

Abstract
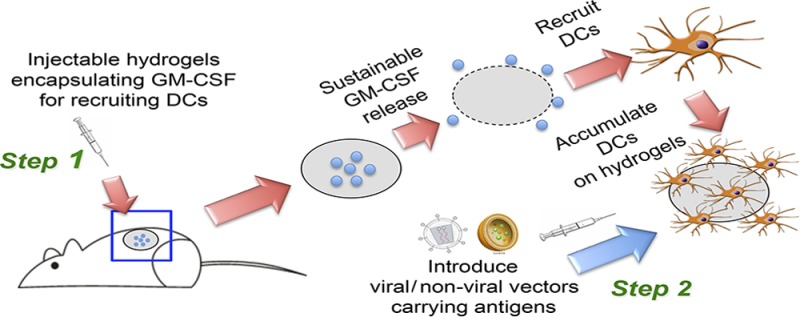
Attempts to develop cell-based cancer vaccines have shown limited efficacy, partly because transplanted dendritic cells (DCs) do not survive long enough to reach the lymph nodes. The development of biomaterials capable of modulating DCs in situ to enhance antigen uptake and presentation has emerged as a novel method toward developing more efficient cancer vaccines. Here, we propose a two-step hybrid strategy to produce a more robust cell-based cancer vaccine in situ. First, a significant number of DCs are recruited to an injectable thermosensitive mPEG–PLGA hydrogel through sustained release of chemoattractants, in particular, granulocyte-macrophage colony-stimulating factor (GM-CSF). Then, these resident DCs can be loaded with cancer antigens through the use of viral or nonviral vectors. We demonstrate that GM-CSF-releasing mPEG–PLGA hydrogels successfully recruit and house DCs and macrophages, allowing the subsequent introduction of antigens by vectors to activate the resident cells, thus, initiating antigen presentation and triggering immune response. Moreover, this two-step hybrid strategy generates a high level of tumor-specific immunity, as demonstrated in both prophylactic and therapeutic models of murine melanoma. This injectable thermosensitive hydrogel shows great promise as an adjuvant for cancer vaccines, potentially providing a new approach for cell therapies through in situ modulation of cells.
Introduction
Much research has been devoted toward generating efficient systemic immunity against tumors, which commonly evades immune detection in patients. The development of an effective cancer vaccine and immunotherapy involves generating a potent antigen-specific immune response by introducing tumor antigens to antigen-presenting cells (APCs),1 including dendritic cells (DCs) and macrophages. DCs are typically considered key regulators of T- and B-cell immunity based on their superior ability to take up, process, and present antigens compared with other APCs.2,3 Therefore, extensive efforts have been made to manipulate DCs to deliver vaccines to achieve protective immunity with optimal efficacy.
There is a growing attempt to enhance immune responses by immunizing cancer patients with their own DCs that have been isolated and activated ex vivo.4,5 This strategy generates potent activated DCs since high efficiency and specificity are achievable under defined in vitro conditions. While initial results are promising, the complexity, added cost, and labor associated with this form of customized cell therapy remain obstacles for the clinical translation of this method. More importantly, a rapid decline in viability and function of the vast majority of transplanted DCs results in limited numbers of DCs able to successfully home to lymph nodes.6,7 To address some of these concerns, DC-based vaccine development has been focused on in vivo direct DC targeting strategies. Direct injection of viral vectors containing the cancer antigen has been shown to induce immune responses against tumors.8−10 This strategy requires less labor to get more APCs home to the lymph nodes and trigger an immune response and is, therefore, more cost-effective. However, the low number of DCs at the site of antigen delivery generally results in a small pool available to prime the immune response, again limiting vaccine efficacy.
To enhance antigen uptake to DCs, vaccine delivery systems need to maximize either the amount of antigen reaching the APCs or the number of antigen-loaded APCs homing to local lymph nodes. These steps cannot be improved with the current system as antigen degradation during the process of formulating the encapsulation materials is the main limiting step in the vaccine development process.11 An alternative strategy would develop materials designed to create a microenvironment where host DCs can be recruited and allowed to proliferate and differentiate in situ. To this end, various chemokine-releasing materials have been explored to recruit DCs at the site of immunization.12−16 Compared to traditional biomaterials, such as degradable polymers,17 thermosensitive hydrogels have become increasingly attractive as drug delivery systems as a result of their minimally invasive injectable design.18 More recently, a thermosensitive gelling carrier, monomethoxypoly(ethylene glycol)-co-poly(lactic-co-glycolic acid) copolymer (mPEG–PLGA hydrogel), has been developed for sustainable delivery of antibiotics to target tissues.19 This sol-to-gel drug delivery system could provide several advantages, including a 100% encapsulation rate, near-linear sustained release of drugs and in situ gelling at the target tissue. Importantly, it has the potential to serve as a vaccine delivery system that can create a microenvironment where DCs can be recruited and subsequently loaded with immunogens.
In this study, we proposed a two-step hybrid strategy which involves recruiting and modulating DCs by cytokine-carrying thermosensitive mPEG–PLGA hydrogels followed by injecting vaccine vectors to load antigens to these resident DCs (Scheme 1). This mPEG–PLGA hydrogel provides a sustained release profile of granulocyte-macrophage colony-stimulating factor (GM-CSF), which can mediate recruitment, proliferation, and maturation of DCs and macrophages at the site of inoculation.20,21 We also show that both viral and nonviral vectors carrying antigens can be introduced to further modulate the resident cells, enhancing their ability to induce cytotoxic T lymphocyte (CTL) immune responses. These enhancements resulted in improved protection against the tumor growth shown in both prophylactic and therapeutic tumor models. These studies demonstrate the potential of injectable thermosensitive hydrogels carrying cytokines to serve as an adjuvant to enhance antigen uptake efficiency of APCs in a localized in vivo environment, offering many new options for delivery of cancer vaccines.
Scheme 1. Schematic Illustration of the Two-Step Hybrid Strategy for a Cancer Vaccine.
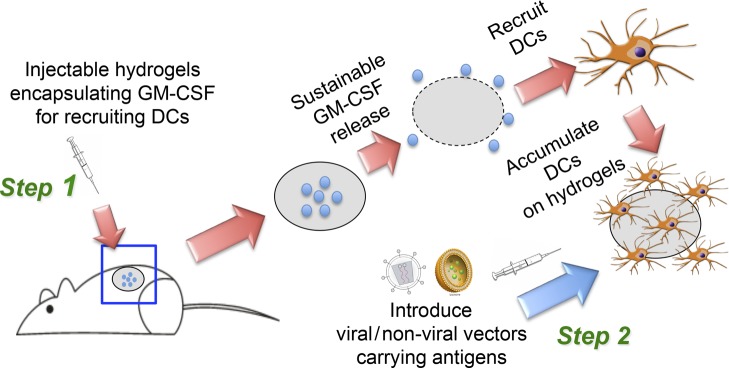
Step 1: Sustainable release of GM-CSF from the injectable thermosensitive mPEG–PLGA hydrogels recruits host dendritic cells (DCs) to the site of administration. Step 2: Viral or nonviral vectors carrying immunogens can be delivered in situ to the resident DCs in hydrogels to enhance antigen uptake efficiency, thereby improving anticancer immunity.
Experimental Section
Mice, Cell Lines, and Reagents
Six- to eight-week-old female C57BL/6J mice were purchased from the Charles River Laboratories. All animal procedures were performed in accordance with the guidelines set by the National Institutes of Health, Caltech, and USC on the Care and Use of Animals. B16 and B16-OVA tumor cells (B16–F10, ATCC number: CRL-6475) were maintained in a 5% CO2 environment with Dulbecco’s modified Eagle’s medium (DMEM; Mediatech, Inc., Manassas, VA) supplemented with 10% FBS (Sigma-Aldrich, St. Louis, MO) and 2 mM of l-glutamine (Hyclone Laboratories, Inc., Omaha, NE). Poly(ethylene glycol) monomethyl ether (mPEG; Mn, 550 g/mol), d,l-lactide and glycolide, and stannous 2-ethylhexanoate (stannous octoate) were purchased from Sigma (St. Louis, MO).
GM-CSF Production
To generate the construct pET302nt-GM-CSF for expression of GM-CSF, the cDNA of murine GM-CSF was cloned into the pET302nt vector (Life Technologies/Invitrogen, Grand Island, NY). The plasmids were transformed to the host Escherichia coli BL21 (DE3; Life Technologies/Invitrogen). The bacteria were grown in luria broth (LB) containing 100 μg/mL of ampicillin at 37 °C until OD600 reached 0.6. Isopropyl-p-d-thiogalactopyranoside (IPTG, 1 mM) was added to the culture medium to induce protein expression. Cells were harvested 6 h postinduction and the recombinant protein was isolated from inclusion bodies by washing them with 2 M urea buffer 4 times and dissolving them in 8 M urea. After renaturation through dialysis in gradient urea buffer, the recombinant GM-CSF was purified by the Ni2+-IDA column. The resulting protein was lyophilized and stored in cold (−80 °C).
Synthesis of mPEG–PLGA Diblock Copolymers
A series of monomethoxypoly(ethylene glycol)-co-poly(lactic-co-glycolic acid) (mPEG–PLGA) diblock copolymers were synthesized using a previously reported protocol.19 Briefly, mPEG (3 g, MW 550) was mixed with lactide (6.2 g) and glycolide (1.42 g) in a four-neck reactor with a mechanical stirrer. N2 gas flow was maintained to keep the reactor dry. The catalyst, stannous 2-ethylhexanoate (4.3 μL), was added to the reactor, and polymerization occurred at 160 °C for 5 h. The copolymer aqueous solution was purified by dialysis (MWCO 1000) for 3 days at 4 °C and freeze-dried by lyophilization for 5 days. The lyophilized hydrogel was dissolved in deionized water overnight in a cold room to make 15, 20, or 25% (w/v) hydrogel solution. To prepare GM-CSF encapsulated hydrogels, various amounts of GM-CSF were added to mPEG–PLGA copolymer aqueous solution and mixed by pipetting on ice. The hydrogel-GM-CSF mixture was then transferred to a 1 mL syringe with a 21G needle and kept at 37 °C for 3 min for gelation. The gelled hydrogel could be further used for study.
In Vitro GM-CSF Release Kinetics Study
Fluorescein (FITC)-labeled GM-CSF was added to 15, 20, or 25% (w/v) mPEG–PLGA copolymer aqueous solution to form a hydrogel. The resulting hydrogels were plated on 24-well tissue culture plates (Corning, Cambridge, MA) and incubated at 37 °C with PBS. To obtain the release kinetics of GM-CSF from hydrogel, the releasing media were collected to measure FITC fluorescence, and fresh PBS was replaced at regular time intervals for continuous monitoring of protein release. FITC fluorescence (excitation 488 nm, emission 515 nm) was measured by a Shimadzu RF-5301PC spectrofluorometer (Japan).
BMDC and BMDM Generation
Bone marrow-derived DCs (BMDCs) were generated according to a previously described procedure.22 Briefly, bone marrow from the femurs and tibias of male C57BL/6 mice was grown in RPMI 1640 with 10% fetal bovine serum, 2 mM l-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 0.05 mM 2-ME, and 20 ng/mL GM-CSF (J558L supernatant) after the red blood cells were lysed. Cultures were initiated by placing 1 × 107 bone marrow cells in 10 mL of medium onto 100 mm Petri dishes (Falcon 1029 plates; BD Labware, Franklin Lakes, NJ). On day 3, another 10 mL of J558L conditioned media were added. On day 6, suspended cells were collected.
Macrophages were derived from BM precursors as described before.23 Briefly, bone marrow cells (1 × 107 cells/mL) were cultured in a volume of 10 mL in a 100 mm Petri dish in RPMI 1640 supplemented with 20% fetal bovine serum and 30% L929 conditioned medium as a source of macrophage colony-stimulating factor (M-CSF). After 3 days of culture, an additional 10 mL of differentiation media were added. At day 7, macrophages were detached with ice-cold PBS and characterized by FACS, using the pan-macrophage marker F4/80.
Virus Production
The lentiviral backbone plasmid FUW-TfROVA was constructed by insertion of cDNA consisting of the first 118 amino acids of the membrane-anchoring domain of murine transferrin receptor fused downstream with truncated chicken ovalbumin (OVA, amino acids 139–386) into FUW.24 FUW is a HIV-1-derived lentiviral plasmid composed of an internal human ubiquitin-C promoter to drive transgene expression and woodchuck responsive element to improve stability of the RNA transcript.25 A previously reported procedure of transient transfection of 293T cells to produce the DC-LV-OVA vector was used in this study.22 Lentiviral vectors were generated by transfecting 293T cells by using a standard calcium phosphate precipitation technique. Briefly, 293T cells were seeded in a 6 cm culture dish in DMEM medium supplemented with fetal bovine serum (Sigma, St. Louis, MO, 10%), l-glutamine (10 mL/L), and penicillin and streptomycin (100 units/mL). After 18–20 h, 293T cells with confluence of 90% were transfected with appropriate plasmids per plate of 5 μg FUW-TfROVA, together with 2.5 μg each of SVGmu and the packaging vector plasmids (pMDLg_pRRE and pRSV-Rev).22 The viral supernatants were harvested 48 and 72 h after transfection and concentrated by using ultracentrifugation (Optima L-80 K preparative ultracentrifuge, Beckman Coulter) for 90 min at 50000 × g. Particle pellets were then resuspended in an appropriate volume of ice-cold PBS for in vivo injection.
In Vitro Transwell Migration Assay
The transwell plates with 0.45 μm pore filters on the bottom of the upper compartment26,27 were used to study BMDC and BMDM migration toward GM-CSF. Hydrogel encapsulating various concentrations of GM-CSF was placed in the lower compartment of the transwell, while 0.5 million bone marrow-derived dendritic cells (BMDCs) or bone marrow-derived macrophages (BMDMs) were seeded in the upper compartment. After a 24 h incubation, the cells that migrated into the lower compartment were collected and counted.
In Vivo DC Recruitment by Hydrogel
Either empty hydrogel or hydrogel encapsulating 5 μg of GM-CSF was subcutaneously (s.c.) injected into mice at day 0 and collected at day 3, 7, 14, or 21. Hydrogel areas were excised for histological detection or FACS analysis of recruited DCs and macrophages. The total number of recruited cells was counted after digesting the hydrogels with collagenases. CD11c+ and F4/80+ cells were counted by FACS analysis. Single-cell suspensions were incubated with antimouse CD16/CD32 Fc blocking antibody and then stained with fluorophore-conjugated monoclonal antibodies against specific BMDC and BMDM surface markers, including CD86, CD11c, and F4/80. All antibodies were purchased from BioLegend (San Diego, CA). Stained cells were assayed using a BD LSRII flow cytometer (BD Biosciences), and acquired data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Immunohistochemical Analysis
Injected empty hydrogel or hydrogel encapsulating GM-CSF samples were excised 7 days postinjection. The samples were fixed with 4% formaldehyde, frozen, cryosectioned, and mounted onto glass slides. After blocking and permeabilization, the slides were stained with biotinylated antimouse CD11c (BioLegend, San Diego, CA), followed by incubation with streptavidin-conjugated HRP for 30 min. After incubation, the slides were washed 3 times with PBS and then developed with the DAB substrate (Abcam, Cambridge, MA). The images were acquired by light microscopy.
For fluorescence staining of DCs and macrophages, the slides were stained with rat anti-F4/80 (BioLegend, San Diego, CA) or FITC-conjugated antimouse CD11c (BioLegend), followed by Alexa647-conjugated antirat IgG (Invitrogen, Carlsbad, CA) and counterstained with DAPI (Invitrogen). Fluorescence images were acquired by a Yokogawa spinning-disk confocal scanner system (Solamere Technology Group, Salt Lake City, UT) using a Nikon Eclipse Ti-E microscope. Illumination powers at 405, 491, 561, and 640 nm solid-state laser lines were provided by an AOTF (acousto-optical tunable filter)-controlled laser-merge system with 50 mW for each laser. All images were analyzed using Nikon NIS-Elements software.
In Vivo DC Migration Assays
For DC migration assay in C57/BL/6 mice, mice were injected s.c. in the hind legs with hydrogel containing saline or 5 μg of GM-CSF. Seven days postinjection, FITC (25 μg, Sigma-Aldrich, St. Louis, MO) and MPL (25 μg, Sigma-Aldrich, St. Louis, MO) mixtures were injected into the same site as the implanted hydrogel. At day 10, draining popliteal lymph node samples were harvested, and the number of CD11c+FITC+ cells and F4/80+FITC+ cells in each sample was determined using FACS.
Immunization
For immunization with DC-targeted lentiviral vectors (DC-LV), mice were injected with replication-defective DC-LV-OVA (5 × 106 transduction units (TU)) at the rear footpad 7 days postinjection with empty hydrogel or GM-CSF-encapsulated hydrogel. To optimize the time point of vector administration, mice were injected with DC-LV-OVA at the rear footpad 3, 7, 10, or 14 days postinjection with hydrogels. The splenocytes from immunized or control mice were excised at day 21. For immunization with adjuvant, either CpG or MPL (25 μg) was administered at the base of tail when DC-LV-OVA was injected. The splenocytes from immunized or control mice were excised 14 days postimmunization.
For immunization with nonviral vectors, mice were injected with OVA protein (50 μg) and MPL (25 μg) at the rear footpad 7 days postinjection with empty hydrogel or GM-CSF-encapsulated (5 μg) hydrogel. Splenocytes were pooled for an ELISPOT assay to analyze IL-2 secretion 7 days postimmunization.
IFN-γ Intracellular Cytokine Staining (ICCS)
Splenocytes from immunized or control mice were pooled and incubated with the OVA257–264 peptide (SIINFEKL; 1 μg/mL) in the presence of costimulatory anti-CD28 antibody (2 μg/mL, BD Biosciences) for 2 h at 37 °C in a 96-well round-bottom plate in RPMI medium supplemented with 10% FBS (Sigma), 10 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM glutamine. Brefeldin A (BFA, Sigma, St. Louis, MO) was added (10 μg/mL) to wells to inhibit cytokine exporting for another 4 h. Surface staining was performed by incubating restimulated cells with antimouse CD16/CD32 Fc blocking antibody, followed by antimouse CD8 and antimouse CD4 antibodies. Cells were then permeabilized in 100 μL Cytofix/Cytoperm solution (BD Biosciences) at 4 °C for 10 min, washed with Perm/Wash buffer (BD Biosciences), followed by intracellular staining with PE-conjugated antimouse IFN-γ at 4 °C for 15 min. The flow cytometry analysis was carried out using the MACSQuant analyzer from Miltenyi Biotec.
IL-2 ELISPOT Assay
ELISPOT assays to detect IL-2 were performed using a kit from Millipore (Billerica, MA). Briefly, antimouse IL-2 antibody (10 μg/mL in PBS) was used as the capture antibody and plated with 100 μL/well on 96-well MultiScreen-IP plates overnight at 4 °C. The plate was decanted and blocked with RPMI medium containing 10% FBS at 37 °C for 2 h. Splenocytes from mice were plated at 5 × 105 cells/well in complete medium together with the CD4 epitope OVA323–339 peptide (ISQAVHAAHAEINEAGR; 10 μg/mL). After 18 h incubation at 37 °C, cells were lysed, and plates were detected by biotinylated anti-IL-2 antibody (BD Biosciences) for 2 h at room temperature. Plates were further washed and incubated with streptavidin-alkaline phosphate conjugate for 45 min at room temperature. After washing, spots were identified by adding BCIP/NBTplus substrate (Millipore), and the number of IL-2 producing cells was quantified by an ELISPOT reader.
Antitumor Immunity Experiment
In the tumor prophylactic model, mice were immunized 7 days after implantation of empty or GM-CSF hydrogel. A total of 10 days postimmunization, mice were inoculated with 1 million B16-OVA tumor cells. A tumor size of 2000 mm3 was used as a surrogate end point of survival. In the therapeutic tumor model, mice were challenged with 0.1 million B16-OVA cells and implanted with hydrogels 1 day later. Two immunizations were given on days 7 and 9.
Statistics
The data were represented as mean ± SEM of the indicated number of measurements. Statistical significance was calculated by using Prism (GraphPad Inc.). The differences between two groups were determined with Student’s t test. The differences among three or more groups were determined with a one-way ANOVA. P < 0.05 is considered statistically significant.
Results
In Vitro Recruitment of DCs and Macrophages by GM-CSF Hydrogel
As illustrated in Scheme 1, our goal was to develop an injectable biomaterial with high cytokine encapsulation efficiency to attract DCs to a defined injection site where antigens could be introduced, thereby enhancing antigen uptake to DCs. To accomplish this, we adapted a previously reported procedure19 to generate a thermosensitive hydrogel, consisting of a diblock copolymer of mPEG (polyethylene glycol) and FDA-approved PLGA (lactic acid and glycolic acid) polymer. This polymeric formulation is in the solution phase at 4 °C and shows a fast sol–gel transition at 37 °C, within 5 min (Figure 1A). This simple formulation procedure allows almost 100% encapsulation efficiency of cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF) (Figure 1B). Additionally, linearly sustained release profiles of GM-CSF can be achieved in 15, 20, and 25 wt % aqueous solutions of the mPEG–PLGA diblock copolymer, as shown in Figure 1B. To examine whether the hydrogel-released GM-CSF remained sufficiently functional to recruit DCs and macrophages toward the hydrogel area, an in vitro transwell cell migration assay was developed. Briefly, hydrogels encapsulating various amounts of GM-CSF were placed in the lower compartment of the transwell, while the bone marrow-derived dendritic cells (BMDCs) and bone marrow-derived macrophages (BMDMs) were seeded in the upper compartment. The chemotaxis of BMDCs and BMDMs in response to GM-CSF gradient was determined by counting the cells that migrated to the lower compartment. As shown in Figure 1C,D, significantly more migrating BMDCs and BMDMs were found for hydrogels containing 5 μg of GM-CSF compared to lower doses of GM-CSF (p < 0.05), indicating that the GM-CSF released from hydrogel enhanced DC and macrophage recruitment in a dose-dependent manner.
Figure 1.

In vitro recruitment of dendritic cells and macrophages by GM-CSF-loaded hydrogels. (A) mPEG–PLGA copolymer formulations are an injectable solution at 4 °C, but turn to gel after incubation at 37 °C for 5 min. (B) The release profiles of FITC-labeled GM-CSF from hydrogel formulations (15, 20, and 25 wt %; n = 3). (C) In vitro chemotaxis using a transwell migration assay. Chemotaxis of BMDCs and BMDMs in response to hydrogels encapsulating various doses of GM-CSF was determined by counting the number of BMDCs and BMDMs migrating to the lower compartment of transwell (n = 3).
In Vivo Recruitment of DCs and Macrophages by GM-CSF Hydrogel
Since it was shown that the sustained release of GM-CSF from hydrogels successfully attracts BMDCs and BMDMs, we next proposed that the diffusion of this factor through the surrounding tissue could effectively recruit host DCs and macrophages. To examine this hypothesis, C57BL/6J mice were injected once s.c. with hydrogels containing 5 μg of GM-CSF or empty hydrogels. Implanted materials were collected from each group at days 3, 7, 14, or 21 postinjection to either count the number of recruited cells or to identify recruited cell types by FACS and histology assay. As shown in Figure 2A, some yellowish color and blood vessels were formed on the GM-CSF hydrogel-injected area, while the empty hydrogel maintained its clear appearance, indicating that cells were recruited around the GM-CSF hydrogels. The thermosensitive hydrogel decreased in size with the passage of time, indicating that it is a biodegradable polymer. On day 7, the quantitative data (Figure 2B) show that the GM-CSF hydrogel recruited 3–4 million cells, consisting of ∼0.5 million CD11c+ DCs, as shown in Figure 2D. Histology examination of the hydrogels also showed a significant number of CD11c+ dendritic cells in the GM-CSF hydrogel-injected mice, indicating that GM-CSF from hydrogel retains its cell-recruiting function in vivo. About 0.5 million F4/80+ macrophages also accumulated around the GM-CSF hydrogel (Figure 2E), suggesting that sustained release of GM-CSF can recruit macrophages, as well as DCs, in vivo. Analysis of DCs and macrophages specifically (CD11c+CD86+ and F4/80+CD86+, respectively) showed that GM-CSF increased not just the total resident cell number, but also the percentage of mature DCs and macrophages (Figure 2F). Importantly, it was shown that most recruited DCs were inactive DCs (CCR7–CD11c+) and that some portions of plasmacytoid DCs (PDCA+CD11c+) were also recruited. Immunofluorescent staining (Figure 2G) on the collected hydrogels further confirmed the effects of GM-CSF encapsulation on in vivo DC and macrophage recruitment.
Figure 2.

In vivo recruitment and migration of dendritic cells and macrophages in response to hydrogels loaded with GM-CSF (5 μg). (A) Degradation of empty hydrogel (Emp-hydrogel) and hydrogel loaded with GM-CSF (GM-hydrogel) at various time points after injection. (B) Total number of cells collected from Emp-hydrogel or GM-hydrogel at indicated time points after injection (n = 3). (C) Histologic appearance of Emp-hydrogel and GM-hydrogel with CD11c staining 7 days postinjection. (D, E) The number of CD11c+ DCs (D) and F4/80+ macrophages (E) recruited by GM-CSF-loaded hydrogel at indicated time points after injection. (F) Flow cytometric analysis of CD11c+CD86+, F4/80+CD86+, CCR7+CD11c+, and PDCA-1+CD11c+ cells from a representative mouse from each group 7 days after Emp-hydrogel or GM-hydrogel injection. (G) Representative images of hydrogel sections of mice stained for CD11c and F4/80, showing recruitment of DCs and macrophages in GM-hydrogel 7 days postinjection. (H) GM-hydrogel increases the migration of DCs and macrophages to the draining LNs. Flow cytometric analysis of CD11c+FITC+ cells and F4/80+FITC+ cells from a representative mouse from each group 3 days after injection of FITC and MPL (as adjuvant).
The number of DCs and macrophages recruited by GM-CSF-encapsulated hydrogels peaked at day 7 and significantly decreased by 14 days postinjection (Figure 2D,E), suggesting that the recruited cells might be dispersed once the release of GM-CSF is decreased, allowing them to home to the lymph nodes over time. The ability of these recruited DCs and macrophages to migrate to the lymph nodes was then tested. Either GM-CSF hydrogel or empty hydrogel was injected into mice, and fluorescein isocyanate (FITC) was injected to the same site 7 days later. DCs and macrophages recruited to the hydrogel area ingested the FITC label, thus, allowing their subsequent movements to be tracked. Lymph nodes were harvested at day 10 and analyzed for FITC-labeled cells. As shown in Figure 2H, FITC-positive DCs and macrophages were significantly enhanced in the draining lymph nodes of mice injected with GM-CSF-encapsulated hydrogel, revealing the superior ability of GM-CSF hydrogel to recruit and subsequently disperse the resident DCs and macrophages to home to the lymph nodes, where B- and T-cell immunities can be induced.
GM-CSF Hydrogel System Enhanced Antigen-Specific Immune Response
The local recruitment of immature DCs and macrophages by GM-CSF hydrogel allows for larger numbers of cells to be further modulated with antigen delivery vectors in situ. To test this hypothesis, previously reported SVGmu-pseudotyped lentiviral vectors encoding ovalbumin (OVA),22,28 which can target antigen delivery to DCs, were introduced to the site of the hydrogel after DC recruitment. First, the optimal time gap between injection of GM-CSF hydrogel and immunization with vectors carrying OVA (DC-LV-OVA) was determined (Figure 3A). Significantly more inflammatory cytokine interferon-γ (IFN-γ) was produced by CD8+ T cells from mice immunized by DC-LV-OVA 7 days postinjection of GM-CSF hydrogel than at other times (Figure 3B,C), suggesting that this immunization schedule was optimal for further study. The optimal dose of GM-CSF exposure to recruited DCs was also evaluated by measuring the degree of immune responses in mice injected with hydrogels encapsulating various amounts of GM-CSF. As shown in Figure 3D,E, the highest immune response was found in mice injected with hydrogel encapsulating 5 μg of GM-CSF. A higher concentration of GM-CSF (15 and 45 μg) in hydrogel led to a decrease in IFN-γ production, suggesting that an optimal GM-CSF concentration is required to recruit and subsequently disperse the activated DCs to lymph nodes for immune response induction.
Figure 3.

Optimization of the two-step hybrid strategy to induce antigen-specific immune responses. (A) Schematic representation showing immunization procedures. Mice were immunized with DC-LV-OVA at different time points after GM-CSF hydrogel injection. (B) Flow cytometric analysis of IFN-γ+ cells within the CD8+ T-cell population in mouse splenocytes. (C) Quantitative data of the percentage of IFN-γ+ cells within the CD8+ T-cell population. (D, E) The effect of different doses of GM-CSF encapsulated in hydrogels on DC-LV-OVA-based vaccine-specific T cell immune responses in vivo. Seven days after injection of hydrogels with different doses of GM-CSF, mice were immunized with DC-LV-OVA. A total of 14 days later, splenocytes were collected, and OVA-specific CD8+ T cells were analyzed by intracellular staining for IFN-γ expression after stimulation with OVA257–264 peptide for 6 h. The FACS data are representative of four analyzed mice (D). Statistical data showing the percentage of IFN-γ+ cells within the CD8+ T cell population (E).
We next asked whether the activation and maturation of DCs with molecular adjuvant could further enhance the immune response in this GM-CSF hydrogel system. To test this possibility, mice were immunized with DC-LV-OVA 7 days postinjection with GM-CSF (5 μg) hydrogel. The effect of two adjuvants, monophosphoryl Lipid A (MPL) and CpG, on the maturation of BMDCs was first examined in vitro. Significant enhancement in the expression of surface markers, including CD54, I-Ab, and CD 86, was observed after incubating BMDCs with the adjuvants overnight (Figure 4A). To test the effect in vivo, either CpG or MPL was injected into the hydrogel inoculation site at day 8, and the degree of immune response was evaluated by measuring IFN-γ production in CD8+ T cells taken from mice 14 days postimmunization (Figure 4B). As shown in Figure 4C,D, the GM-CSF hydrogel enabled a significant enhancement in immune responses induced by DC-LV-OVA with CpG or MPL compared to empty hydrogel. The data suggest that DCs recruited by GM-CSF could be further activated and maturated by the addition of adjuvant to enhance antigen-specific immune responses. Moreover, the ability of GM-CSF hydrogel to serve as a microenvironment for DC recruitment and programming by nonviral antigen delivery vectors was investigated. As shown in Figure 4E, immunization with OVA protein and MPL resulted in a 3-fold increase in IFN-γ production of CD8+ cytotoxic T cells in mice bearing GM-CSF hydrogels over those bearing empty hydrogel. Taken together, these data suggest that GM-CSF hydrogel can recruit DCs and allow in situ DC programming by antigen delivery vectors and adjuvant to increase T cell priming.
Figure 4.

GM-hydrogel allows for further modulation of resident DCs by adjuvants to enhance immune responses against specific antigen (OVA). (A) In vitro effects of adjuvants MPL and CpG on maturation of BMDCs. BMDCs were stimulated with 1 μg/mL MPL or 5 μM CpG overnight. The BMDCs were collected for staining of CD11c, I-Ab, CD54, and CD86. The result was analyzed by flow cytometry, and the expression of surface markers I-Ab, CD54, and CD86 was gated on CD11c+ DCs. (B) Schematic diagram showing the procedures. Seven days after injection of hydrogels with 5 μg of GM-CSF, mice were immunized with DC-LV-OVA. One day after immunization, the mice were injected with adjuvants (CpG or MPL). Two weeks after immunization, splenocytes were collected, and OVA-specific CD8+ T cells were analyzed by intracellular staining of IFN-γ expression. (C, D) GM-CSF hydrogels enable further enhancement of lentiviral vector-mediated immune responses with adjuvants. The FACS data are representative of four analyzed mice (C). Statistical data showing the percentage of IFN-γ+ cells within the CD8+ T cell population (D). (F) The effect of GM-CSF-loaded hydrogels on nonviral vector-mediated immune response. Seven days after injection of hydrogels with 5 μg of GM-CSF, mice were immunized with OVA protein and MPL. Seven days after immunization, splenocytes were pooled for an ELISPOT assay to analyze IL-2 secretion following stimulation with peptide for 18 h (n = 3).
Ability of Two-Step Hybrid Strategy As Cancer Vaccines in Tumor Model
The utility of this two-step hybrid strategy (illustrated in Scheme 1) that first recruits DCs and then programs them within the GM-CSF hydrogel as a cancer vaccine was evaluated in a murine melanoma model. In the prophylactic tumor model, mice were immunized with DC-LV-OVA alone or with MPL adjuvant 7 days after injection of hydrogels, as illustrated in Figure 5A. A total of 10 days after immunization, mice were inoculated with B16-OVA tumor cells, which stably express the model tumor antigen OVA. Using a tumor size of 2000 mm3 as a surrogate end point of survival, none of the PBS-injected (control) mice survived for more than 30 days (Figure 5B). Mice injected with empty hydrogels showed moderately improved immune protection compared to mice injected with PBS, suggesting that the residence provided by hydrogels could be beneficial to enhance antigen uptake efficiency of APCs. However, a longer overall survival (p < 0.001) was observed in mice injected with GM-CSF hydrogels compared to mice injected with empty hydrogels, indicating the benefit of recruiting DCs with GM-CSF. The benefit of GM-CSF-encapsulated hydrogel was further confirmed by a significantly longer median survival time in GM-CSF-treated mice that received both DC-LV-OVA and MPL treatment compared to empty hydrogel-injected mice with the same immunization condition (p < 0.001).
Figure 5.

Two-step hybrid strategy confers potent antitumor immunity. (A) Schematic diagram showing the immunization and tumor challenge procedure in the prophylactic model. (B) Kaplan–Meier survival plot of mice treated with PBS (Ctrl), empty hydrogel scaffolds (Emp-gel), GM-CSF hydrogel scaffolds (GM-gel), followed by immunization with DC-LV-OVA only or with adjuvant MPL (Emp-gel + MPL, GM-gel + MPL; n = 10). A tumor size of 2000 mm3 was used as a surrogate end point of survival, P < 0.001. (C) Schematic diagram showing tumor inoculation on day 1 and hydrogel implantation 1 day later, followed by two immunizations in the therapeutic model. (D) Tumor growth was plotted as mean ± SEM (n = 8) as a function of days after B16-OVA tumor challenge (** indicates P < 0.01).
We also evaluated the two-step hybrid strategy as a cancer vaccine in a therapeutic tumor model, as illustrated in Figure 5C. Mice were challenged with B16-OVA tumor cells 1 day posthydrogel injection. Two immunizations were administered on days 7 and 9, respectively. Compared to the PBS-injected group, significantly slower tumor growth was observed, even in empty hydrogel-injected mice, confirming the previous finding that hydrogel itself could provide a residence for APCs and could, consequently, benefit antitumor immunity. Moreover, injecting GM-CSF-loaded hydrogel, which provides sustained release of GM-CSF capable of recruiting DCs and macrophages for further programming, resulted in marked tumor suppression (p < 0.01). The effect of the two-step hybrid strategy on antitumor immunity could be further enhanced by modulating recruited DCs with the adjuvant MPL, as demonstrated by the slower tumor growth in GM-CSF hydrogel-injected mice receiving both lentiviral vaccine and MPL treatment compared to that of the vaccine only group (p < 0.01).
Discussion
Development of an effective cancer vaccine and immunotherapy involves generating a potent antigen-specific immune response by delivering tumor antigens to APCs, especially DCs.1,2,4−7 Cell-based therapies often fail to induce a strong systemic immunity because only a limited number of transplanted DCs remain sufficiently functional to home to lymph nodes.6,7 To overcome the problems associated with the ex vivo strategy, an in vivo direct DC targeting strategy has been proposed to deliver antigens by viral8,9,25 or nonviral29,30 vectors. However, this strategy is hampered by the low number of targetable DCs and low efficiency of antigen uptake to DCs. Therefore, in the present study, a two-step hybrid strategy was presented. An injectable thermosensitive hydrogel network that can house DCs and macrophages is used to deliver DC chemoattractants (GM-CSF) that enhance their recruitment and migration (step 1 in Scheme 1). This is followed by programming of recruited DCs with antigen delivery vectors (step 2 in Scheme 1), allowing for efficient antigen uptake to DCs and therefore enhancing the immune response. This strategy allows us to program DCs in situ, not only bypassing the complication and cost of ex vivo strategy, but also improving antigen uptake to DCs. We also demonstrated that this two-step hybrid strategy improves antitumor immunity, as shown in both prophylactic and therapeutic tumor models (Figure 5B,D), potentially providing a new means of delivering potent cancer vaccines.
Biomaterials have been extensively explored to deliver chemoattractants and create a physical microenvironment for recruited cells.12−14,16,31 For instance, PLG scaffolds have been recently reported for controlled-release of chemoattractants to recruit DCs in situ; however, their application is limited by the low survival rate of transplanted cells. In this study, we evaluate an injectable thermosensitive mPEG–PLGA hydrogel as a GM-CSF delivery system to successfully recruit, activate, and direct DCs to the lymph nodes. The sol-to-gel delivery system provides 100% encapsulation efficiency and sustained release of GM-CSF, which can attract large numbers of immature DCs and macrophages to the injection site. The hydrogel can also provide a residence for recruited DCs as a result of its gel structure and its release of activated DCs during gel degradation. Moreover, the low phase transition temperature of this mPEG–PLGA hydrogel can protect encapsulated molecules from denaturing or aggregating.19
GM-CSF has attracted considerable interest as an immune adjuvant by its ability to increase the maturation and function of DCs and macrophages.20,21,32,33 However, systemic administration of GM-CSF has been associated with side effects, such as induction of histologically abnormal liver and spleen,34 limiting the clinical application of this adjuvant. As an alternative, local GM-CSF delivery using such biomaterials as polymer or gel has been proposed to enhance vaccine efficacy and, hence, avoid the side effects associated with systemic delivery.14,31,35 Here, we demonstrated the potential of thermosensitive mPEG–PLGA hydrogels as GM-CSF carriers for local recruitment of DCs and macrophages. Most of the encapsulated GM-CSF was released from hydrogels within 5 days (Figure 1B), resulting in a peak number of DCs and macrophages residing in the hydrogels at day 7 postinjection (Figure 2D,E). Moreover, optimizing the GM-CSF dose (5 μg) in combination with immunization increased immune response by nearly 2-fold over nonoptimal GM-CSF exposures (15 and 45 μg; Figure 4B), indicating that high doses of GM-CSF might reduce the ability of recruited DCs to home to lymph nodes for immunity induction. Therefore, decreasing the release rate of GM-CSF over time would be desirable to allow the recruited and programmed DCs and macrophages to migrate and home to lymph nodes.
Overall, our results highlight an alternative two-step hybrid approach to current cancer vaccines. First, sustainable release of chemoattractants from mPEG–PLGA hydrogels is able to recruit immature DCs. Next, in situ programming of resident DCs by antigen delivery vectors results in their efficient maturation and activation. More broadly, this study demonstrated a new application of injectable thermosensitive hydrogels that may potentially serve as a temporary residence for in situ cell programming to treat a variety of diseases. For instance, local sustained delivery of some osteoinductive growth factors by these injectable and biodegradable hydrogels could be beneficial for the reconstruction of large bone defects.
Conclusions
We developed a two-step vaccine procedure that allows continuous recruitment of DCs to the site of administration by the sustainable release of GM-CSF from an injectable thermosensitive hydrogel, followed by in situ programming of these DCs by antigen delivery vectors. We demonstrated that primary APCs, including DCs and macrophages, migrated efficiently to hydrogels in response to gel-released GM-CSF and that they could be further modulated by vectors carrying antigens, leading to potent antitumor immunity. This method could be a powerful alternative to current cell therapies by allowing modulation or reprogramming host cells in situ.
Acknowledgments
This work was supported by National Institutes of Health Grants (R01AI068978, R01CA170820, and P01CA132681), a translational acceleration grant from the Joint Center for Translational Medicine and the National Cancer Institute (P30CA014089).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Trombetta E. S.; Mellman I. Cell Biology of Antigen Processing In Vitro and In Vivo. Annu. Rev. Immunol. 2005, 23, 975–1028. [DOI] [PubMed] [Google Scholar]
- Banchereau J.; Steinman R. Dendritic Cells and the Control of Immunity. Nature 1998, 392, 245–252. [DOI] [PubMed] [Google Scholar]
- Mellman I.; Steinman R. M. Dendritic Cells: Specialized and Regulated Antigen Processing Machines. Cell 2001, 106, 255–258. [DOI] [PubMed] [Google Scholar]
- Banchereau J.; Palucka A. K. Dendritic Cells As Therapeutic Vaccines against Cancer. Nat. Rev. Immunol. 2005, 5, 296–306. [DOI] [PubMed] [Google Scholar]
- Nestle F. O.; Farkas A.; Conrad C. Dendritic-Cell-Based Therapeutic Vaccination against Cancer. Curr. Opin. Immunol. 2005, 17, 163–169. [DOI] [PubMed] [Google Scholar]
- Steinman R. M.; Banchereau J. Taking Dendritic Cells into Medicine. Nature 2007, 449, 419–426. [DOI] [PubMed] [Google Scholar]
- Gilboa E. DC-Based Cancer Vaccines. J. Clin. Invest. 2007, 117, 1195–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckpot K.; Aerts J. L.; Thielemans K. Lentiviral Vectors for Cancer Immunotherapy: Transforming Infectious Particles into Therapeutics. Gene Ther. 2007, 14, 847–862. [DOI] [PubMed] [Google Scholar]
- Dullaers M.; Van Meirvenne S.; Heirman C.; Straetman L.; Bonehill A.; Aerts J. L.; Thielemans K.; Breckpot K. Induction of Effective Therapeutic Antitumor Immunity by Direct in Vivo Administration of Lentiviral Vectors. Gene Ther. 2006, 13, 630–640. [DOI] [PubMed] [Google Scholar]
- Shiver J. W.; Emini E. A. Recent Advances in the Development of HIV-1 Vaccines Using Replication-Incompetent Adenovirus Vectors. Annu. Rev. Med. 2004, 55, 355–372. [DOI] [PubMed] [Google Scholar]
- O’Hagan D. T.; Valiante N. M. Recent Advances in the Discovery and Delivery of Vaccine Adjuvants. Nat. Rev. Drug Discovery 2003, 2, 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Jain S.; Larman H. B.; Gonzalez S.; Irvine D. J. Directed Cell Migration via Chemoattractants Released from Degradable Microspheres. Biomaterials 2005, 26, 5048–5063. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Winans A. M.; Irvine D. J. Modular Injectable Matrices Based on Alginate Solution/Microsphere Mixtures That Gel in Situ and Co-Deliver Immunomodulatory Factors. Acta Biomater. 2009, 5, 969–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali O. A.; Huebsch N.; Cao L.; Dranoff G.; Mooney D. J. Infection-Mimicking Materials to Program Dendritic Cells In Situ. Nat. Mater. 2009, 8, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto T.; Huang E. K.; Paek H. J.; Morita A.; Matsue H.; Valentini R. F.; Takashima A. Induction of Tumor-Specific Protective Immunity by In Situ Langerhans Cell Vaccine. Nat. Biotechnol. 2002, 20, 64–69. [DOI] [PubMed] [Google Scholar]
- Singh A.; Suri S.; Roy K. In Situ Crosslinking Hydrogels for Combinatorial Delivery of Chemokines and siRNA-DNA Carrying Microparticles to Dendritic Cells. Biomaterials 2009, 30, 5187–5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Fox V.; Lei Y.; Hu B.; Joo K. I.; Wang P. Synthetic Niches for Differentiation of Human Embryonic Stem Cells Bypassing Embryoid Body Formation. J. Biomed. Mater. Res., Part B: Appl. Biomater. 2014, 102, 1101–1112. [DOI] [PubMed] [Google Scholar]
- Nagahama K.; Hashizume M.; Yamamoto H.; Ouchi T.; Ohya Y. Hydrophobically Modified Biodegradable Poly(ethylene glycol) Copolymers that Form Temperature-Responsive Nanogels. Langmuir 2009, 25, 9734–9740. [DOI] [PubMed] [Google Scholar]
- Peng K.-T.; Chen C.-F.; Chu I.-M.; Li Y.-M.; Hsu W.-H.; Hsu R. W.-W.; Chang P.-J. Treatment of Osteomyelitis with Teicoplanin-Encapsulated Biodegradable Thermosensitive Hydrogel Nanoparticles. Biomaterials 2010, 31, 5227–5236. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Liu C. H.; Roberts A. I.; Das J.; Xu G.; Ren G.; Zhang Y.; Zhang L.; Yuan Z. R.; Tan H. S.; Das G.; Devadas S. Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) and T-Cell Responses: What We Do and Don’t Know. Cell Res. 2006, 16, 126–133. [DOI] [PubMed] [Google Scholar]
- Warren T. L.; Weiner G. J. Uses of Granulocyte-Macrophage Colony-Stimulating Factor in Vaccine Development. Curr. Opin. Hematol. 2000, 7, 168–173. [DOI] [PubMed] [Google Scholar]
- Yang L.; Yang H.; Rideout K.; Cho T.; Joo K. i.; Ziegler L.; Elliot A.; Walls A.; Yu D.; Baltimore D.; Wang P. Engineered Lentivector Targeting of Dendritic Cells for in Vivo Immunization. Nat. Biotechnol. 2008, 26, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerpohl H. G.; Lohmann-Matthes M. L.; Fischer H. Studies on the Activation of Mouse Bone Marrow-Derived Macrophages by the Macrophage Cytotoxicity Factor (MCF). Eur. J. Immunol. 1976, 6, 213–217. [DOI] [PubMed] [Google Scholar]
- Rowe H. M.; Lopes L.; Ikeda Y.; Bailey R.; Barde I.; Zenke M.; Chain B. M.; Collins M. K. Immunization with a Lentiviral Vector Stimulates Both CD4 and CD8 T Cell Responses to an Ovalbumin Transgene. Mol. Ther. 2006, 13, 310–319. [DOI] [PubMed] [Google Scholar]
- Lois C.; Hong E. J.; Pease S.; Brown E. J.; Baltimore D. Germline Transmission and Tissue-Specific Expression of Transgenes Delivered by Lentiviral Vectors. Science 2002, 295, 868–872. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Joo K.-I.; Wang P. Endocytic Processing of Adeno-Associated Virus Type 8 Vectors for Transduction of Target Cells. Gene Ther. 2013, 20, 308–317. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Kim Y. J.; Ji M.; Fang J.; Siriwon N.; Zhang L. I.; Wang P. Enhancing Gene Delivery of Adeno-Associated Viruses by Cell-Permeable Peptides. Mol. Ther. Methods Clin. Dev. 2014, 1, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Tai A.; Joo K.-I.; Wang P. Visualization of DC-SIGN-Mediated Entry Pathway of Engineered Lentiviral Vectors in Target Cells. PLoS One 2013, 8, e67400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y. J.; James E.; Shastri N.; Fréchet J. M. In Vivo Targeting of Dendritic Cells for Activation of Cellular Immunity Using Vaccine Carriers Based on pH-Responsive Microparticles. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 18264–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nchinda G.; Kuroiwa J.; Oks M.; Trumpfheller C.; Park C. G.; Huang Y.; Hannaman D.; Schlesinger S. J.; Mizenina O.; Nussenzweig M. C.; Uberla K.; Steinman R. M. The Efficacy of DNA Vaccination is Enhanced in Mice by Targeting the Encoded Protein to Dendritic Cells. J. Clin. Invest. 2008, 118, 1427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou H. Y.; Lin X. Z.; Pan W. Y.; Wu P. Y.; Chang C. M.; Lin T. Y.; Shen H. H.; Tao M. H. Hydrogel-Delivered GM-CSF Overcomes Nonresponsiveness to Hepatitis B Vaccine through the Recruitment and Activation of Dendritic Cells. J. Immunol. 2010, 185, 5468–75. [DOI] [PubMed] [Google Scholar]
- Mach N.; Dranoff G. Cytokine-Secreting Tumor Cell Vaccines. Curr. Opin. Immunol. 2000, 12, 571–575. [DOI] [PubMed] [Google Scholar]
- Salgia R.; Lynch T.; Skarin A.; Lucca J.; Lynch C.; Jung K.; Hodi F. S.; Jaklitsch M.; Mentzer S.; Swanson S.; Lukanich J.; Bueno R.; Wain J.; Mathisen D.; Wright C.; Fidias P.; Donahue D.; Clift S.; Hardy S.; Neuberg D.; Mulligan R.; Webb I.; Sugarbaker D.; Mihm M.; Dranoff G. Vaccination with Irradiated Autologous Tumor Cells Engineered to Secrete Granulocyte-Macrophage Colony-Stimulating Factor Augments Antitumor Immunity in Some Patients with Metastatic Non-Small-Cell Lung Carcinoma. J. Clin. Oncol. 1993, 21, 624–630. [DOI] [PubMed] [Google Scholar]
- Burke B.; Pridmore A.; Harraghy N.; Collick A.; Brown J.; Mitchell T. Transgenic Mice Showing Inflammation-Inducible Overexpression of Granulocyte Macrophage Colony-Stimulating Factor. Clin. Vaccine Immunol. 2004, 11, 588–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen C. L.; Bui J. T.; Demcheva M.; Vournakis J. N.; Cole D. J.; Gillanders W. E. Sustained Release of Granulocyte-Macrophage Colony-Stimulating Factor from a Modular Peptide-Based Cancer Vaccine Alters Vaccine Microenvironment and Enhances the Antigen-Specific T-Cell Response. J. Immunother. 2001, 24, 420–429. [PubMed] [Google Scholar]