aspergilloma.
Figure 1.

Case 1: The fluorine-18-L-dihydroxyphenylalanine (18F-DOPA) positron emission tomography (PET) CT scan showed increased 18F-DOPA uptake in an adenoma in the right adrenal gland.
Case 2: Twenty-four hour urinary cortisol and cortisol metabolites were elevated, as were ACTH levels. MRI of the pituitary gland was normal. Sinus petrosus catherisation ruled out excess ACTH production from the pituitary.
Metaiodobenzyl guanidine (MIBG) scintigraphy was negative and the FDG PET-CT and CT scan with contrast showed an adrenal mass in the right adrenal gland with a moderately increased metabolism (figure 2). The mass had a cystic appearance and measured about 3 cm in diameter. The HU score was not indicated on the radiology report, but the contrast washout was 60%. Night-urinary catecholamines were elevated. To rule out other neuroendocrine tumours, the patient underwent somatostatin scintigraphy, which showed no significant pathological findings.
Figure 2.
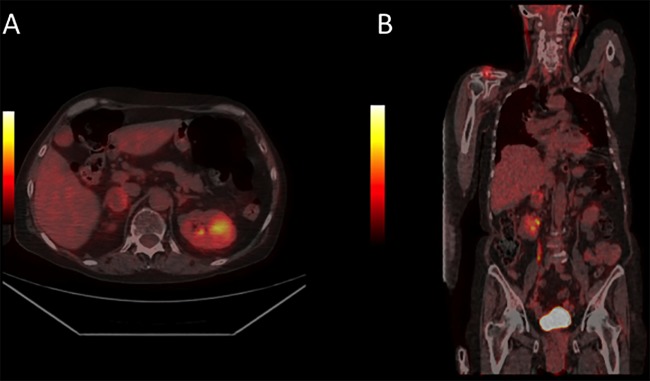
Case 2: The fluorodeoxyglucose (FDG) positron emission tomography (PET) CT scan showed moderate FDG uptake in the right adrenal gland, indicating this to be the location of the phaeochromocytoma. The scan showed no other pathologies.
Differential diagnosis
In both cases, the patients presented with typical clinical signs of CS and elevated urinary cortisol. Since ACTH levels were elevated, the pituitary or ectopic ACTH-producing tumour needed to be ruled out. In Case 1, a pulmonary mass was detected in a patient with a history of tobacco use and pulmonary malignancy was excluded.
Treatment
Case 1: The patient was stabilised clinically and biochemically. The patient needed aggressive antibiotic treatment to treat infection, potassium for hypokalaemia, and insulin due to steroid-induced diabetes mellitus. Preoperatively, the patient was treated with an α-adrenergic-blocking agent (phenoxybenzamine). Both adrenal glands were surgically removed.
Case 2: The patient was treated preoperatively with phenoxybenzamine and underwent a right adrenalectomy.
Outcome and follow-up
Case 1: Owing to bilateral adrenalectomy, the patient was treated with hydrocortisone. Five days postoperatively, ACTH levels had decreased from 96 to 2 pmol/L. The patient had opportunistic infections and a prolonged stay in the intensive care unit. She died 2 months after surgery due to septic shock.
The pathology report showed a phaeochromocytoma with positive immunohistochemical markers for chromogranin A, CD56, neuron-specific enolase, synaptophysin, and S100 indicative of phaeochromocytoma.
DNA screening showed no mutations in the SDHB/C/D genes but a von Hippel-Lindau (VHL) missense mutation (p.PRO25Leu). This mutation was described as non-pathogenic for VHL disease.11 The excised tissue was not checked for loss of heterozygosity. Clinical evaluation for other manifestations of VHL disease was not performed, as the genetic results became available postmortem.
Case 2: ACTH levels decreased from 16pmol/L preoperatively to 3 pmol/L 3 months postoperatively. At present, the patient has been followed for 3.5 years in our outpatient clinic with no biochemical or clinical signs of relapse of CS or phaeochromocytoma. A PET-CT scan performed 42 months after surgery showed no pathological findings.
The pathology report showed a phaeochromocytoma with positive synaptophysin and melan-A. There was scattered positive staining for ACTH-producing cells in the medullar tissue. The patient declined screening for DNA mutations.
Discussion
Ectopic ACTH-producing tumours are rare. In a literature review by Ballav et al,6 5.2% of patients with ectopic ACTH-producing tumours had ACTH-producing phaeochromocytomas. To date, 30 patient cases, including our own two cases, have been reported in the English language literature, and one case has been reported in Spanish.6 12 13 The mean age at presentation ranged from 12 to 74 years and most cases were female.6 12
ACTH-secreting phaeochromocytomas cause hypertension and hypokalaemia more frequently than do ectopic ACTH-producing tumours.6 This could be due to higher ACTH and thus cortisol levels.6 Both of our cases had hypokalaemia, which is a frequent sign of cortisol excess and is often seen in patients with ACTH-secreting phaeochromocytomas,6 and both had right-sided adrenal tumours. Nineteen of the previously presented cases had unilateral phaeochromocytomas, most frequently on the left side.6 12 Kim et al14 found that in a case series of 348 patients with adrenal masses, 62% of patients had a left adrenal mass.
With early diagnosis and preoperative conditioning, most patients will have a complete remission of their disease.6 Both our cases were diagnosed prior to surgery, but this is not always the case.12 Case 1 died postoperatively due to complications that were most likely linked to severe CS. She had several comorbidities and severe Aspergillus, Epstein-Barr virus and cytomegalovirus infections, indicating an impaired immunocompetence. It can be discussed whether or not bilateral adrenalectomy was the proper course of action. The patient had bilateral adrenal masses with a high HU index, and PET signalling from the right adrenal gland. Seeing that a high HU can indicate malignancy, a bilateral adrenalectomy was chosen. Case 2 has been followed for 5 years without relapse. We do not know the HU index of the adrenal adenoma in this patient. A high washout can indicate a benign adrenal mass, but it is unknown if this is true for phaeochromocytomas.
Genetic screening was performed in Case 1, whereas Case 2 declined genetic testing. Approximately 40% of cases of phaeochromocytomas have genetic germ line defects.10 These can be grouped into five major genetic groups with an autosomal dominant mode of inheritance.15 Buffet et al15 found mutations in 22.4% of their 1620 patients tested. Recent guidelines suggest that genetic screening should be considered in all patients with phaeochromocytoma.10
Postoperatively, immunohistochemical markers indicated phaeochromocytoma, but positive staining for ACTH-producing cells was only found in Case 2. In 2012, Cassarino et al13 reported a case with suspected ACTH-secreting phaeochromocytoma and negative immunohistochemistry for ACTH-secreting cells. The negative immunohistochemistry can be due to a corticotropin-releasing hormone-secreting tumour or because the tumour secretes high molecular weight ACTH or small ACTH-derived peptides that are not recognised by the antibodies typically used for immunohistochemistry. However, this is not routinely tested for.13 Ectopic ACTH-producing tumours need to be stained with antibodies that capture ACTH and other POMC-splicing products; thus, it is not unusual for such a tumour to be negative when stained with the usual anti-ACTH antibodies.
In conclusion, ACTH-producing phaeochromocytomas should be suspected in patients with CS adrenal masses and elevated ACTH levels.
Learning points.
Patients with Cushing's syndrome, elevated adrenocorticotropic hormone levels, adrenal mass and no pituitary adenoma should be screened for phaeochromocytoma.
Phaeochromocytomas need preoperative treatment with phenoxybenzamine.
Up to 40% of phaeochromocytoma cases have mutations; thus, genetic testing should be offered to patients with phaeochromocytoma.
Footnotes
Contributors: LF wrote the case report. MSA reviewed and approved the final manuscript. DG had the idea to write the case report, and also reviewed and finally approved the manuscript. ALN had nuclear medicine expertise. She reviewed and finally approved the manuscript.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab 2003;88:5593–602 [DOI] [PubMed] [Google Scholar]
- 2.Cushing H. The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism). 1932. Obesity Res 1994;2:486–508 [DOI] [PubMed] [Google Scholar]
- 3.Newell-Price J, Bertagna X, Grossman AB, et al. Cushing's syndrome. Lancet 2006;367:1605–17 [DOI] [PubMed] [Google Scholar]
- 4.Lindholm J, Juul S, Jorgensen JO, et al. Incidence and late prognosis of Cushing's syndrome: a population-based study. J Clin Endocrinol Metab 2001;86:117–23 [DOI] [PubMed] [Google Scholar]
- 5.Ilias I, Torpy DJ, Pacak K, et al. Cushing's syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab 2005;90:4955–62 [DOI] [PubMed] [Google Scholar]
- 6.Ballav C, Naziat A, Mihai R, et al. Mini-review: phaeochromocytomas causing the ectopic ACTH syndrome. Endocrine 2012;42:69–73 [DOI] [PubMed] [Google Scholar]
- 7.Lenders JW, Eisenhofer G, Mannelli M, et al. Phaeochromocytoma. Lancet 2005;366:665–75 [DOI] [PubMed] [Google Scholar]
- 8.Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected phaeochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc 1981;56:354–60 [PubMed] [Google Scholar]
- 9.Prejbisz A, Lenders JW, Eisenhofer G, et al. Mortality associated with phaeochromocytoma. Horm Metab Res 2013;45:154–8 [DOI] [PubMed] [Google Scholar]
- 10.Lenders JW, Duh QY, Eisenhofer G, et al. Phaeochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014;99:1915–42 [DOI] [PubMed] [Google Scholar]
- 11.Pettman RK, Crowley A, Riddell C, et al. VHL P25L is not a pathogenic von Hippel-Lindau mutation: a family study. Mol Diagn Ther 2006;10:239–42 [DOI] [PubMed] [Google Scholar]
- 12.Li XG, Zhang DX, Li X, et al. Adrenocorticotropic hormone-producing phaeochromocytoma: a case report and review of the literature. Chin Med J (Engl) 2012;125:1193–6 [PubMed] [Google Scholar]
- 13.Cassarino MF, Ambrogio AG, Pagliardini L, et al. ACTH-secreting phaeochromocytoma with false-negative ACTH immunohistochemistry. Endocr Pathol 2012;23:191–5 [DOI] [PubMed] [Google Scholar]
- 14.Kim J, Bae KH, Choi YK, et al. Clinical characteristics for 348 patients with adrenal incidentaloma. Endocrinol Metab (Seoul) 2013;28:20–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buffet A, Venisse A, Nau V, et al. A decade (2001–2010) of genetic testing for phaeochromocytoma and paraganglioma. Horm Metab Res 2012;44:359–66 [DOI] [PubMed] [Google Scholar]