

Abstract
Interactions between α-helices within the hydrophobic environment of lipid bilayers are integral to the folding and function of transmembrane proteins; however, the major forces that mediate these interactions remain debated, and our ability to predict these interactions is still largely untested. We recently demonstrated that the frequent transmembrane association motif GASright, the GxxxG-containing fold of the glycophorin A dimer, is optimal for the formation of extended networks of Cα–H hydrogen bonds, supporting the hypothesis that these bonds are major contributors to association. We also found that optimization of Cα–H hydrogen bonding and interhelical packing is sufficient to computationally predict the structure of known GASright dimers at near atomic level. Here, we demonstrate that this computational method can be used to characterize the structure of a protein not previously known to dimerize, by predicting and validating the transmembrane dimer of ADCK3, a mitochondrial kinase. ADCK3 is involved in the biosynthesis of the redox active lipid, ubiquinone, and human ADCK3 mutations cause a cerebellar ataxia associated with ubiquinone deficiency, but the biochemical functions of ADCK3 remain largely undefined. Our experimental analyses show that the transmembrane helix of ADCK3 oligomerizes, with an interface based on an extended Gly-zipper motif, as predicted by our models. The data provide strong evidence for the hypothesis that optimization of Cα–H hydrogen bonding is an important factor in the association of transmembrane helices. This work also provides a structural foundation for investigating the role of transmembrane association in regulating the biological activity of ADCK3.
Introduction
A fundamental event in the folding and oligomerization of membrane proteins is the association of the transmembrane (TM) helices.1,2 After the TM helices have been inserted in the membrane, helix–helix association is required to achieve the final fold and oligomeric state of the protein. A favorite system for investigating the rules that govern TM helix association are the single-span membrane proteins,3−7 primarily because a variety of methods are available for measuring their oligomerization (including FRET,8−11 sedimentation equilibrium analytical ultracentrifugation,12,13 in vivo assays in biological membranes,14−16 SDS-PAGE,17,18 and steric trapping19,20). Conversely, assessing the folding energetics of multispan membrane proteins still represents a tremendous challenge.21,22
In addition to being a tractable system, the single-span membrane proteins attract interest because of their biological importance. These proteins comprise the most numerous class of membrane proteins, constituting about half of the total.23−25 Rather than acting as mere membrane anchors for soluble domains, as it was once assumed, the oligomerization of single TM domains actively plays roles in assembly, signal transduction, ion conduction and regulation in a wide variety of biological processes.7
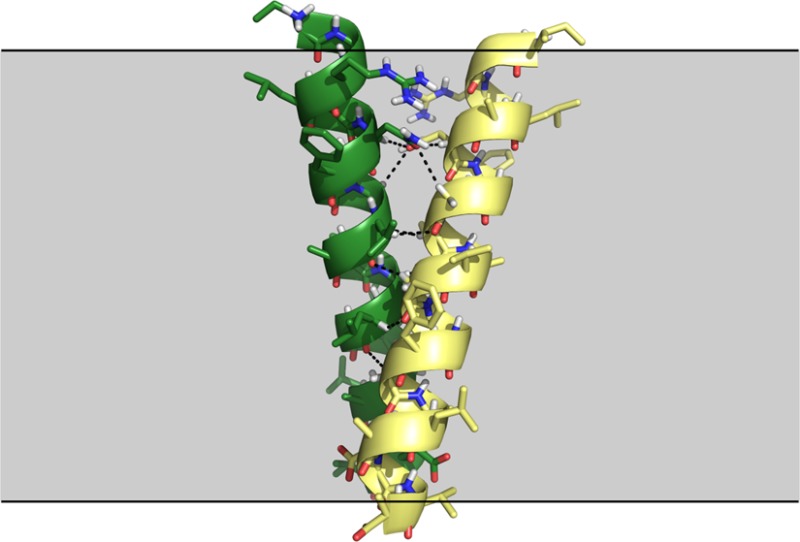
To investigate the basis of oligomerization in TM helices, our group and others have pursued a strategy based on the analysis of frequently occurring association motifs.26−32 One of the most important motifs is GASright27 (Figure 1), which is best known as the fold of a widely studied model system for TM association, the glycophorin A TM dimer.33 GASright gets its name from its right-handed crossing angle (Figure 1b), and from the characteristic small amino acids at its interface (GAS: Gly, Ala, Ser),27 which are arranged to form GxxxG and GxxxG-like patterns (GxxxA, AxxxG, etc.).28,29 In many ways GASright parallels the important coiled coil, a frequently occurring interaction motif and model for folding and association for soluble proteins.34−36 Like the coiled coil, GASright is characterized by a specific geometry (a short interhelical distance and a crossing angle near −40°), it has a distinctive sequence signature (the GxxxG patterns), and is one of the most common oligomerization motifs, if not the most common.27
Figure 1.
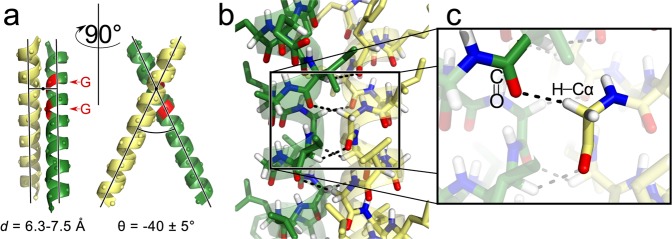
Structural features of the GASright TM association motif. (a) The GASright motif (which is best known as the fold of the TM region of glycophorin A) is a right-handed helical dimer with a short interhelical distance d and a right-handed crossing angle θ of approximatively −40°. The GxxxG sequence pattern near the crossing point (marked in red in the green helix) allows the backbones to come into close contact. (b) The contact enables the formation of networks of interhelical hydrogen bonds between Cα–H donors and carbonyl oxygen acceptors (shown in detail in (c)).
In a recent computational analysis of transmembrane dimer geometry, we proposed that the primary role of GxxxG in GASright is to promote the formation of networks of stabilizing hydrogen bonds between Cα–H donors and carbonyl oxygen acceptors on opposed helices37 (Figure 1b,c). More specifically, we proposed that the small amino acids perform two distinct functions: the first is to create permissive steric conditions, allowing the two helices to come in backbone contact, thus bringing the Cα–H donors and carbonyl acceptors in proximity. The second function, which is performed exclusively by Gly, is to increase the number of hydrogen bonds by donating with the second Hα, which corresponds to the side chain R-group in all other amino acids. To perform these functions, the small amino acids are required to be present at specific positions. The formation of this network of hydrogen bonds is also dependent on the specific crossing angle of GASright (−40°), which precisely aligns Cα–H donors spaced at i, i+1 on one helix against carbonyl acceptors spaced at i, i+3 on the opposing helix (see Figure 4 in Mueller et al.37). Overall, GASright appears geometrically optimized for interhelical Cα–H formation.37
Cα–H hydrogen bonds are commonly observed in proteins.38 Carbons are generally weak donors, but the Cα in proteins is activated by the electron-withdrawing amide groups on both sides, and quantum calculations indicate that the energy of Cα–H hydrogen bonds may be as much as one-third to half of that of canonical donors in vacuum.39,40 Therefore, they are likely to be stabilizing factors in proteins embedded in the hydrophobic milieu of the membrane, particularly when they occur in multiple instances at the same interface, as in the GASright motif (Figure 1).41 An IR-based investigation of the CD2 stretching mode of a Cα–H donor in the transmembrane domain of glycophorin A produced an estimated contribution of −0.88 kcal/mol for the hydrogen bond.42 Conversely, a folding study of the multispan membrane protein bacteriorhodopsin in which a Cα–H···O side chain hydroxyl acceptor (Thr-24) was mutated indicated that this particular bond was not stabilizing.43 Subsequent computational work suggested that the orientation of the groups can determine whether an interaction may be strongly favorable or unfavorable.44,45
The exact contribution of hydrogen bonds to membrane protein folding and association − whether the donor is a Cα–H or a more “canonical” N–H or O–H group − is still unresolved.46 A governing assumption maintains that donors and acceptors buried in the membrane would not pay a significant desolvation penalty upon helix association, and therefore the formation of hydrogen bonds should contribute appreciably to the stability of membrane proteins. Indeed, polar residues can promote interaction of transmembrane helices.13,47,48 Yet, the limited number of experimental observations made to date seem to indicate that the contribution of hydrogen bonding in the membrane may be, surprisingly, of the same magnitude observed for water-soluble proteins.46
Despite the scarce experimental evidence regarding the contribution of Cα–H hydrogen bonds to TM interactions, the hypothesis that they drive folding and oligomerization remains compelling. In particular, the fact that the prevalent GASright motif corresponds to the only interhelical geometry that maximizes formation of Cα—H···O=C networks, strongly suggests that these bonds are indeed a major contributor to association.37 Under these premises, we hypothesized that a computational structural search based on the simultaneous optimization of side chain packing and Cα–H hydrogen bonding may be able to predict the structure of GASright dimers. The resulting program, named CATM, was tested against the small database of known GASright homodimeric structures.37 We found that CATM predicts these known structures at near atomic precision. The finding provides further indirect support that Cα–H hydrogen bonding is likely to be a structural determinant of GASright dimers.37 The positive result also indicates that CATM may be a powerful tool for assisting the experimental investigation of GASright homodimers of unknown structure.
To test the ability of our methods to predict ab initio the structure of unknown GASright dimers, here we investigate ADCK3, a human mitochondrial protein that is a member of the highly conserved UbiB protein kinase-like family.49 UbiB family members account for approximately one-quarter of microbial PKL sequences,50 are ubiquitous among eukaryotes,50 and are strongly associated with lipid metabolism.51−53 Most organisms have a UbiB family member that is required for the biosynthesis of coenzyme Q (CoQ, ubiquinone). Deletion of the Escherichia coli gene ubiB(54) or the yeast gene coq8(55) completely halts CoQ biosynthesis. Similarly, mutations to human ADCK3 are known to cause CoQ deficiency and cerebellar ataxia,56−59 and mutations to human ADCK4 were recently shown to cause CoQ deficiency and a steroid-resistant nephrotic syndrome.60 Our knowledge of the molecular mechanism by which UbiB family proteins enable CoQ biosynthesis is limited, primarily because the endogenous substrates of UbiB proteins have not yet been discovered. However, we do know that coq8p in yeast somehow stabilizes a complex of CoQ biosynthesis enzymes.61 CoQ biosynthesis occurs within the context of cellular membranes, either the plasma membrane of prokaryotes or the inner mitochondrial membrane of eukaryotes, and the responsible enzymes are either integral membrane proteins or peripherally associated membrane proteins.61 ADCK3, which contains a predicted TM domain, is also likely to associate with membranes, but this hypothesis has not yet been tested. Biochemical characterization of the ADCK3 TM domain would provide an important foundation for understanding how it enables CoQ biosynthesis.
The potential functional importance of the ADCK3 TM region is underlined by the existence of a mutation at the putative edge of the TM domain (R213W) that disrupts CoQ biosynthesis and causes cerebellar ataxia in human patients.57 Furthermore, dimerization of single-span TM domains is known to be central to the regulation of some kinase families, such as the receptor tyrosine kinases.62,63 However, it was unknown whether the predicted TM helix of ADCK3 could actually insert into biological membranes and whether the TM helix can self-associate to potentially drive dimerization of ADCK3.
Here, we demonstrate experimentally that the TM domain of ADCK3 inserts into membranes and self-associates. Using extensive mutagenesis, we also show that the interaction interface is consistent with the structural models predicted by CATM, which involves an extended Gly-zipper motif30 (i.e., a series of Gly amino acids separated at i, i+4). The experimental and computational data also indicates that the Gly-zipper interface is potentially compatible with alternative conformations of the TM domain, opening the possibility that conformational changes of the TM dimer may be important for ADCK3 function.
Methods
Vectors and Strains
All oligonucleotides were purchased in desalted form from Integrated DNA Technologies and used without purification. The expression vectors pccKAN, pccGpA-wt, and pccGpA-G83I, and malE deficient E. coli strain MM39 were kindly provided by Dr. Donald M. Engelman.14 The genes encoding the TM domain of ADCK3 (214-LANFGGLAVGLGFGALA-230) and ADCK4 (92-LANFGGLAVGLGLGVLA-108) were cloned into the NheI-BamHI restriction sites of the pccKAN vector. Site directed mutations to produce single amino acid variants in the TM domain of ADCK3 were introduced with the QuikChange kit (Stratagene).
Expression of Chimeric Proteins in MM39 Cells
The TOXCAT constructs were transformed into MM39 cells. A freshly streaked colony was inoculated into 3 mL of LB broth containing 100 μg/mL ampicillin and grown overnight at 37 °C. 30 μL of overnight cultures were inoculated into 3 mL of LB broth and grown to an OD420 of approximately 0.8–1.1 (OD600 of 0.4 to 0.6) at 37 °C. After recording the optical density, 1 mL of cells was spun down for 10 min at 17000g and resuspended in 500 mL of sonication buffer (25 mM Tris-HCl, 2 mM EDTA, pH 8.0). Cells were lysed by probe sonication at medium power for 8 s over ice. An aliquot was removed from each sample and stored in SDS-PAGE loading buffer for immunoblotting. The lysates were then cleared by centrifugation at 17000g, and the supernatant was kept on ice for chloramphenicol acetyltransferase (CAT) activity assay.
MalE Complementation Assay
To confirm proper membrane insertion and orientation of the TOXCAT constructs, overnight cultures were plated on M9 minimal medium plates containing 0.4% maltose as the only carbon source and grown at 37 °C for 48 h.14
Chloramphenicol Acetyltransferase (CAT) Spectrophotometric Assay
CAT activity was measured as described.64,65 Briefly, 1 mL of buffer containing 0.1 mM acetyl CoA, 0.4 mg/mL 5,5′-dithiobis(2-nitrobenzoic acid) or Ellman’s reagent, and 0.1 M Tris-HCl pH 7.8, were mixed with 40 μL of cleared cell lysates and the absorbance at 412 nm was measured for 2 min to establish basal enzyme activity rate. After addition of 40 μL of 2.5 mM chloramphenicol in 10% ethanol, the absorbance was measured for an additional 2 min to determine CAT activity. The basal CAT activity was subtracted and the value was normalized by the cell density measured as OD420. All measurements were determined at least in duplicate and the experiments were repeated at least twice.
Quantification of Expression by Immunoblotting
Protein expression was confirmed by immunoblotting. The cell lysates (10 μL) were loaded onto a NuPAGE 4–12% Bis-Tris SDS-PAGE gel (Invitrogen) and then transferred to PVDF membranes (VWR) for 1 h at 100 millivolts. Blots were blocked using 5% bovine serum albumin (US Biologicals) in TBS-Tween buffer (50 mM Tris, 150 mM NaCl, 0.05% Tween 20) for 2 h at 4 °C, incubated with biotinylated anti-Maltose Binding Protein antibodies (Vector laboratories) overnight at 4 °C, followed by peroxidase-conjugated streptavidin (Jackson ImmunoResearch) for 2 h at 4 °C. Blots were developed with the Pierce ECL Western Blotting Substrate Kit and chemiluminescence was measured using an ImageQuant LAS 4000 (GE Healthsciences).
Computational Modeling
The structure of ADCK3-TM was predicted with CATM,37 which is distributed with the open source MSL C++ library v. 1.266 at http://msl-libraries.org. The computational mutagenesis was performed on all ADCK3 models by applying the same point mutations measured experimentally in the context of a fixed backbone, followed by side chain optimization. Side chain mobility was modeled using the energy-based conformer library applied at the 95% level.67 Energies were determined using the CHARMM 22 van der Waals function68 and the hydrogen bonding function of SCWRL 4,69 as implemented in MSL,66 with the following parameters for Cα donors, as reported previously: B = 60.278; D0 = 2.3 Å; σd = 1.202 Å; αmax = 74.0°; βmax = 98.0°.37 The relative energy of each mutant was calculated as
where EWT,dimer and Emut,dimer are the energies of the wild type and mutant sequence, respectively, in the dimeric state, and EWT,monomer and Emut,monomer are the energies of the wild type and mutant sequence, respectively, in a side chain optimized monomeric state with the same sequence. As reported previously,65 the effect of each mutation was classified in four categories (analogous to the experimental mutagenesis) using the following criterion: category 0, “WT-like”, ΔEmut < 2 kcal/mol; category 1, “Mild”, 2 ≤ ΔEmut < 4; category 2, “Severe”, 4 ≤ ΔEmut < 8; category 3, “Disruptive”, ΔEmut ≥ 8. The numerical category values were averaged to calculate the average position-dependent disruption value.
Results and Discussion
ADCK3 Is Predicted to Have a TM Helix
The protein kinase-like domain of ADCK3 is preceded on the N-terminal side by a region of undefined function. A predicted TM helix within this region is annotated in UniProt for the close homologue ADCK4,70 providing a potential anchor for the protein at the inner mitochondrial membrane (Figure 2a). UniProt does not report a predicted TM domain for the corresponding region of ADCK3, but the sequence of the putative TM segment is highly conserved between the two proteins. The same general domain organization and function is also predicted for the yeast homologue Coq8p. Given that ADCK3 and ADCK4 are localized to the mitochondrial matrix,71 the TM domain would position their catalytic kinase domains on the matrix face of the inner membrane, the same localization of the enzymes involved in the biosynthesis of coenzyme Q.61,71 Therefore, it is important to verify the TM domain experimentally and to investigate its potential functional role.
Figure 2.
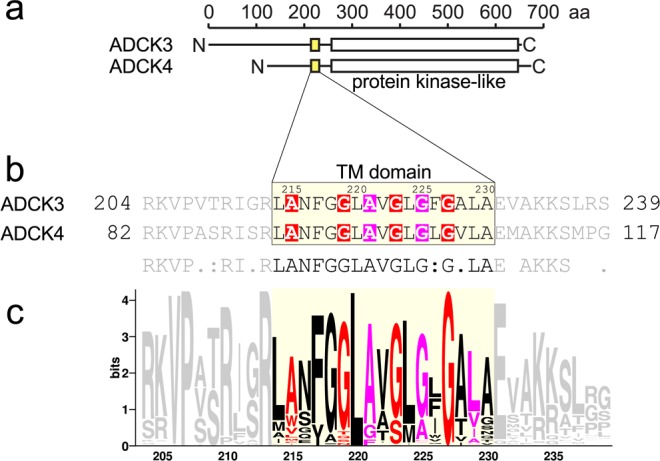
The transmembrane domain of ADCK3 has a conserved Gly-zipper motif. (a) Domain organization of ADCK3 homologues, which are proteins associated with the mitochondrial inner membrane. They are predicted to contain a TM domain (yellow) and a protein kinase-like domain (white). (b) The sequence alignment of the TM domains of ADCK3, ADCK4 (yellow box). The TM domains of ADCK3 and ADKC4, which differ only at two positions, contain a number of GxxxG-like motifs, including an extended Gly-zipper motif (red) and a second AxxxG motif which is off-register by two positions (magenta). (c) Sequence logo of the alignment of 400 sequences homologous to ADCK3 from a broad range of eukaryotic species highlights conservation in the TM domain and in the N-terminal side of the juxta-membrane region. All Gly positions in the Gly-zipper (red) appear strongly conserved. The most conserved positions in the TM region are L220 and G227. Identifiers of the sequences used for the alignment are provided in Supplementary Text S1 (SI).
The sequences of the putative TM domains of ADCK3 and ADCK4 are aligned in Figure 2b. As summarized in Table 1, these sequences have low hydrophobicity and a relative short length (17 amino acids), and thus are not well recognized by prediction servers. The TM domain of ADCK3, which contains one polar amino acid (Asn 216), is recognized as a borderline TM sequence by most servers. Specifically, the segment is not recognized by TMHMM72 and E(z),73 but the segment is predicted as transmembrane by MemBrain74,75 and HMMTOP,76,77 and Phobius78,79 and ΔG prediction80 recognize it with low confidence. ADCK4 shares over 50% sequence identify with ADCK3 but their TM domains are almost identical, differing only at two positions (Figure 2b). Because of these two substitutions (and primarily because of the A228 V substitution), the hydrophobicity of the TM domain of ADCK4 is higher (as calculated with either the Wimley-White octanol scale81 or the “biological” scale80,82) and is sufficient to be predicted by most servers, except E(z), with good confidence (Table 1).
Table 1. Prediction of the Transmembrane Domain of the ADCK3 Homologs.
| name | sequence | ΔGOcta | ΔGAppb | TMPREDc | Phobiusd | TMHMMe | ΔG predictorf | MemBraing | E(z)h |
|---|---|---|---|---|---|---|---|---|---|
| ADCK3 | LANFGGLAVGLGFGALA | –0.28 | +2.11 | yes | 50% | no | yes (+1.80) | possible (70%) | no |
| ADCK4 | LANFGGLAVGLGLGVLA | –0.78 | +1.91 | yes | 90% | 40% | yes (+1.69) | yes (80%) | no |
Wimley–White octanol scale (kcal/mol).81
Phobius78,79 at http://phobius.sbc.su.se.
ΔG predictior80 at http://dgpred.cbr.su.se (in paretheses the ΔGApp for the predicted TM segment, kcal/mol).
MemBrain74,75 at http://www.csbio.sjtu.edu.cn/bioinf/MemBrain.
E(z) potential73 at http://ez.degradolab.org/ez/original.
In interpreting these prediction data, it is important to consider that TM prediction servers are trained against a majority of proteins that are inserted in the membrane via a translocon mediated mechanism. The sequence requirements for translocon mediated insertion in an eukaryotic system are well understood.80,82 A recent analysis in a bacterial system shows good overall correspondence to the mammalian system, but the hydrophobicity threshold appears to be distinctly lower.83 Much less is known about the requirements for membrane insertion of mitochondrial integral membrane proteins that are encoded in the nucleus, such as ADCK3. There is, however, good indication that the hydrophobicity threshold for these proteins should be even lower, to avoid mistargeting of these proteins to the endoplasmic reticulum and to facilitate their translocation to the mitochondrion.84−89 Based on the above considerations, it is highly probable that the predicted TM segments of ADCK3 and ADCK4 are indeed bona fide TM domains.
The TM Domain of ADCK3 Has Conserved GxxxG-like Motifs
As shown in Figure 2b, the predicted TM regions of ADCK3 and ADCK4 are very rich in small amino acids such as Gly, Ala and Ser (9 in each). The sequences contain a number of GxxxG and GxxxG-like (AxxxG) helix association patterns, which appear to be evolutionarily conserved (Figure 2c). In particular, they contain an extended Gly-zipper motif,30 i.e., a series of small amino acids (215-AxxxGxxxGxxxG-227) spaced at i, i+4, highlighted in red in Figure 2b. They also contain an additional AxxxG motif (magenta), which is off-frame by two positions with respect to the Gly-zipper. This spacing projects the two motifs on opposite helical faces.
CATM Predicts That the TM Domain of ADCK3 Can Form a GASright Homodimer
GxxxG-like patterns can drive helix–helix association.28 They occur with high frequency in TM helices,29 both in multispan proteins and in oligomerizing single-span membrane proteins,41 and are often important for biological function.4 The presence of GxxxG-like motifs in the putative TM sequence of ADCK3 raised the question of whether this domain oligomerizes. To investigate this question, we analyzed the sequence with CATM,37 a program for the structural prediction of GASright motifs, an important and common class of GxxxG-mediated dimers.27
As shown in Figure 3, CATM predicts five alternative models for the TM sequence of ADCK3. The figure schematically depicts the geometrical features of the dimers. The position of the crossing point between the two helices is marked (dot), and the interfacial positions that surround this crossing point are highlighted by a green parallelogram. All the positions that are involved in intermonomer contacts at the interface are highlighted in either yellow, or in red if they belong to the Gly-zipper motif. The scores of the top models of ADCK3 in CATM (−59.8, −50.8, and −47.7 for Models 1, 2, and 3 respectively) are comparable to the scores obtained for the five known structures of GASright motifs (which range between −56 and −38), which CATM is able to predict at near atomic precision.37
Figure 3.

CATM predicts multiple modes of interaction along the Gly-zipper motif of ADCK3. Schematic representation of the five models of GASright homodimers generated by CATM for ADCK3-TM. The crossing point is marked by a black dot. The four positions that surround the crossing point are marked by a green parallelogram and are underlined in the sequence. The positions involved in interhelical packing at the dimer interface are highlighted: in red are the interfacial positions that belong to the extended Gly-zipper motif of ADCK3; all other interfacial positions are highlighted in yellow. The table summarizes the geometry of the five models: interhelical distance d; crossing angle θ; vertical (Z′) and axial (ω′) coordinates of the crossing point within the parallelogram of closest approach; and energy score E. For the geometric definitions, see Figure S2 (SI) and Mueller et al.37
Notably, the extended Gly-zipper is involved at the helix–helix interface in all models (Figure 3). Model 1 and 2 are related geometries whose crossing points fall in the quadrilateral defined by Gly 219, Leu 220, Gly 223 and Leu 224 (AxxxGLxxGLxxG). These two models differ by the position of the crossing point and, most importantly, by their crossing angle, which is near the canonical −40° of GASright motifs for Model 2, and narrower for Model 1 (−27.1°). The smaller crossing angle causes Model 1 to have a more extended interface, which is reflected also by the more extensive van der Waals interaction of Model 1. Both models have 12 interhelical Cα–H hydrogen bonds, although Model 2 has a better overall hydrogen bonding score (Tables S1 and S2 (SI)).
The other three predicted models cross at different sections of the Gly-zipper. Model 3 and Model 5 are variations that cross within the N-terminal side of the zipper (ANxxGLxxGxxxG). Conversely, Model 4 crosses on the C-terminal side of the zipper (AxxxGxxxGLxxGA). CATM does not produce any model mediated by the off-frame AxxxG motif of ADCK3 (magenta in Figure 2b). The coordinates of all ADCK3 models are available as Supporting Information and for download at http://seneslab.org/ADCK3_models.
ADCK3-TM Self-Associates Strongly in E. coli Membranes
To investigate the structural predictions of CATM, we assessed the dimerization of ADCK3-TM and ADCK4-TM experimentally using TOXCAT, a widely used assay for TM association in biological membranes.14 This assay involves the biological expression in the membrane of E. coli of a chimeric construct that fuses the TM domain of interest with the ToxR transcriptional activator of Vibrio cholera (Figure 4a). TM helix association leads to the dimerization of the ToxR domain, resulting in expression of the reporter gene chloramphenicol acetyltransferase (CAT). The expression level of CAT (measured by its enzymatic activity) is compared to that of a stable dimer, Glycophorin A (GpA), and to a monomeric GpA variant (GpA-G83I) as standards.
Figure 4.

ADCK3-TM and ADCK4-TM associate strongly in TOXCAT. (a) TOXCAT is an in vivo assay based on a construct in which the transmembrane domain under investigation is fused to the ToxR transcriptional activator of V. cholerae. Transmembrane association results in the expression of a reporter gene in E. coli cells, which can be quantified. (b) malE complementation assay. The TOXCAT construct containing the TM domain of ADCK3 and ADCK4 can use maltose as a carbon source, demonstrating correct insertion. GpA: Glycophorin A positive control; no TM: pcckan plasmid without TM insert, negative control. (c) TOXCAT assay of ADCK3 and ADCK4. ADCK3 shows approximately 150% of the CAT activity of the strong transmembrane dimer of Glycophorin A (GpA). The monomeric G83I mutant (GpA*) is used as a negative control. Data reported as average and standard deviation over four replicate experiments. Expression levels were controlled by immunoblotting.
We first tested whether the constructs inserted correctly in the plasma membrane of E. coli, using a complementation test in the malE deficient strain MM39. The ADCK3-TM and ADCK4-TM TOXCAT constructs supported growth in minimal media with maltose as the sole carbon source (Figure 4b), indicating that the fusion proteins are recognized as TM domains and are expressed in the bacterial inner membrane in the correct orientation, with the MBP moiety positioned on the periplasmic side.
To examine whether ADCK3 oligomerizes in TOXCAT, we quantified the enzymatic activity of the reporter gene CAT, as an indirect measure of its expression. As shown in Figure 4c, the CAT activities of the ADCK3-TM and ADCK4-TM constructs are higher than the activity of the GpA standard, which is a stable homodimer. These results indicate that the TM domain of ADCK3 and ADCK4 form strong homo-oligomers in TOXCAT.
Large Scale Mutagenesis Demonstrates That the Gly Zipper Motif Is Important for Association
To assess experimentally the interaction interface of the ADCK3-TM oligomer and validate the computational predictions, we performed large scale mutagenesis along the entire span of the TM segment, and measured their self-association in TOXCAT. Each position was individually changed to a variety of large and small hydrophobic amino acids. The expectation is that the changes at interfacial positions are more likely to perturb oligomerization than changes at lipid exposed positions, as commonly observed (for example17,90−93). A total of 53 mutants were generated and analyzed in TOXCAT.
The TOXCAT data is shown in Figure S1 (SI) and is schematically represented in Figure 5a. To compute an overall position-dependent sensitivity to mutation, we applied a classification scheme for the variants’ phenotypes using four categories (dashed lines in Figure S1 (SI)), labeled as “WT-like” (>80% of wild type CAT activity), “Mild” (50–80%), “Severe” (20–50%) and “Disruptive” (0–20%). These scores were then averaged to obtain a position specific “average disruption”. Position-based averaging reduces some of the natural variability of the biological assay and the method has been reliable in identifying the most sensitive positions at the helix–helix binding interface.65,90,93,94 The position-dependent “average disruption” is also plotted in numerical form in Figure 5b.
Figure 5.

Position specific “average disruption” suggests that the Gly-zipper is at the helical interface. (a) “MacKenzie plot” summarizing the effect of all mutations of ADCK3-TM measured in TOXCAT. The color coding of the GxxxG motifs in the sequence corresponds to Figure 1. The data has been subdivided in three categories as in the legend. The raw TOXCAT data is shown in Figure S1 (SI). A calculated average disruption score for each position is displayed at the bottom of the scheme. (b) The same average disruption plotted numerically (0 = as TW; 3 = disruptive). The mutagenesis reveals two positions that are essential for self-association, G223 and G227, which are the last two position of ADCK3′s Gly-zipper (red).
A majority of the variants had CAT activity levels similar or higher compared to the wild type sequences (Figure 5b). However, a number of variants showed dramatically reduced activity in a position specific fashion. In particular, all variants of the two C-terminal Gly residues of the Gly-zipper (G223 and G227) have the strongest disruptive phenotypes. Interestingly, the next most sensitive positions are L220 and L224, which are also predicted to be interfacial in almost all CATM models (Figure 3). Conversely, the N-terminal positions of the zipper are either mildly affected by mutation (G219) or appear completely tolerant (A215).
The off-frame 221-AxxxG-225 (magenta) is also relatively insensitive to mutation. Substitution for a large Leu at these two positions has only a mild effect, and the Ile variants are completely tolerated. This is consistent with the CATM predictions, which do not identify any model in which this motif is at the interface.
A position of interest for self-association was Asn 216. Polar residues can drive TM helix oligomerization through the formation of hydrogen bonds, and have been found to be important for the association of model peptides13,47 and of biological systems,95−98 including in the context of GASright motifs.94,99 In addition, some polarity of position 216 appears to be relatively conserved, as the main substitutions of N216 in a sequence alignment (Figure 2c) are Gly, Ser, Gln and Glu. However, neither the computational nor the experimental analysis suggest that N216 is important for self-association. Asn 216 can be mutated to Ala, Leu or Phe in TOXCAT without reduction of self-association. CATM is in agreement with the experimental data, as it does not identify any potential strong polar interaction (i.e., N–H···O hydrogen bonds) involving the side chain of N216, although the side chain carbonyl oxygen (Oδ1) acts as a Cα–H bond acceptor in most models.
Overall the data indicates that the interface of the ADCK3-TM oligomer is mediated by Gly-zipper motif and, in particular, by the C-terminal side of this interaction motif.
Computational Mutagenesis Suggests Potential Alternative Conformations for ADCK3-TM
In order to identify the structure most consistent with the TOXCAT data, we performed a mutational analysis of the five models generated by CATM. Using a protocol developed previously to analyze similar mutational data,65 we created in silico the same set of variants that were tested experimentally, and computed an analogous position-dependent “average disruption” index based on the interaction energies.
The experimental and theoretical disruption patterns are compared in Figure 6. Given that all CATM structures interact through portions of the extended Gly-zipper, the computed patterns have similar periodicity across all models, with disruption peaking at position G219, G223 and/or G227. Models 1, 3, and 5 (Figure 6a,c,e) show high sensitivity to mutation on the N-terminal side of the TM domain, in disagreement with the experimental observations. In these three models, mutations to G219 are completely disruptive, whereas the position is only mildly sensitive in TOXCAT. Models 1 and 5 are also very sensitive at positions A215 and N216, which are completely tolerant experimentally.
Figure 6.

Computational mutagenesis identifies compatible models. Comparison of the mutagenesis obtained in TOXCAT (same as Figure 5b) with the computational mutagenesis performed on the five CATM models (a–e). The comparison suggests that Model 2 is the best fit to the experimental data, followed by Model 4. (f) A linear combination of Model 2 (60%) and Model 4 (40%) produces an excellent fit to the data, suggesting that the TM of ADCK3 may be in equilibrium between at least two conformations in the TOXCAT system.
The structures of Models 2 and 4 are compared in Figure 7. Model 2 (Figure 6b) and Model 4 (Figure 6d) are in better agreement with the experimental data and represent two possible structural solutions for the ADCK3 TM dimer. The disruption for both models peak at G223 and G227, which are also the two most disruptive positions in TOXCAT. However, Model 4 appears insensitive at position G219 (which is mildly sensitive experimentally) and it is extremely disruptive at position A228 (which is insensitive experimentally). In addition to being a better match, Model 2 also has lower energy, better packing and a larger number of hydrogen bonds (Tables S2 and S4 (SI)). Therefore, Model 2 appears to be the best structural candidate for the ADCK3 TM dimer.
Figure 7.
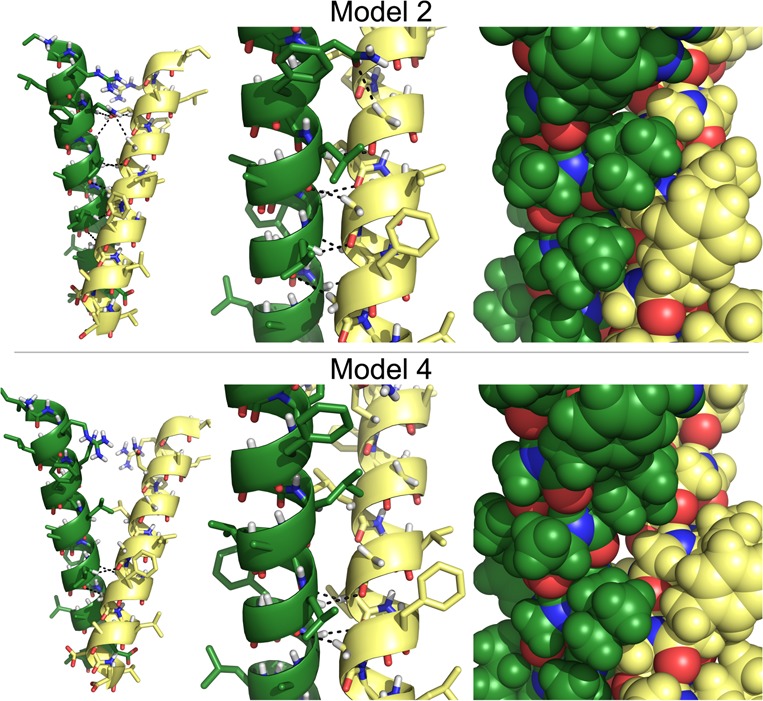
Structural Models 2 and 4. Comparison of the structures of CATM Models 2 and 4 for ADCK3. From left to right, entire TM helix, detail of the interface, and same conformation in full atom spheres. Model 2 has lower energy, a larger number of hydrogen bonds (12 in Model 2 versus 4 in Model 4) and more extended and complementary packing.
In a recent analysis of known GASright structures, we demonstrated that CATM is capable of capturing alternative conformations of biological importance.37 Therefore, an additional possibility is that the TM domain of ADCK3 may be in equilibrium between two or more structures. We observed that a linear combination of the mutagenesis profiles of the two models that best fit the data, 60% of Model 2 and 40% of Model 4, improves the fit with the TOXCAT data, producing an excellent correspondence between the two experiments (Figure 6f). This interpolation is not necessarily quantitative, but it suggests that a conformational equilibrium would be compatible with the data. If such an equilibrium occurs in the biological context, it would postulate that the TM domain of ADCK3 may be a switchable element, a trait that could be important for regulation or signaling, as observed in a number of other single-span TM proteins.7 In this framework, the Gly-zipper would provide a dynamic interface for structural changes that could potentially affect either the distance of the helical termini or the relative rotation of the helices.
Conclusions
We have presented a computational and experimental analysis of the structural organization of the TM domain of the mitochondrial kinase ADCK3. While more experiments are necessary to fully test CATM, the work provides a first practical demonstration of the applicability of the program to the characterization of a TM dimer of unknown structure. It also confirms the ability of the algorithm (which is based on optimization of van der Waals and Cα–H hydrogen bonding) to correctly predict GASright motifs.
We have experimentally demonstrated that the TM domain of ADCK3 self-associates in E. coli membranes. While the specific oligomeric state could not be determined by TOXCAT, the evidence suggests that ADCK3-TM is likely dimeric. Although Gly-zipper motifs can be involved in the formation of higher-oligomeric complexes,30 the good agreement between the experimental and computational mutagenesis supports the homodimeric hypothesis. Moreover, such oligomeric state is also consistent with a large body of structural evidence, which shows that kinases frequently form dimeric complexes (for example100−102), while higher oligomers are rarely observed.
The analysis reveals a number of leads that may be biologically important. The helix–helix interaction interface was determined and the mutagenesis identified a number of disruptive interfacial mutations that will be useful for follow-up functional studies. The computational prediction of alternative models in which the helices adopt a different crossing point along the Gly-zipper interface raises also the hypothesis that the TM domain of ADCK3 may possibly undergo conformational changes.
Indirectly, the work also provides important insight about ADCK4. All the amino acids that participate at the dimerization interface of ADCK3 are identical in ADCK4. The two positions that differ between the two sequences (F228L and A230 V, Figure 2) are insensitive to variation when they are mutated individually (Figure 5). The computational predictions obtained for ADCK3 and ADCK4 are nearly identical, and it is thus expected that both TM domains dimerize with the same structure. Because the two interfaces are compatible with each other, it also possible that the TM domains could associate to drive formation of a heterodimeric complex between ADCK3 and ADCK4. These hypotheses need to be investigated in a biological context; the present analysis provides the theoretical foundation necessary for testing in vivo the role of these TM domains.
Acknowledgments
The work was supported by startup funds from the University of Wisconsin–Madison and by National Institutes of Health Grant R01GM099752 and National Science Foundation Grant CHE-1415910 to A.S.; D.J.P. and J.A.S. were supported by a Searle Scholar Award, a Shaw Scientist Award and by National Institutes of Health Grants R01DK098672 and U01GM094622 to D.J.P.; J.A.S was supported by an NIH Ruth L. Kirschstein National Research Service Award F30AG043282; B.K.M. acknowledges the support of the NLM Grant 5T15LM007359 to the CIBM Training Program. We are grateful to Sabareesh Subramaniam for assistance in the computational modeling and Jennifer Peotter for assistance in the preparation of the TOXCAT constructs.
Supporting Information Available
Summaries of the features of the five CATM models, PDB files of the models, TOXCAT mutagenesis data, definition of the geometric parameters, and identifiers of the sequences of ADCK3 homologues. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Popot J. L.; Engelman D. M. Biochemistry (Moscow) 1990, 29, 4031. [DOI] [PubMed] [Google Scholar]
- Engelman D. M.; Chen Y.; Chin C.-N.; Curran A. R.; Dixon A. M.; Dupuy A. D.; Lee A. S.; Lehnert U.; Matthews E. E.; Reshetnyak Y. K.; Senes A.; Popot J.-L. FEBS Lett. 2003, 555, 122. [DOI] [PubMed] [Google Scholar]
- Rath A.; Deber C. M. Annu. Rev. Biophys. 2012, 41, 135. [DOI] [PubMed] [Google Scholar]
- Senes A.; Engel D. E.; DeGrado W. F. Curr. Opin. Struct. Biol. 2004, 14, 465. [DOI] [PubMed] [Google Scholar]
- Mackenzie K. R. Chem. Rev. 2006, 106, 1931. [DOI] [PubMed] [Google Scholar]
- MacKenzie K. R.; Fleming K. G. Curr. Opin. Struct. Biol. 2008, 18, 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore D. T.; Berger B. W.; DeGrado W. F. Structure 2008, 16, 991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher L. E.; Engelman D. M.; Sturgis J. N. J. Mol. Biol. 1999, 293, 639. [DOI] [PubMed] [Google Scholar]
- Merzlyakov M.; You M.; Li E.; Hristova K. J. Mol. Biol. 2006, 358, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M.; Li E.; Wimley W. C.; Hristova K. Anal. Biochem. 2005, 340, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khadria A.; Senes A. Methods Mol. Biol. 2013, 1063, 19. [DOI] [PubMed] [Google Scholar]
- Fleming K. G. J. Mol. Biol. 2002, 323, 563. [DOI] [PubMed] [Google Scholar]
- Choma C.; Gratkowski H.; Lear J. D.; DeGrado W. F. Nat. Struct. Biol. 2000, 7, 161. [DOI] [PubMed] [Google Scholar]
- Russ W. P.; Engelman D. M. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner E.; Langosch D. Proteins 2006, 65, 803. [DOI] [PubMed] [Google Scholar]
- Schneider D.; Engelman D. M. J. Biol. Chem. 2003, 278, 3105. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A.; Flanagan J. M.; Treutlein H. R.; Zhang J.; Engelman D. M. Biochemistry (Moscow) 1992, 31, 12719. [DOI] [PubMed] [Google Scholar]
- Rath A.; Glibowicka M.; Nadeau V. G.; Chen G.; Deber C. M. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.; Blois T. M.; Cao Z.; Bowie J. U. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.; Chang Y.-C.; Bowie J. U. Methods Mol. Biol. 2013, 1063, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie J. U. Nature 2005, 438, 581. [DOI] [PubMed] [Google Scholar]
- Chang Y.-C.; Bowie J. U. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin E.; von Heijne G. Protein Sci. 1998, 7, 1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin I. T.; Brunger A. T. Biochim. Biophys. Acta 1998, 1429, 113. [DOI] [PubMed] [Google Scholar]
- Hubert P.; Sawma P.; Duneau J.-P.; Khao J.; Hénin J.; Bagnard D.; Sturgis J. Cell Adhes. Migr. 2010, 4, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F.; Poulsen B. E.; Ip W.; Deber C. M. Biopolymers 2011, 96, 340. [DOI] [PubMed] [Google Scholar]
- Walters R. F. S.; DeGrado W. F. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 13658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ W. P.; Engelman D. M. J. Mol. Biol. 2000, 296, 911. [DOI] [PubMed] [Google Scholar]
- Senes A.; Gerstein M.; Engelman D. M. J. Mol. Biol. 2000, 296, 921. [DOI] [PubMed] [Google Scholar]
- Kim S.; Jeon T.-J.; Oberai A.; Yang D.; Schmidt J. J.; Bowie J. U. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 14278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doura A. K.; Kobus F. J.; Dubrovsky L.; Hibbard E.; Fleming K. G. J. Mol. Biol. 2004, 341, 991. [DOI] [PubMed] [Google Scholar]
- Unterreitmeier S.; Fuchs A.; Schäffler T.; Heym R. G.; Frishman D.; Langosch D. J. Mol. Biol. 2007, 374, 705. [DOI] [PubMed] [Google Scholar]
- MacKenzie K. R.; Prestegard J. H.; Engelman D. M. Science 1997, 276, 131. [DOI] [PubMed] [Google Scholar]
- Harbury P. B.; Zhang T.; Kim P. S.; Alber T. Science 1993, 262, 1401. [DOI] [PubMed] [Google Scholar]
- Harbury P. B.; Tidor B.; Kim P. S. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz S. F.; Bryson J. W.; DeGrado W. F. Curr. Opin. Struct. Biol. 1995, 5, 457. [DOI] [PubMed] [Google Scholar]
- Mueller B. K.; Subramaniam S.; Senes A. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz S.; Trievel R. C. J. Biol. Chem. 2012, 287, 41576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas R.; Garza J.; Dixon D. A.; Hay B. P. J. Am. Chem. Soc. 2000, 122, 4750. [Google Scholar]
- Scheiner S.; Kar T.; Gu Y. J. Biol. Chem. 2001, 276, 9832. [DOI] [PubMed] [Google Scholar]
- Senes A.; Ubarretxena-Belandia I.; Engelman D. M. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbely E.; Arkin I. T. J. Am. Chem. Soc. 2004, 126, 5362. [DOI] [PubMed] [Google Scholar]
- Yohannan S.; Faham S.; Yang D.; Grosfeld D.; Chamberlain A. K.; Bowie J. U. J. Am. Chem. Soc. 2004, 126, 2284. [DOI] [PubMed] [Google Scholar]
- Mottamal M.; Lazaridis T. Biochemistry (Moscow) 2005, 44, 1607. [DOI] [PubMed] [Google Scholar]
- Park H.; Yoon J.; Seok C. J. Phys. Chem. B 2008, 112, 1041. [DOI] [PubMed] [Google Scholar]
- Bowie J. U. Curr. Opin. Struct. Biol. 2011, 21, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F. X.; Cocco M. J.; Russ W. P.; Brunger A. T.; Engelman D. M. Nat. Struct. Biol. 2000, 7, 154. [DOI] [PubMed] [Google Scholar]
- Herrmann J. R.; Fuchs A.; Panitz J. C.; Eckert T.; Unterreitmeier S.; Frishman D.; Langosch D. J. Mol. Biol. 2010, 396, 452. [DOI] [PubMed] [Google Scholar]
- Leonard C. J.; Aravind L.; Koonin E. V. Genome Res. 1998, 8, 1038. [DOI] [PubMed] [Google Scholar]
- Kannan N.; Taylor S. S.; Zhai Y.; Venter J. C.; Manning G. PLoS Biol. 2007, 5, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T.; Ozbalci C.; Brügger B.; Rapaport D.; Dimmer K. S. J. Cell Sci. 2013, 126, 3563. [DOI] [PubMed] [Google Scholar]
- Martinis J.; Glauser G.; Valimareanu S.; Kessler F. Plant Physiol. 2013, 162, 652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist P. K.; Poliakov A.; Giacomelli L.; Friso G.; Appel M.; McQuinn R. P.; Krasnoff S. B.; Rowland E.; Ponnala L.; Sun Q.; van Wijk K. J. Plant Cell 2013, 25, 1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon W. W.; Davis D. E.; Ha H. T.; Jonassen T.; Rather P. N.; Clarke C. F. J. Bacteriol. 2000, 182, 5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do T. Q.; Hsu A. Y.; Jonassen T.; Lee P. T.; Clarke C. F. J. Biol. Chem. 2001, 276, 18161. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C.; Tazir M.; López L. C.; Quinzii C. M.; Assoum M.; Drouot N.; Busso C.; Makri S.; Ali-Pacha L.; Benhassine T.; Anheim M.; Lynch D. R.; Thibault C.; Plewniak F.; Bianchetti L.; Tranchant C.; Poch O.; DiMauro S.; Mandel J.-L.; Barros M. H.; Hirano M.; Koenig M. Am. J. Hum. Genet. 2008, 82, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollet J.; Delahodde A.; Serre V.; Chretien D.; Schlemmer D.; Lombes A.; Boddaert N.; Desguerre I.; de Lonlay P.; de Baulny H. O.; Munnich A.; Rötig A. Am. J. Hum. Genet. 2008, 82, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerards M.; van den Bosch B.; Calis C.; Schoonderwoerd K.; van Engelen K.; Tijssen M.; de Coo R.; van der Kooi A.; Smeets H. Mitochondrion 2010, 10, 510. [DOI] [PubMed] [Google Scholar]
- Horvath R.; Czermin B.; Gulati S.; Demuth S.; Houge G.; Pyle A.; Dineiger C.; Blakely E. L.; Hassani A.; Foley C.; Brodhun M.; Storm K.; Kirschner J.; Gorman G. S.; Lochmüller H.; Holinski-Feder E.; Taylor R. W.; Chinnery P. F. J. Neurol., Neurosurg. Psychiatry 2012, 83, 174. [DOI] [PubMed] [Google Scholar]
- Ashraf S.; Gee H. Y.; Woerner S.; Xie L. X.; Vega-Warner V.; Lovric S.; Fang H.; Song X.; Cattran D. C.; Avila-Casado C.; Paterson A. D.; Nitschké P.; Bole-Feysot C.; Cochat P.; Esteve-Rudd J.; Haberberger B.; Allen S. J.; Zhou W.; Airik R.; Otto E. A.; Barua M.; Al-Hamed M. H.; Kari J. A.; Evans J.; Bierzynska A.; Saleem M. A.; Böckenhauer D.; Kleta R.; El Desoky S.; Hacihamdioglu D. O.; Gok F.; Washburn J.; Wiggins R. C.; Choi M.; Lifton R. P.; Levy S.; Han Z.; Salviati L.; Prokisch H.; Williams D. S.; Pollak M.; Clarke C. F.; Pei Y.; Antignac C.; Hildebrandt F. J. Clin. Invest. 2013, 123, 5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C. H.; Xie L. X.; Allan C. M.; Tran U. C.; Clarke C. F. Biochim. Biophys. Acta 2014, 1841, 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon M. A.; Schlessinger J.; Ferguson K. M. Cold Spring Harbor Perspect. Biol. 2014, 6, a020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E.; Hristova K. Biochemistry (Moscow) 2006, 45, 6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw W. V. Methods Enzymol. 1975, 43, 737. [DOI] [PubMed] [Google Scholar]
- LaPointe L. M.; Taylor K. C.; Subramaniam S.; Khadria A.; Rayment I.; Senes A. Biochemistry (Moscow) 2013, 52, 2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulp D. W.; Subramaniam S.; Donald J. E.; Hannigan B. T.; Mueller B. K.; Grigoryan G.; Senes A. J. Comput. Chem. 2012, 33, 1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S.; Senes A. Proteins 2012, 80, 2218. [DOI] [PubMed] [Google Scholar]
- MacKerell; Bashford D.; Bellott; Dunbrack; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiórkiewicz-Kuczera J.; Yin D.; Karplus M. J. Phys. Chem. B 1998, 102, 3586. [DOI] [PubMed] [Google Scholar]
- Krivov G. G.; Shapovalov M. V.; Dunbrack R. L. Proteins 2009, 77, 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucleic Acids Res. 2013, 41, D43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee H.-W.; Zou P.; Udeshi N. D.; Martell J. D.; Mootha V. K.; Carr S. A.; Ting A. Y. Science 2013, 339, 1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A.; Larsson B.; von Heijne G.; Sonnhammer E. L. J. Mol. Biol. 2001, 305, 567. [DOI] [PubMed] [Google Scholar]
- Senes A.; Chadi D. C.; Law P. B.; Walters R. F. S.; Nanda V.; Degrado W. F. J. Mol. Biol. 2007, 366, 436. [DOI] [PubMed] [Google Scholar]
- Shen H.; Chou J. J. PLoS One 2008, 3, e2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Jang R.; Zhang Y.; Shen H.-B. Bioinformatics 2013, 29, 2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnády G. E.; Simon I. J. Mol. Biol. 1998, 283, 489. [DOI] [PubMed] [Google Scholar]
- Tusnády G. E.; Simon I. Bioinformatics 2001, 17, 849. [DOI] [PubMed] [Google Scholar]
- Käll L.; Krogh A.; Sonnhammer E. L. L. J. Mol. Biol. 2004, 338, 1027. [DOI] [PubMed] [Google Scholar]
- Käll L.; Krogh A.; Sonnhammer E. L. L. Nucleic Acids Res. 2007, 35, W429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessa T.; Meindl-Beinker N. M.; Bernsel A.; Kim H.; Sato Y.; Lerch-Bader M.; Nilsson I.; White S. H.; von Heijne G. Nature 2007, 450, 1026. [DOI] [PubMed] [Google Scholar]
- Wimley W. C.; Creamer T. P.; White S. H. Biochemistry (Moscow) 1996, 35, 5109. [DOI] [PubMed] [Google Scholar]
- Hessa T.; Kim H.; Bihlmaier K.; Lundin C.; Boekel J.; Andersson H.; Nilsson I.; White S. H.; von Heijne G. Nature 2005, 433, 377. [DOI] [PubMed] [Google Scholar]
- Ojemalm K.; Botelho S. C.; Stüdle C.; von Heijne G. J. Mol. Biol. 2013, 425, 2813. [DOI] [PubMed] [Google Scholar]
- Tong J.; Dolezal P.; Selkrig J.; Crawford S.; Simpson A. G. B.; Noinaj N.; Buchanan S. K.; Gabriel K.; Lithgow T. Mol. Biol. Evol. 2011, 28, 1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supekova L.; Supek F.; Greer J. E.; Schultz P. G. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie C.; Suzuki H.; Sakaguchi M.; Mihara K. Mol. Biol. Cell 2002, 13, 1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley D. O.; Clifton R.; Whelan J. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie C.; Suzuki H.; Sakaguchi M.; Mihara K. J. Biol. Chem. 2003, 278, 41462. [DOI] [PubMed] [Google Scholar]
- Daley D. O.; Whelan J. Genome Biol. 2005, 6, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Arkin I. T.; Engelman D. M.; Brünger A. T. Nat. Struct. Biol. 1995, 2, 154. [DOI] [PubMed] [Google Scholar]
- Fleming K. G.; Engelman D. M. Proteins 2001, 45, 313. [DOI] [PubMed] [Google Scholar]
- Jenei Z. A.; Warren G. Z. L.; Hasan M.; Zammit V. A.; Dixon A. M. FASEB J. 2011, 25, 4522. [DOI] [PubMed] [Google Scholar]
- Li R.; Gorelik R.; Nanda V.; Law P. B.; Lear J. D.; DeGrado W. F.; Bennett J. S. J. Biol. Chem. 2004, 279, 26666. [DOI] [PubMed] [Google Scholar]
- Sulistijo E. S.; Jaszewski T. M.; MacKenzie K. R. J. Biol. Chem. 2003, 278, 51950. [DOI] [PubMed] [Google Scholar]
- Fleming K. G.; Engelman D. M. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 14340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley A. M.; Fleming K. G. J. Mol. Biol. 2007, 370, 912. [DOI] [PubMed] [Google Scholar]
- Li E.; You M.; Hristova K. J. Mol. Biol. 2006, 356, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrie C. M.; Sulistijo E. S.; MacKenzie K. R. J. Mol. Biol. 2010, 396, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulistijo E. S.; Mackenzie K. R. Biochemistry (Moscow) 2009, 48, 5106. [DOI] [PubMed] [Google Scholar]
- Cobb M. H.; Goldsmith E. J. Trends Biochem. Sci. 2000, 25, 7. [DOI] [PubMed] [Google Scholar]
- West A. H.; Stock A. M. Trends Biochem. Sci. 2001, 26, 369. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A.; Schlessinger J. Cell 2010, 141, 1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.