

Abstract
Genetic analysis indicates that Escherichia coli possesses two independent pathways for oxidation of phosphite (Pt) to phosphate. One pathway depends on the 14-gene phn operon, which encodes the enzyme C-P lyase. The other pathway depends on the phoA locus, which encodes bacterial alkaline phosphatase (BAP). Transposon mutagenesis studies strongly suggest that BAP is the only enzyme involved in the phoA-dependent pathway. This conclusion is supported by purification and biochemical characterization of the Pt-oxidizing enzyme, which was proven to be BAP by N terminus protein sequencing. Highly purified BAP catalyzed Pt oxidation with specific activities of 62–242 milliunits/mg and phosphate ester hydrolysis with specific activities of 41–61 units/mg. Surprisingly, BAP catalyzes the oxidation of Pt to phosphate and molecular H2. Thus, BAP is a unique Pt-dependent, H2-evolving hydrogenase. This reaction is unprecedented in both P and H biochemistry, and it is likely to involve direct transfer of hydride from the substrate to water-derived protons.
Unlike the other major elements in living organisms, P is commonly considered to be a redox conservative element. Accordingly, the P centers found in the vast majority of biological intermediates, including inorganic phosphate (Pi), organic phosphate esters, and phosphoanhydrides, are fully oxidized (+5 valence state). Nevertheless, it is now clear, although not widely appreciated, that many organisms are capable of metabolizing reduced P compounds, indicating that P redox reactions are biochemically possible. A wide array of prokaryotic (and some eukaryotic) organisms can synthesize or degrade reduced P compounds (1, 2). Moreover, the widespread occurrence of this trait strongly suggests that a selective advantage is conferred on organisms with the ability to metabolize reduced P compounds. Examination of reduced P metabolism has revealed a wealth of unusual biology and biochemistry. The bacterium Desulfotignum phosphitoxidans gains energy for growth by oxidation of phosphite (Pt) coupled to either sulfate reduction or acetogenesis while fixing CO2 as its sole C source (3, 4). Many other organisms can use reduced P compounds as sole P sources (5–8). For example, Pseudomonas stutzeri is capable of oxidizing hypophosphite (P valence, +1) to phosphate by means of a Pt (P valence, +3) intermediate (9). The following two enzymes catalyze these reactions: 2-oxoglutarate/hypophosphite dioxygenase catalyzes the oxidation of hypo-Pt to Pt (10), and Pt/NAD oxidoreductase catalyzes the oxidation of Pt to phosphate (11). The latter reaction is the most thermodynamically favorable reduction of NAD that is known, and this trait makes this enzyme particularly useful as a cofactor-regenerating catalyst for enzyme-based synthetic strategies (12). P biochemistry can be found also in more familiar organisms, such as Escherichia coli. As shown below, E. coli has two pathways for the oxidation of Pt. Our examination of these pathways demonstrates that thoroughly characterized biochemistry still contains surprises. Here, we demonstrate that E. coli alkaline phosphatase, which is thought to be the subject of more citations than any other enzyme, catalyzes the oxidation of Pt to phosphate and molecular H2, an enzymatic reaction that is unprecedented in both H and P metabolism.
Materials and Methods
Bacterial Strains and Growth Conditions. Bacterial strains used in this study are described in the text, and detailed genotypes are shown in Table 1, which is published as supporting information on the PNAS web site. Media used in the study are reported in refs. 9 and 13. Antibiotics were used at the following concentrations: 50 μg/ml kanamycin, 100 μg/ml ampicillin, 100 μg/ml streptomycin (for rpsL strains), 100 μg/ml streptomycin/100 μg/ml spectinomycin (for aadA-encoding plasmids) (14), and 12.5 μg/ml or 6 μg/ml chloramphenicol for plasmid cloning or for single-copy integrants of conditional replication, integration, and modular (CRIM) plasmids (15), respectively. The Pt-oxidation phenotype was scored by growth on 0.4% glucose/Mops medium with 0.5 mM Pt as the sole P source.
Isolation and Characterization of E. coli Mutants with Defects in Pt Oxidation. BW14894 was mutagenized by using the EZ::TN Transposon Insertion System (Epicentre Technologies, Madison, WI), as recommended by the manufacturer, or by using λ::Tn5seq1, as described in ref. 16. Mutants that showed no growth or decreased growth on glucose/Mops/Pt medium but grew well on glucose/Mops/Pi medium were isolated, and linkage between the transposon insertion and the Pt– phenotype was confirmed after crossing the transposon insertion into WM1584 by P1 transduction as described in ref. 17. Each transposon insertion along with flanking chromosomal DNA was cloned by selection for the transposon-encoded antibiotic resistance marker by using the in vivo miniMu method with pMW11, as described in refs. 14 and 18. The transposon-insertion sites were determined by DNA sequence analysis of both end junctions using primers appropriate for the particular transposon used. The following sequencing primers were used for λ::Tn5seq1: 5′-CATACGATTTAGGTGACACTATAG-3′ and 5′-TAATACGACTCACTATAGGG-3′. Standard FP1 and RP1 primers (Epicentre Technologies) were used for the EZ::TN Transposon systems.
DNA Methods. Standard methods were used throughout for isolation and manipulation of plasmid DNA (19). DNA sequencing was carried out by the W. M. Keck Center for Comparative and Functional Genomics (University of Illinois at Urbana–Champaign). The results were compared by using blast (20) with the sequences available in the GenBank database to identify the mutated genes (January 15, 2004).
Activity Assays. Phosphatase activity was assayed by using the chromogenic substrate p-nitrophenylphosphate (pNPP). Phosphatase activity was determined at 37°C in 50 mM N-2-hydroxyethylpiperazine-N′-3-propanesulfonic acid (EPPS) buffer (pH 8.0) with 0.12 mM pNPP. Reactions were started by addition of pNPP. For the comparison of mammalian phosphatases and bacterial phosphatase activities, assays were performed at room temperature in 1 M Tris·Cl, pH 8.0/0.12 mM pNPP. Reactions were monitored by continuously following production of p-nitrophenol (ε = 15,460 M–1·cm–1 at 410 nm). Qualitative assays were used to follow some protein purifications. These were performed in 96-well microplates by mixing 10 μl of each fraction (or an appropriate dilution) with 5 μl of 0.4% pNPP in 50 mM Tris·Cl (pH 8.0). Cell extracts used for determination of bacterial alkaline phosphatase (BAP) activity in transposon-induced mutants were prepared as follows. Cells were grown to saturation in 5 ml of glucose/Mops medium with 100 μM phosphate (to induce BAP synthesis), harvested by centrifugation, washed twice, and then resuspended in 1 ml of Tris·Cl buffer (50 mM, pH 8.0). Cells were lysed by sonication, and debris was removed by centrifugation at 14,000 × g for 10 min at 4°C.
Pt oxidation was assayed by measuring Pt-dependent phosphate production by using a sensitive (nanomole detection level) malachite green dye-binding assay (21), as modified in ref. 11, with the additional modification of omitting the addition of sodium citrate. Activity was determined by assaying the level of phosphate present at sequential time points over a 30-min reaction in 50 mM Mops buffer (pH 7.0) with 10 mM sodium Pt (pH 7.0) at 37°C. Pt oxidation activity was assessed qualitatively during protein purifications by mixing 10 μl from each fraction (or an appropriate dilution) with 5 μl of 0.5 M Pt (pH 7.0) and incubating overnight at 37°C before adding 240 μl of the malachite green reagent. A green color indicates the production of phosphate from the reactions.
H2O2 was detected with Amplex Red and horseradish peroxidase, as described in ref. 22. O2 concentrations were measured as recommended by the manufacturer by using an O2-measurement controller Digital Model 10 (Rank Brothers, Cambridge, U.K.).
Purification of BAP. BAP was purified from BW14894 grown in glucose/Mops medium with 500 μM Pt. Subsequent steps were conducted at 4°C. Cells were harvested by centrifugation, and periplasmic proteins were released by osmotic shock as described in ref. 23, concentrated to ≈1 mg/ml, and brought to 60% saturation by using a saturated ammonia sulfate solution. Precipitated proteins were removed by centrifugation at 20,000 × g for 30 min. The supernatant was concentrated by ultrafiltration and loaded onto a 10 × 100-mm POROS 20-μm HP2 hydrophobic-interaction column (PerSeptive Biosystems, Framingham, MA) equilibrated with buffer A [50 mM Tris·Cl/2.5 M (NH4)2SO4/10% glycerol, pH 8.0]. Proteins were eluted by using a linear gradient (0–100%) with buffer B (50 mM Tris·Cl/10% glycerol, pH 8.0) at a flow rate of 1 ml/min over 50 column volumes. Fractions with the highest phosphatase activity were pooled, concentrated, and desalted by dialysis overnight by using a 30-kDa cut-off Slide-A-Lyzer Cassette (Pierce) against 500 ml of buffer B with two buffer changes. The desalted protein was then loaded to a 10 × 100-mm POROS HQ anion-exchange column (PerSeptive Biosystems) equilibrated with buffer B and subsequently eluted with a linear gradient of 0–0.5 M NaCl in buffer B over 50 column volumes at a flow rate of 1 ml/min. The purest fractions (as judged by SDS/PAGE analysis) were pooled. An ultrafiltration cell (Amicon) equipped with a membrane of 30-kDa molecular cut-off was used to concentrate proteins throughout the purification.
Site-Specific Mutagenesis. The phoA gene, with its native promoter, was amplified by PCR (35 cycles of 95°C for 4 min, 94°C for 1 min, 50°C for 1 min, and 72°C for 1 min) from genomic DNA of BW14894. The primers were designed to introduce a SpeI site upstream (5′-GGCGGCGCACTAGTTTAATCTTTTCAACAGCTGTC) and an XbaI site downstream (5′-GGCGGCGCTCTAGAAAATTCACTGCCGGGCGCGGT-3′) of phoA. The resulting PCR product was digested with SpeI and XbaI; cloned into the same sites of the conditional replication, integration, and modular (CRIM) plasmid pCAH63 (15) to create pKY1; and then integrated in single copy into the chromosome of BW14893, as described in ref. 15. The Ser-102–Ala substitution mutation was introduced into phoA gene with the QuikChange Site-Directed Mutagenesis Kit (Stratagene) as recommended by the manufacturer by using pKY1 as the template to create pKY2. The following synthetic oligonucleotides were used: 5′-GACTACGTCACCGACGCCGCTGCATC AGCAACC-3′ and 5′-GGTTGCTGATGCAGCGGCGTCGGTGACGTAGTC-3′. The cloned inserts of pKY1 and pKY2 were verified by DNA sequencing.
Construction of hyp Mutants. A ΔhypABCDE::aph mutant was constructed by homologous recombination, which was facilitated by the phage λ recombinase system, as described in ref. 24, by using the following mutagenic primers: 5′-ATGCACGAAATAACCCTCTGCCAACGGGCACTGGAAGTGTAGGCTGGAGCTGCTTCG-3′ and 5′-TTAGCATATACGCGGAAGCGGTTCGGCGTGTGGTAATTCCGGGGATCCGTCGACCTG-3′. The following sequencing primers were used to verify the mutation: 5′-TCGGCTTTCTCGCTTATTTC-3′, 5′-AAGCGGTGCAAAAGGTCAT-3′, K1, K2, and Kt (24).
31P Nuclear Magnetic Resonance for Phosphate Production. Proton-coupled and decoupled spectra were acquired from a Unity 500 (Varian) equipped with a 5-mm QUAD probe (Nalorac Cryogenics, Martinez, CA) at the Varian Oxford Instruments Center for Excellence in the Nuclear Magnetic Resonance Laboratory at the University of Illinois at Urbana–Champaign. We conducted 200 acquisitions for each sample with an acquisition time of 0.655 s and pulse width of 4.4 μs. D2O was added to all samples at 30% final concentration, and an external reference of 85% phosphoric acid set at 0 ppm was used.
Gas Chromatography Analysis for H2 Production. To assay H2 production in vitro, 50 mM sodium Pt (pH 7.0) was incubated overnight in 3 ml of Mops buffer (50 mM, pH 7.0) with 0.35 mg of BAP. The reactions, together with no enzyme controls and no substrate controls, were set up in triplicate in sealed vials. For in vivo H production, WM3924 was grown to saturation in 5 ml of glucose/Mops medium with either 500 μM Pt or 500 μM Pi in sealed culture tubes. The gas composition in the headspace was analyzed by Isotech Laboratories (Champaign, IL) by using thermal-conductivity detection after separation over custom-designed AGC400 and AGC100 gas chromatographs (Chandler Engineering, Tulsa, OK).
Other Methods. Protein concentrations were determined with Coomassie Plus reagent (Pierce), according to the manufacturer's protocols, with BSA (2 mg/ml) as the standard. SDS/PAGE was carried out as described by Laemmli (25) in 12.5 polyacrylamide slab gels. Proteins were visualized by staining with Commassie blue or Silver Stain Plus (Bio-Rad)
Results and Discussion
Two Pathways for Pt Oxidation in E. coli. Genetic studies have shown that the enzyme C-P lyase, encoded by the phn operon, is capable of oxidizing Pt (26). However, in a recent study (9), we were surprised to observe that some E. coli phn mutants remained capable of oxidizing Pt. Examination of the genotype of these strains suggested that the phoA locus might be involved in Pt oxidation. To test this hypothesis directly, we examined E. coli strains that differed solely in the phn and phoA loci for their ability to oxidize Pt, as demonstrated by their ability to grow on media with Pt as the sole P source. Because phosphate is required for growth, organisms cannot grow on this medium unless they have the capacity to oxidize Pt to phosphate. Strains that were either phoA+ or phn+ grew on Pt medium (regardless of whether the other locus was mutated), whereas strains with mutations in both phn and phoA did not (Fig. 1). Therefore, E. coli has two pathways for the oxidation of Pt: one that is phn-dependent and one that is phoA-dependent.
Fig. 1.

Two pathways for Pt oxidation in E. coli. Growth on media containing Pt as the sole P source requires oxidation of Pt to phosphate. Strains differing only in the phn and phoA loci, which encode C-P lyase and BAP, respectively, were streaked on 0.4% glucose/Mops medium with either 0.5 mM phosphate or 0.5 mM Pt as sole P sources. All strains grew on phosphate medium (positive control). Strains that are phoA+ and/or phn+ are capable of growth on Pt medium, indicating that they can oxidize Pt; however, strains deleted for both phn and phoA cannot grow on Pt medium, indicating that they are incapable of Pt oxidation. Thus, two independent pathways for Pt oxidation exist in E. coli: one depends on phoA and the other depends on phn. Because the amount of P required for growth is relatively small, the contaminating levels of phosphate found in many media components allow a slight background level of growth of all strains on these media. To control for this variable, the strains in question were always compared with suitable positive and negative controls on the same plate. The following strains were used: BW13711 [ΔlacX74, phn(EcoB)], BW14894 (Δlac X74, Δphn 33–30), BW14322 [Δlac X74, ΔphoA532, phn(EcoB)], and BW14893 (Δlac X74, ΔphoA532, Δphn 33–30). The phn(EcoB) allele is phenotypically Phn+ (13).
Isolation and Analysis of Mutants Defective for phoA-Dependent Pt Oxidation. The E. coli phoA gene encodes the secreted, periplasmic enzyme BAP. BAP is an exhaustively studied, nonspecific phosphomonoesterase that allows E. coli to use phosphate esters as alternative sources of P (27, 28). Consistent with the known enzymatic function of BAP, our original hypothesis regarding the phoA-dependent pathway was that Pt would be oxidized to a phosphate ester by as-yet-unknown enzymes and then hydrolyzed by BAP to release the free phosphate needed for growth. In an attempt to identify the genes encoding these putative unknown enzymes, we examined ≈30,000 transposon-induced mutants of the Δphn strain (a number sufficient to saturate all nonessential genes on the chromosome) for their ability to use Pt as the sole P source. We isolated 22 mutants that grew poorly, or not at all, on Pt medium. The transposon-insertion sites in each mutant were cloned and sequenced to identify the genes responsible for the Pt phenotype. The loci identified included phoA (seven isolates), the phoBR operon (eight isolates), dsbA, cpxA, lpp (two isolates), ygiT, ygjM, and yhjA.
Examination of the known functions for these genes led to the unexpected conclusion that BAP is the only enzyme involved in Pt oxidation via the phoA-dependent pathway. Accordingly, both phoA and phoB mutants are phenotypically BAP–. [PhoB is a transcriptional activator required for phoA expression (28).] PhoR mutants constitutively express phoA but at a much lower level than is seen in the wild type under phosphate-starvation conditions (28). DsbA catalyzes the formation of disulfide bonds in periplasmic proteins and is required for synthesis of fully active BAP (which contains four essential disulfide bonds per dimer), whereas CpxA is required for normal regulation of dsbA transcription (29, 30). Lpp mutants are known to leak periplasmic proteins, including BAP, into the surrounding medium and, thus, have lower levels of periplasmic proteins than wild-type strains (31). Although functions have yet to be proposed for the other three genes, it seemed to be possible that lower BAP activity might be responsible for their Pt phenotype as well. To test this possibility, BAP activity was assayed in representative mutants of each gene identified in the genetic screen. Each of the mutations, including those in ygiT, ygjM, and yhjA, resulted in lower levels of BAP activity, ranging from 6–76% of the wild-type levels. Moreover, the levels of BAP activity roughly correlate to the levels of growth observed in Pt medium: the lower the BAP activity, the slower the growth rate in Pt medium (see Table 2, which is published as supporting information on the PNAS web site).
Pt Oxidation Is Catalyzed by BAP. Because genetic approaches involving transposon-induced mutants cannot identify essential genes, we also used a biochemical strategy to identify the protein(s) required for oxidation of Pt to phosphate. By using a sensitive malachite green dye-binding assay (21), we were able to detect Pt-dependent phosphate production (i.e., Pt oxidation) in extracts of Pt-grown Δphn E. coli strains. This assay was used to follow a four-step purification of the Pt-oxidizing enzyme. In each step, only a single fraction with Pt-oxidizing activity was identified, which, after the fourth step, was comprised of a single protein, estimated to be ≈99% pure by visual inspection of stained denaturing polyacrylamide gels. N terminus protein sequencing identified this protein as the product of the phoA gene, namely BAP. Under optimal conditions, highly purified BAP from five independent preparations catalyzed Pt oxidation with specific activities ranging from 62 to 242 milliunits/mg (measured as phosphate production by means of the malachite green assay) and phosphate ester hydrolysis with specific activities of 41–61 units/mg (measured with the chromogenic substrate p-nitrophenyl-phosphate). Interestingly, the pH optimum for the Pt oxidation reaction is substantially lower (pH 7) than it is for phosphate ester hydrolysis (pH > 8.5) (ref. 27 and K.Y. and W.W.M, unpublished data).
Pi and Molecular H2 Are the Products of BAP-Catalyzed Pt Oxidation. Oxidation of Pt to phosphate releases two electrons; however, exogenous electron acceptors were not included in the assays described above. Moreover, addition of standard electron acceptors, including FAD, FMN, NAD, NADP, and various redox active dyes, failed to stimulate Pt oxidation in cell extracts (data not shown). A plausible explanation for this result was that Pt was not oxidized to phosphate during the reaction and that some other reduced P product was being detected by the dye-binding assay. This possibility was excluded by using 31P nuclear magnetic resonance spectroscopy, which confirmed the formation of Pi in the reaction, Fig. 2A. Subsequently, we considered the possibility that molecular O2 was the missing electron acceptor according to Eq. 1.
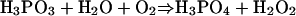 |
[1] |
Despite the production of significant levels of phosphate in our assays, however, we were unable to detect either the consumption of O2 (by means of sensitive O2 electrode measurements) or the production of H2O2 (by means of a sensitive horseradish peroxidase assay). Moreover, Pt oxidation was observed at similar rates under strictly anaerobic conditions in assays that included only BAP, Pt, and water. These data strongly suggested that protons derived from water are the missing electron acceptor for the Pt oxidation reaction and that the other product of the reaction was likely to be molecular H2, according to Eq. 2.
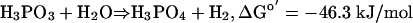 |
[2] |
In support of this hypothesis, we detected Pt-dependent H2 production both in vitro and in vivo. In vitro assays were conducted aerobically in sealed vials by using highly purified BAP. After incubation, the headspace was analyzed for H2, and the aqueous portion was assayed for phosphate. As shown in Fig. 2B, stoichiometric amounts of phosphate and H2 were produced from Pt in the presence of BAP, whereas neither phosphate nor H2 was produced in control reactions containing either BAP or Pt alone.
Fig. 2.

The products of the BAP-catalyzed Pt oxidation are phosphate and molecular H2.(A) The proton-decoupled 31P nuclear magnetic resonance spectra and peak positions are indicated for 5 mM Pt and 5 mM phosphate controls, as well as for an overnight reaction that initially contained 5 mM Pt plus 500 μg of purified BAP. A peak for unreacted Pt and a new phosphate peak are evident in the spectrum of the enzymatic reaction, but no other products were produced. The slight shift in the position of the phosphate peak between the control and the BAP-catalyzed reaction product is due to minor pH differences: addition of phosphate stock solution to the assay increased the height of the Pi peak but did not produce extra peaks. Proton-coupled spectra are completely consistent with this interpretation (data not shown). (B) BAP-catalyzed Pt oxidation produces stoichiometric amounts of phosphate and molecular H2. In vitro reactions containing the indicated components were incubated overnight at 37°C in sealed vials. The headspace atmosphere was then assayed for H2 by gas chromatography with thermal conductivity detection, whereas the aqueous phase was assayed for phosphate by using the malachite green assay. Reactions (3 ml) were conducted in 50 mM Mops (pH 7.0) with 50 mM Pt (pH 7.0) and 387 μg of purified BAP, as indicated. Triplicate controls containing either BAP or Pt alone were performed, but neither phosphate nor H2 was detected under these conditions. In vivo results show the amount of H2 produced during growth of WM3924 (Δlac X74, Δphn 33–30, ΔhypABCDE) in 0.4% glucose/Mops broth containing 2.5 μmol of the indicated P source. Because this level of P (500 μM) is growth limiting, the P source is expected to be completely assimilated when the cultures reach saturation. After overnight growth at 37°C in sealed vials, the headspace was assayed for H2 by gas chromatography with thermal conductivity detection. Both the in vitro and in vivo experiments were conducted aerobically (i.e., O2 was present during Pt oxidation).
Measurement of H2 in vivo is complicated by the fact that E. coli has multiple hydrogenases that can both produce and consume H2. To circumvent this problem, we constructed a ΔhypABCDE mutant, which cannot produce any active hydrogenases because of its inability to mature the precursor proteins (32). The hyp mutant, grown aerobically in sealed tubes, also produced roughly stoichiometric amounts of H2 in cultures grown with growth-limiting levels of Pt, whereas no H2 was detected in cultures grown with limiting levels of phosphate or the phosphate ester phosphoserine (Fig. 2B).
Pt Oxidation Is Not a Common Feature of All Alkaline Phosphatases. Efficient Pt oxidation appears to be a unique feature of E. coli alkaline phosphatase. Neither Bacillus subtilis, which is known to produce multiple highly active phosphatases (33), nor P. stutzeri, which is known to produce a phosphatase distinct from BAP (in strains that lack the Pt dehydrogenase system) (A. K. White, S. Neuhaus, M. M. Wilson, and W.W.M., unpublished data), can use Pt as a sole P source (Fig. 3A), although both organisms grow on media with phosphoserine as the sole P source (data not shown). Thus, the phosphatases produced by these organisms are incapable of oxidizing Pt at rates that are sufficient to support growth. We also assayed commercial preparations of calf intestinal phosphatase (CIP) and shrimp alkaline phosphatase (SAP) for their ability to produce phosphate from Pt (Fig. 3B). Although small amounts of phosphate were produced by the eukaryotic phosphatases after overnight incubation, only E. coli BAP was able to catalyze the reaction at significant rates. This finding is particularly surprising because the eukaryotic enzymes are actually much better phosphatases, with specific activities up to 40-fold higher than BAP (27). These enzymes all share significant homology (25–35% identity) with the E. coli BAP; furthermore, most of the active-site residues, including Ser-102 (which forms a covalent phospho-enzyme intermediate during the phosphatase reaction), are conserved (27).
Fig. 3.

Pt oxidation is unique to E. coli alkaline phosphatase. (A) The ability of B. subtilis and P. stutzeri to oxidize Pt, as demonstrated by growth on media with Pt as the sole P source, was tested, as described in Fig. 1. Neither organism grew on the Pt medium, whereas both organisms grew on phosphate medium. Thus, despite the fact that both organisms have active phosphatases, neither can oxidize Pt at rates sufficient to support growth. The P. stutzeri WM3617 (ΔptxA-htxP, Δphn) contains mutations that eliminate the two characterized Pt oxidation pathways of this organism but that do not effect phosphatase expression (9, 10). (B) BAP (E. coli alkaline phosphatase), SAP (shrimp alkaline phosphatase; Roche Applied Science, Mannheim, Germany) and CIP (calf intestinal phosphatase; Sigma) were assayed for Pt oxidation as described above. We used 56 μg of the indicated phosphatase in each assay, which corresponds to 3 BAP, 32 SAP, and 65 CIP phosphatase units, as measured by pNPP hydrolysis. Despite the much higher phosphatase activities of the eukaryotic enzymes, only BAP catalyzed Pt oxidation at significant rates. The average of two trials is plotted.
It seems to be plausible that Pt oxidation is catalyzed by BAP by a mechanism similar to that used for phosphate ester hydrolysis (Fig. 4A). Accordingly, Pt may be hydrolyzed by BAP with hydride anion as the formal leaving group. In support of this idea, a phoA mutant with the active-site Ser-102 residue changed to alanine is unable to use Pt as sole P source, indicating that this amino acid is required for phosphate ester hydrolysis (27) and for Pt oxidation (Fig. 4B). Although direct hydride transfer from a substrate to an aqueous proton is biochemically unprecedented, this reaction is thermodynamically reasonable given the strong reducing potential of the phosphate–Pt couple (Eo′ = –0.650 V). Thus, the H-producing reaction quite favorable: ΔGo′ = –46.3 kJ/mol (calculated from redox potentials in ref. 34). If the hydrolytic model for Pt oxidation proves to be correct, it would be a very unusual enzymatic reaction. Studies have shown that BAP is also able to hydrolyze phosphodiesters (35), phosphoamides (36), sulfate esters (37), and thiophosphate (38). However, these reactions occur at rates that are considerably slower than that of the Pt hydrolysis reaction despite the fact that they involve much better leaving groups. Also, they are not redox reactions. The analogous reaction involving hydrolysis of alkylphosphonic acids is not catalyzed by BAP, as shown both biochemically (39) and by the inability of Δphn strains to use these compounds as sole P sources (13).
Fig. 4.

Pt oxidation by BAP may occur by means of hydrolysis with hydride anion as the leaving group. (A) The chemical mechanism for phosphate ester hydrolysis by BAP involves nucleophilic attack by an activated serine residue (Ser-102) on the phosphate ester to form a phosphoserine enzyme intermediate. The alkoxide leaving group rapidly acquires a proton from solution to form the corresponding alcohol. It seems to be likely that Pt oxidation occurs by means of a similar mechanism with hydride anion as the leaving group. (B) The role of Ser-102 in Pt oxidation was tested by examining whether a mutant carrying a Ser-102–Ala mutation in the phoA gene could grow on Pt media, as described in Fig. 1. The mutant failed to grow on Pt medium, demonstrating the requirement for the active-site Ser-102 in Pt oxidation. The host strain was BW14893 (Δlac X74, ΔphoA532, Δphn 33–30), WM3610 and WM3611 carry single copy integrants of plasmids pKY1 and pKY2, which encode the wild-type phoA gene (phoA+) or phoA-S102A mutant (Ser-102–Ala), respectively.
Pt dehydrogenase (PtxD) had been the only in vitro-characterized enzyme shown to catalyze Pt oxidation (11). Although the details of the BAP reaction remain to be elucidated, the chemical mechanisms of the two enzymes are clearly distinct. Whereas PtxD requires NAD as the electron acceptor for the redox reaction, the reaction catalyzed by BAP requires no exogenous electron acceptors but exploits the large thermodynamic driving force of the reaction to produce a highly reduced product (H2) from water in an essentially irreversible reaction (calculated Keq = 1.1 × 108). All other known H2-producing reactions operate much closer to chemical equilibrium and are typically reversible. Moreover, all other known hydrogenases contain either Fe or Ni, or both, in their active sites (40). This observation includes the so-called “metal-free” H2-forming methylene tetrahydromethanopterin dehydrogenase of methanogenic archaea, which is now known to contain Fe as well (41). In contrast, BAP contains no redox active metals, although both Zn and Mg are required for hydrolytic activity (27). Treatment of BAP with chelators inhibits Pt oxidation, suggesting that metals play a role in the Pt reaction (data not shown).
Finally, the in vivo data presented here suggest that the Pt oxidation reaction is biologically relevant. The observation that many bacteria possess enzymes dedicated to Pt oxidation demonstrates that the trait is under strong selective pressure in microbial populations (4, 5, 9, 42). With this fact in mind, it is interesting to note that although the eukaryotic enzymes are much better phosphatases, they are incapable of Pt oxidation. This observation raises the possibility that E. coli BAP may not have evolved to be the most efficient phosphatase but rather to be a phosphatase with the ability to hydrolyze Pt. This idea is consistent with the curious observation that E. coli BAP is very highly expressed (up to 6% of total cell protein) under phosphate-starvation conditions (28). The traditional explanation for this phenomenon is that some unknown BAP substrate must be poorly hydrolyzed and, therefore, require massive amounts of enzyme to support competitive growth rates. However, because there is little difference in the measured rates of hydrolysis for a wide variety of phosphate ester substrates (39, 43), it seems to be unlikely that this poor substrate could be a phosphate ester. In contrast, the rate of Pt hydrolysis is substantially lower than the rate of phosphate ester hydrolysis, suggesting that Pt may be the substrate that accounts for the extreme level of phoA expression observed in phosphate-starved E. coli.
Clearly, many details of the BAP reaction with Pt remain to be elucidated. Further study of this unique reaction is likely to contribute not only to our understanding of P redox chemistry and phosphoryl-transfer reactions but also to our knowledge of hydride transfer, H2 producing reactions, and the role of reduced P compounds in nature.
Supplementary Material
Acknowledgments
We thank Ralph S. Wolfe, Thomas Rauchfuss, Barry L. Wanner, and Wilfred van der Donk for critical reading of the manuscript. This work was supported by National Institute of General Medical Sciences Grant GM59334.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: BAP, bacterial alkaline phosphatase; Pi, inorganic phosphate; Pt, phosphite; pNPP, p-nitrophenylphosphate.
References
- 1.Ternan, N. G., McGrath, J. W., McMullan, G. & Quinn, J. P. (1998) World J. Microbiol. Biotech. 14, 635–647. [Google Scholar]
- 2.Seto, H. a. K., Tomohisa. (1999) Nat. Prod. Rep. 16, 589–596. [DOI] [PubMed] [Google Scholar]
- 3.Schink, B. & Friedrich, M. (2000) Nature 406, 37. [DOI] [PubMed] [Google Scholar]
- 4.Schink, B., Thiemann, V., Laue, H. & Friedrich, M. W. (2002) Arch. Microbiol. 177, 381–391. [DOI] [PubMed] [Google Scholar]
- 5.Foster, T. L., Winans, L., Jr., & Helms, S. J. (1978) Appl. Environ. Microbiol. 35, 937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Casida, L. E., Jr. (1960) J. Bacteriol. 80, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinen, W. & Lauwers, A. M. (1974) Arch. Microbiol. 95, 267–274. [Google Scholar]
- 8.Malacinski, G. & Konetzka, W. A. (1966) J. Bacteriol. 91, 578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metcalf, W. W. & Wolfe, R. S. (1998) J. Bacteriol. 180, 5547–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White, A. K. & Metcalf, W. W. (2002) J. Biol. Chem. 277, 38262–38271. [DOI] [PubMed] [Google Scholar]
- 11.Costas, A. M., White, A. K. & Metcalf, W. W. (2001) J. Biol. Chem. 276, 17429–17436. [DOI] [PubMed] [Google Scholar]
- 12.Vrtis, J. M., White, A. K., Metcalf, W. W. & van der Donk, W. A. (2002) Angew. Chem. Int. Ed. 41, 3257–3259. [DOI] [PubMed] [Google Scholar]
- 13.Wanner, B. L. & Boline, J. A. (1990) J. Bacteriol. 172, 1186–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groisman, E. A., Castilho, B. A. & Casadaban, M. J. (1984) Proc. Natl. Acad. Sci. USA 81, 1480–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haldimann, A. & Wanner, B. L. (2001) J. Bacteriol. 183, 6384–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nag, D. K., Huang, H. V. & Berg, D. E. (1988) Gene 64, 135–145. [DOI] [PubMed] [Google Scholar]
- 17.Wanner, B. L. (1986) J. Mol. Biol. 191, 39–58. [DOI] [PubMed] [Google Scholar]
- 18.Metcalf, W. W., Steed, P. M. & Wanner, B. L. (1990) J. Bacteriol. 172, 3191–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ausebel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A. & Struhl, K. (1992) Current Protocols in Molecular Biology (Wiley, New York).
- 20.Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990) J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 21.Lanzetta, P. A., Alvarez, L. J., Reinach, P. S. & Candia, O. A. (1979) Anal. Biochem. 100, 95–97. [DOI] [PubMed] [Google Scholar]
- 22.Seaver, L. C. & Imlay, J. A. (2001) J. Bacteriol. 183, 7173–7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nossal, N. G. & Heppel, L. A. (1966) J. Biol. Chem. 241, 3055–3062. [PubMed] [Google Scholar]
- 24.Datsenko, K. A. & Wanner, B. L. (2000) Proc. Natl. Acad. Sci. USA 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laemmli, U. K. (1970) Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 26.Metcalf, W. W. & Wanner, B. L. (1991) J. Bacteriol. 173, 587–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coleman, J. E. (1992) Annu. Rev. Biophys. Biomol. Struct. 21, 441–483. [DOI] [PubMed] [Google Scholar]
- 28.Wanner, B. L. (1996) Escherichia coli and Salmonella: Cellular and Molecular Biology (Am. Soc. Microbiol., Washington, DC).
- 29.Bardwell, J. C., McGovern, K. & Beckwith, J. (1991) Cell 67, 581–589. [DOI] [PubMed] [Google Scholar]
- 30.Pogliano, J., Lynch, A. S., Belin, D., Lin, E. C. & Beckwith, J. (1997) Genes Dev. 11, 1169–1182. [DOI] [PubMed] [Google Scholar]
- 31.Yem, D. W. & Wu, H. C. (1978) J. Bacteriol. 133, 1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacobi, A., Rossmann, R. & Bock, A. (1992) Arch. Microbiol. 158, 444–451. [DOI] [PubMed] [Google Scholar]
- 33.Hulett, F. M., Kim, E. E., Bookstein, C., Kapp, N. V., Edwards, C. W. & Wyckoff, H. W. (1991) J. Biol. Chem. 266, 1077–1084. [PubMed] [Google Scholar]
- 34.Bard, A. J., Parsons, R. & Joran, J. (1985) Standard Potentials in Aqueous Solutions (Dekker, New York).
- 35.O'Brien, P. J. & Herschlag, D. (2001) Biochemistry 40, 5691–5699. [DOI] [PubMed] [Google Scholar]
- 36.Snyder, S. L. & Wilson, I. B. (1972) Biochemistry 11, 3220–3223. [DOI] [PubMed] [Google Scholar]
- 37.Neumann, H. (1968) J. Biol. Chem. 243, 4671–4676. [PubMed] [Google Scholar]
- 38.Chlebowski, J. F. & Coleman, J. E. (1974) J. Biol. Chem. 249, 7192–7202. [PubMed] [Google Scholar]
- 39.Reid, T. W. & Wilson, I. B. (1971) Enzymes 4, 373–415. [Google Scholar]
- 40.Albracht, S. P. (1994) Biochim. Biophys. Acta 1188, 167–204. [DOI] [PubMed] [Google Scholar]
- 41.Lyon, E. J., Shima, S., Buurman, G., Chowdhuri, S., Batschauer, A., Steinbach, K. K. & Thauer, R. (2004) Eur. J. Biochem. 271, 195–204. [DOI] [PubMed] [Google Scholar]
- 42.Adams, F. & Conrad, J. P. (1953) Soil Sci. 75, 361–371. [Google Scholar]
- 43.Heppel, L. A., Harkness D. R. & Hilmoe R. J. (1962) J. Biol. Chem. 237, 841–846. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.