

Abstract
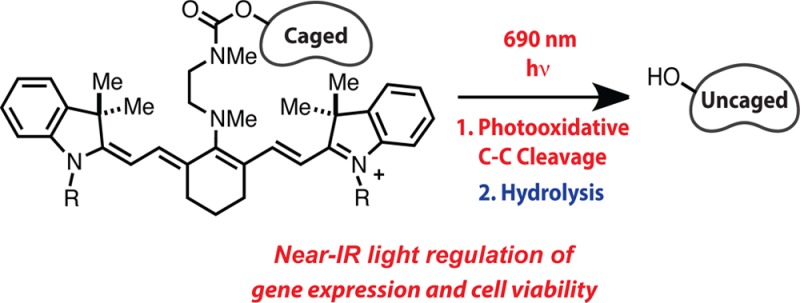
The development of photocaging groups activated by near-IR light would enable new approaches for basic research and allow for spatial and temporal control of drug delivery. Here we report a near-IR light-initiated uncaging reaction sequence based on readily synthesized C4′-dialkylamine-substituted heptamethine cyanines. Phenol-containing small molecules are uncaged through sequential release of the C4′-amine and intramolecular cyclization. The release sequence is initiated by a previously unexploited photochemical reaction of the cyanine fluorophore scaffold. The uncaging process is compatible with biological milieu and is initiated with low intensity 690 nm light. We show that cell viability can be inhibited through light-dependent release of the estrogen receptor antagonist, 4-hydroxycyclofen. In addition, through uncaging of the same compound, gene expression is controlled with near-IR light in a ligand-dependent CreERT/LoxP-reporter cell line derived from transgenic mice. These studies provide a chemical foundation that we expect will enable specific delivery of small molecules using cytocompatible, tissue penetrant near-IR light.
Introduction
Chemical reactions that proceed efficiently in complex biological settings underpin many biomedical methods. Photoremovable protecting groups, most notably those based on o-nitroaryl ring systems, that control or “cage” the activity of small molecules represent one such class of reactions. Since seminal studies over 35 years ago with photocaged ATP, these approaches have found application in diverse fields ranging from cell biology to materials science.1 Nevertheless, the general requirement of UV or blue light is a significant limitation due to associated toxicity and poor tissue penetration. By contrast, light between 650 and 900 nm, often referred to as the near-IR window, is cytocompatible and has significant tissue penetration.2 Moreover, the application of near-IR light has been broadly validated through numerous optical methods, including in vivo fluorescence imaging. For these reasons, expanding the repertoire of uncaging reactions to include approaches initiated by near-IR light is an important objective.
The substantial challenge in identifying near-IR photocages centers on translating modest photonic energy into bond cleavage and small molecule release. Key considerations include removal with readily achievable light intensity, straightforward synthesis, and biological compatibility. Most existing near-IR uncaging relies on two-photon excitation, which requires pulsed laser sources with limited release in only the small focal volume.3−5 A previous single-photon near-IR uncaging approach, suggested by Breslow, employs a photosensitizer and an electron rich olefin attached to the payload.6,7 Irradiation with red or near-IR light forms singlet oxygen, which cleaves the olefinic caging group. An inherent consequence of this design is an intact photosensitizer after uncaging with resulting sensitizer-dependent phototoxicity effects.7b Very recent advances from Lawrence and co-workers using contact-quenching induced scission of a Co–C bond are also quite promising.8 While these methods are proving quite useful, the full potential of near-IR uncaging has almost certainly not yet been realized. Essential to the development of this area is the identification of promising candidate photochemical reactions.
We speculated that the intrinsic photochemistry of the heptamethine cyanine fluorophore scaffold might serve admirably in this context. Heptamethine cyanines are used in numerous fluorescence-based biomedical applications, and find broad application as conjugated labels for in vivo imaging.9,10 One example, indocyanine green, is a clinical diagnostic agent with a long history of well-tolerated human use.11 Although useful, cyanine fluorophores are prone to light-dependent decomposition or photobleaching. The chemical basis of cyanine photobleaching has been shown to be a regioselective photooxidative polyene cleavage reaction through numerous mechanistic studies.12−21 While historically a liability, this reactivity is exploited here as the central component of a near-IR uncaging strategy.
Here we report a near-IR (690 nm) light-initiated uncaging approach based on the C4′-dialkylamine-substituted variant of the heptamethine cyanine fluorophore scaffold (1). The reaction sequence, shown in Figure 1, entails photooxidative cleavage of 1 at the C1–C1′ and C2′−C3′ bonds to afford 2 and 3, which both then hydrolyze (C4′−N) and cyclize to liberate the previously caged molecule. The increased hydrolytic susceptibility of 2 and 3, relative to 1, was predicted by considering that altered π-conjugation would increase the electrophilic reactivity of the key C4′−N bond (i.e., through increased iminium character). Here it is shown that these readily synthesized cyanine derivatives liberate phenol-containing small molecules upon irradiation with 690 nm light. This method is applied to alter gene expression through ligand-dependent genetic recombination and, in a separate demonstration, to release cytotoxic concentrations of a therapeutic agent.
Figure 1.
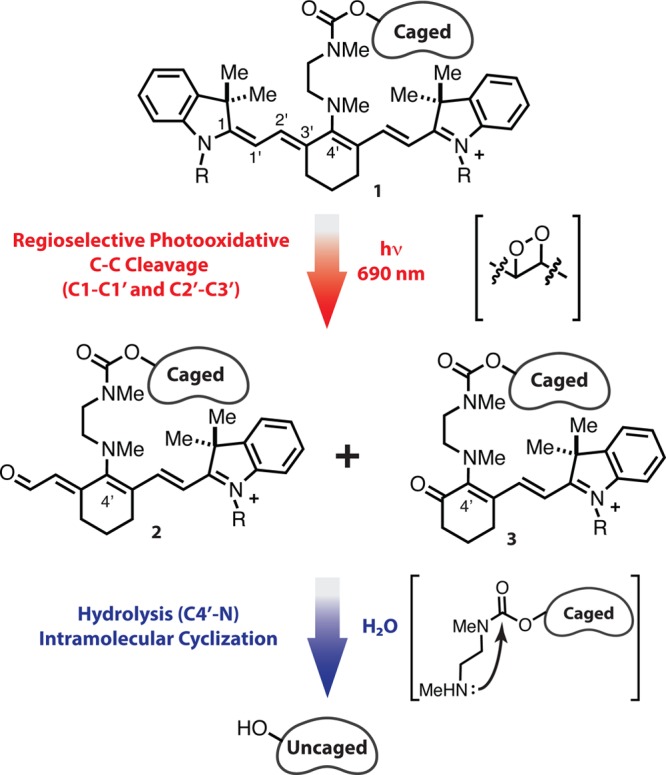
Uncaging reaction sequence of C4′-dialkylamine-substituted heptamethine cyanines.
Results and Discussion
Design and Synthesis
To test this approach, a series of dialkylamine-substituted cyanines were synthesized (Scheme 1).22N-Methyl carbamates 6–11 were designed to unveil phenols through sequential amine release/intramolecular cyclization (Figure 2A).23 Cyanines 6–10 serve as mechanistic probes through release of optically silent free phenol, the fluorophore 4-methylumbelliferone, and the absorbance reporter 4-nitrophenol. We also prepared 11, which is designed to release 4-hydroxycyclofen, a bioactive small molecule. The indoline nitrogen of the cyanine was substituted with either n-propyl or 4-butanesulfonate substituents, the latter being more well tolerated by biological systems.24 Compounds 6–10 were accessed through a protocol comprising initial conjugation of N,N′-dimethylethylenediamine to commercially available IR-780 (4) and IR-783 (5), followed by addition of a chloroformate to the unpurified diamine cyanine intermediate. The 4-hydroxycyclofen conjugate 11 was prepared by adding the corresponding mixed nitrophenyl carbonate to the same cyanine intermediate formed from N,N′-dimethylethylenediamine and 5. We also prepared N-methylethanolamine-substituted cyanines, 12 and 13, which are designed to release only N-methylethanolamine. Cyanine 12 proved useful to evaluate the amine release reaction by NMR and 13 served as a negative control in the cellular studies. These two molecules were prepared by heating 4 or 5 and N-methylethanolamine in MeCN or DMF, respectively. Of note, cyanines 6–11 exhibit optical properties similar to that of other N-substituted heptamethine cyanines, including useful quantum yield of fluorescent emission (Supporting Information Table S1).
Scheme 1. Synthesis of 6–13.

Figure 2.
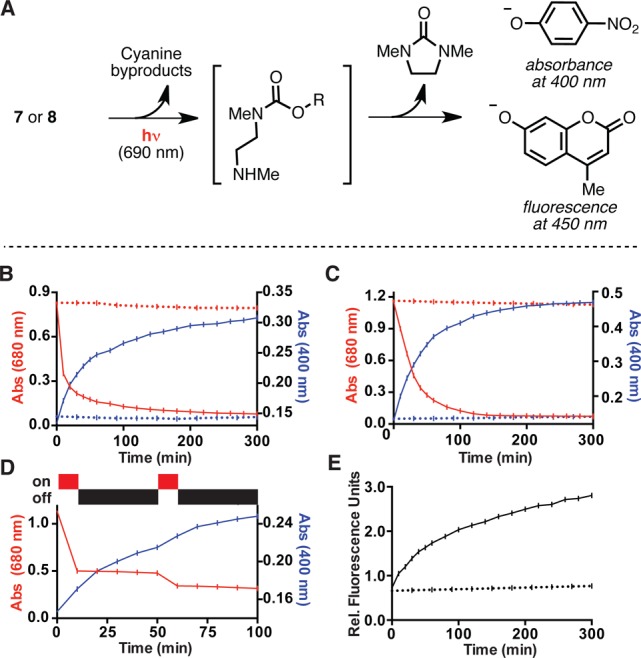
(A) General scheme for phenolate release. (B) Absorbance traces at 400 nm (blue) and 680 nm (red) with (solid line) or without (dashed line) 1 mW/cm2 690 nm irradiation of a 50 μM solution of 8 (50 mM HEPES, pH 7.5 with 5% DMSO). (C) As (B), but with 3 mW/cm2 690 nm irradiation in DMEM (buffered with 50 mM HEPES, pH 7.5, 0.1% DMSO) with 10% FBS. (D) As (B), but with intermittent irradiation. (E) Fluorescence traces (360 nm ex., 460 nm em.) with (solid line) or without (dashed line) 1 mW/cm2 690 nm irradiation of a 50 nM solution of 7 (50 mM HEPES, pH 7.5, 5% DMSO). All data shown are the average of three independent experiments with the standard deviation ≤5% in all cases.
Uncaging Analysis
With cyanines 6–11, the phenol release reaction was investigated under a range of conditions and concentrations. UV–vis analysis indicated that these compounds do not aggregate at micromolar concentrations under a variety of aqueous conditions, including those shown in Figure 2. Irradiation of a 50 μM solution of 8 in pH 7.5 HEPES buffer with 1 mW/cm2 690 (± 20) nm light from a commercial LED source provided an increase in the characteristic nitrophenolate absorbance (400 nm), with concomitant reduction of the cyanine absorbance (680 nm) (Figure 2B). Consistent with the accumulation of intermediate species that convert to the final phenolate product, the timing for disappearance of the cyanine absorbance signal and appearance of the 4-nitrophenolate signal diverge significantly, with half-lives (t1/2) of 8.5 and 40 min, respectively. Indicating that uncaging can occur in complex biological mixtures, similar absorbance profiles were seen in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS), albeit requiring a somewhat higher light intensity of 3 mW/cm2 to achieve a similar release rate (t1/2 for release = 34 min) (Figure 2C). To examine the effect of intermittent irradiation, compound 8 was irradiated for 10 min, maintained in the dark for 50 min, and irradiated for another 10 min (Figure 2D). The profile of the cyanine absorption decrease correlates directly with irradiation, while the nitrophenolate signal increases in interim periods. This result suggests that only irradiation sufficient to disrupt the cyanine absorption is required, and that the subsequent steps that culminate in release occur in the absence of irradiation. Fluorescence was used to examine the release reaction at lower concentrations. Irradiation of a 50 nM solution of 7, or the corresponding butanesulfonate-substituted 10, led to appearance of 4-methylumbelliferone fluorescence (Figure 2E and Supporting Information Figure S1). We also subjected control compounds 6 and 9, which release only phenol, to the conditions above. Verifying that the signals discussed above derive from release of the absorbance and fluorescence reporters, irradiation of 6 and 9 demonstrated that the byproducts of photolysis only minimally absorb at 400 nm and are not fluorescent at the excitation/emission wavelengths of 4-methylumbelliferone (Supporting Information Figure S2).
Our approach is based on the prediction that phenol release is preceded by cleavage of the C4′ secondary amine. Accordingly, we examined the C4′-amine release step in isolation with a simpler N-methylethanolamine-substituted cyanine, which cannot undergo a subsequent cyclization step. Continuous irradiation (690 nm, 5 mW/cm2) of a 1 mM solution of 12 at 690 nm for 24 h at 25 °C in 1:1 D2O:d4-methanol provided substantial conversion (66–70%) to N-methylethanolamine (14) (Scheme 2), as determined by 1H NMR. As seen in previous studies, oxindole 15 and aldehyde 16 were also generated.20,21 Compound 12 is stable in the dark under these conditions with greater than 95% remaining after 14 days, indicating the light dependence of this reactivity.
Scheme 2. Products Observed upon 690 nm Irradiation of 12.
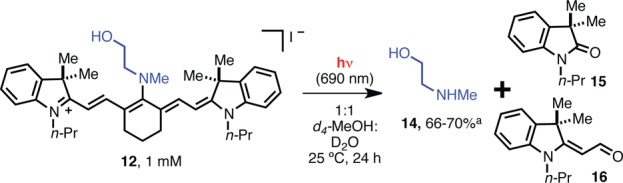
Yield is based on an NMR internal standard and was run in triplicate to provide the range shown.
The proposed release sequence is shown in Scheme 3 with compound 10. Photooxidative cleavage occurs through initial formation of dioxetane intermediates 17 and 18, which thermally decompose to form carbonyl products 19 and 21, respectively. Subsequent hydrolysis of both of these intermediates provides 25, which undergoes rapid intramolecular cyclization to provide the now uncaged phenolate 26. To provide support for this proposal, we have carried out a series of mass spectrometry experiments. Spectral signals for all intermediates shown in Scheme 3 are observed by high-resolution mass spectrometry (HRMS) of an irradiated solution of 10 in H2O. Notably, signals consistent with dioxetanes 17 and 18 are obtained, an observation that has also been made elsewhere.20,21 Singlet oxygen (1O2), generated through energy transfer from the triplet excited state of the cyanine, is generally assumed to be the major reactive oxygen species involved in the photooxidative cleavage,12−21 although light-generated superoxide (O2–) has been suggested to be a competing, albeit likely minor, O2 source.15,18,19 We note that methods, such as chromophore-assisted light inactivation (CALI), using the highly localized nature of singlet oxygen generated by photosensitization find broad use for a variety of biological applications.25
Scheme 3. Proposed Intermediates in Uncaging Sequence of 10.

We sought to provide further support for the notion that the photooxidation intermediates accumulate during irradiation and then undergo subsequent light-independent hydrolysis. Mass spectral ion counts of starting material 10, the indistinguishable dioxetanes, 17/18, the photooxidative cleavage products, 19 and 21, and released 4-methylumbelliferone, 26, were measured at various time points. As shown in Figure 3, 10 is consumed after 30 min of irradiation with concomitant increase in 17/18, 19, and 21. During the ensuing incubation at 37 °C without irradiation, these signals decrease as the signal corresponding to 26 increases. These results, as well as the mass spectral characterization of the direct products of the hydrolysis reaction, 23–25, are noteworthy because, unlike the photooxidative cleavage step, the hydrolysis step has only indirect precedent, and then not under neutral aqueous conditions.26,27 It is plausible that hydrolysis is particularly facile in this case as a consequence of the electron-withdrawing α and γ carbonyls. Further characterization of 23 and 24, for example through chromatographic isolation and spectral analysis, has not been possible to date.
Figure 3.
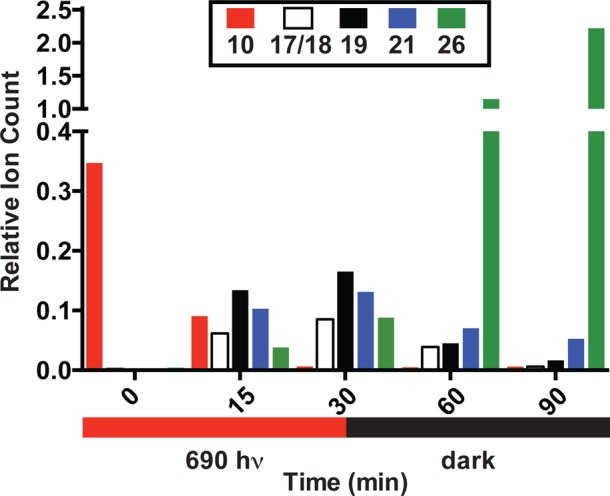
Relative spectral ion counts of 10, 17/18, 19, 21, and 26 over time upon exposing a 20 μM solution of 10 in H2O to 10 mW/cm2 690 nm light for 30 min at rt, and at 37 °C for an additional 60 min without irradiation. Ion counts were determined at each time point relative to an internal standard (phenylalanine).
Cellular Studies
We next examined if this technique could be used to control cellular function using near-IR light. The cyanine caged form of 4-hydroxycyclofen, 11, was used to illustrate two objectives where near-IR uncaging could be of significant benefit: release of a pharmacological agent and regulation of gene expression. In the case of the former, the goal is selective drug delivery to distinct tissue, and, in the latter, to obtain fine spatial control of expression in cellular subpopulations. 4-Hydroxycyclofen is a readily synthesized analog of 4-hydroxytamoxifen that exhibits similarly potent estrogen receptor antagonist/agonist activity.28 We first measured the yield of light-dependent release in biological media. A 10 μM solution of 11 in DMEM with 10% FBS was irradiated using a standard set of conditions also employed in the experiments below (690 nm, 10 mW/cm2, 30 min, 25 °C), followed by 37 °C incubation. Conversion exceeded 40% 1 h after the irradiation and approached 60% after 4 h, as determined by HPLC. The unirradiated control showed minimal release (less than 2%) under otherwise identical conditions (Supporting Information Figure S3).
Tamoxifen and its analogues can exhibit significant cytotoxicity, particularly against breast cancer cell lines that overexpress the estrogen receptor.29 With MCF-7 cells, irradiation of 11 recapitulated the IC50 of 4-hydroxycyclofen (9.4 μM for irradiated 11 vs 10 μM for 4-hydroxycyclofen, Figure 4). By contrast, unirradiated 11 exhibited an IC50 of 150 μM. We also established that the majority of observed cytotoxicity derived from released 4-hydroxycyclofen and not the cyanine irradiation process or byproducts from cyanine uncaging. Cyanine 13, which should only release N-methylethanolamine, exhibited an IC50 of >200 μM upon identical irradiation, and an IC50 of >200 μM in the absence of irradiation. This result is consistent with previous observations suggesting that heptamethine cyanine fluorophores are generally only weakly cytotoxic, even upon protracted irradiation.24 The findings presented here, as well as these previous observations, appear to suggest the products of the cyanine photodecomposition process, 19–24, are relatively well-tolerated, despite the presence of potentially reactive carbonyl groups. As expected with this low energy light, no phototoxicity was observed with irradiation alone.
Figure 4.
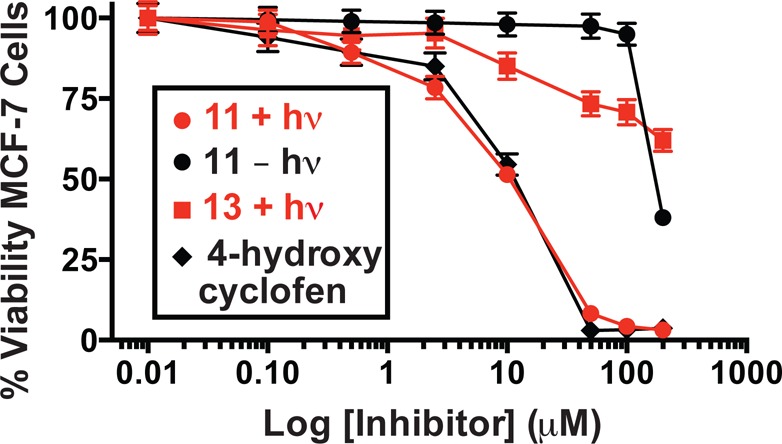
Light-dependent (690 nm, 30 min, 10 mW/cm2) cytotoxicity of 11 and 13 against MCF-7 cells. Error bars represent standard deviation of at least three independent experiments for the percent change in cell viability, relative to control, as measured by MTT assay.
Among various strategies that use light to achieve precise regulation of gene expression, a number of studies over the past 15 years have used UV light-mediated uncaging of small molecules in combination with inducible gene expression systems.30,31 The distinct advantages of near-IR light might prove beneficial as these techniques progress in complex biological settings and organismal contexts. To pursue this, we used a CreER/LoxP-reporter approach in which recombination is initiated by tamoxifen and its analogues, such as 4-hydroxycyclofen.28,32−34 Small molecule binding to the CreER chimera promotes nuclear translocation and site-specific recombination at LoxP sites. We employed a Rosa26-driven dual mT/mG reporter in transgenic mouse embryonic fibroblast (MEFs) cells.35 Here, a ligand-dependent Rosa26CreERT excises a LoxP flanked fluorescent tdTomato reporter (mT), enabling expression of a second downstream locus, Enhanced Green Fluorescent Protein (EGFP, mG), now under Rosa promoter control (Figure 5A).36 We chose this approach because it allows EGFP expression to be used as an easily assessed readout of 4-hydroxycyclofen uncaging.
Figure 5.
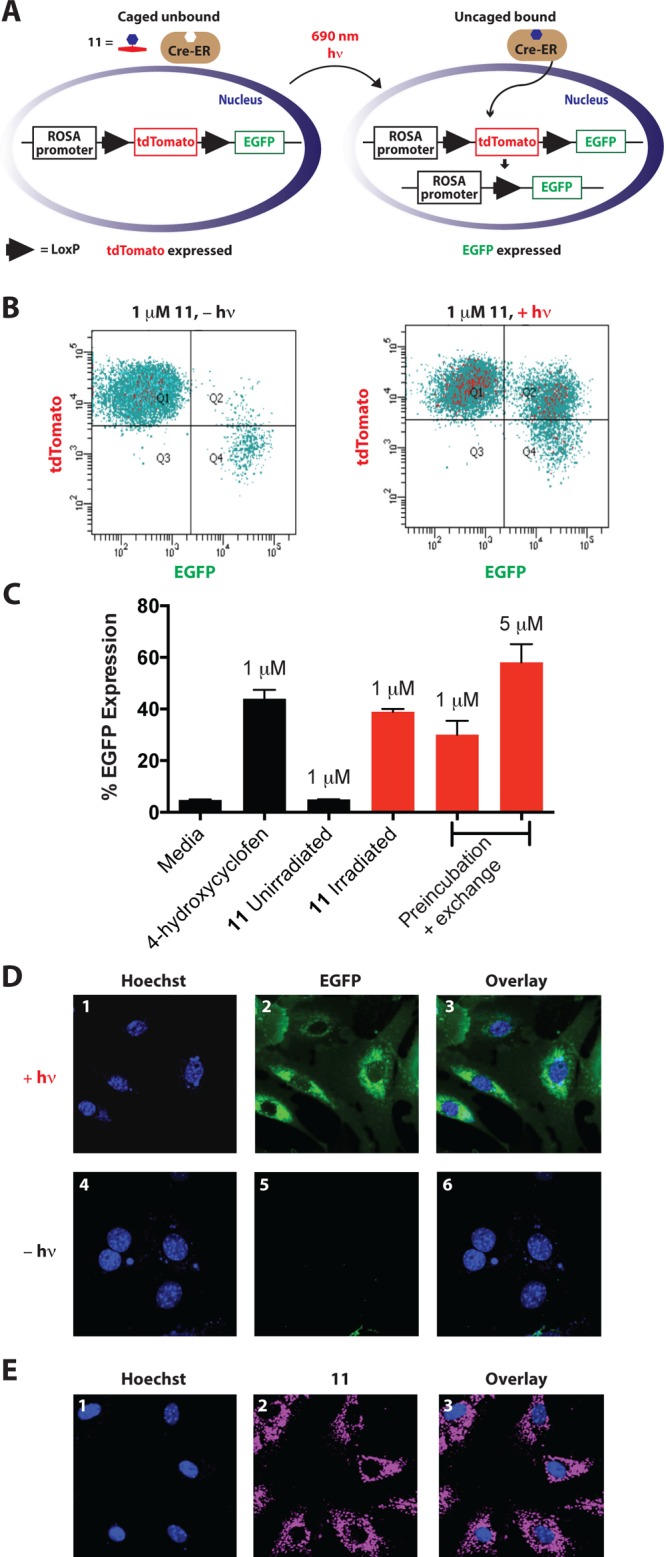
(A) Schematic of light-dependent EGFP expression. (B and C) Flow cytometry of light-dependent EGFP expression in MEFs obtained from Rosa26CreERT;Rosa26mT/mG embryos (n = 3). (D) Confocal microscopy images of live MEF cells treated with 11 (1 μM) and Hoechst 33342. The cells in panels 1–3 were irradiated, and the cells in 4–6 were not. From the left, emission (1 and 4) from Hoescht (nuclear staining), emission (2 and 5) from EGFP, and overlay (3 and 6) are shown. (E) Confocal microscopy images of live MEF cells treated with 11 (1 μM) and Hoechst 33342. From the left, emission (1) from Hoescht (nuclear staining), emission (2) from 11, and overlay (3) are shown.
Irradiation of 11 (1 μM) followed by 48 h incubation significantly increased the proportion of EGFP positive cells, as measured by flow cytometry (Figure 5B). The persistent tdTomato fluorescence after uncaging likely reflects the long half-life of this fluorescent protein.34 As shown in Figure 5C, EGFP expression was similar to the 1 μM 4-hydroxycyclofen control and, critically, only background expression levels were seen without irradiation. We also verified these EGFP expression patterns using confocal microscopy (Figure 5D). We sought to examine the cellular uptake of 11, noting that other disulfonated heptamethine cyanines show intracellular staining, presumably via an endosomal uptake pathway.24b,37,38 In doing so, we were able to take advantage of the fluorescent properties of the cyanine scaffold. After preincubating the cells for 2 h in DMEM and washing, significant intracellular signal derived from cyanine fluorescence was observed using confocal microscopy (Figure 5E). Similar punctate intracellular signal was also observed in another mammalian cell line (HeLa), and this colocalized with LysoTracker staining (Supporting Information Figure S4). Irradiating the preincubated/washed transgenic MEF cells led to meaningful EGFP expression, albeit to a slightly lower level than at the same concentration when media is maintained throughout (1 μM), suggesting that uncaging occurs, at least in part, within endosomal compartments. Indeed, greater EGFP expression was obtained after preincubation and media exchange with a higher concentration of 11 (5 μM). Further studies to define the uptake mechanism and to determine and optimize the location of uncaging are ongoing.
Conclusion
In summary, we have developed a near-IR light-initiated reaction sequence of dialkylamine-substituted heptamethine cyanines that can be used to uncage small molecules. This technique involves easily synthesized compounds, functions over a wide concentration range (nM to mM), and uses easily attainable light intensity. This method was applied both to inhibit cell survival and to alter gene expression. Mechanistically, cleavage from the cyanine scaffold occurs through two discrete stages. In the first stage, which is dependent on light, photooxidative C–C cleavage renders the C–N bond susceptible to hydrolysis by altering the structure, and consequently the reactivity, of the cyanine polyene. The second stage, light-independent C–N bond hydrolysis, provides a liberated secondary amine. Here the secondary amine undergoes an intramolecular cyclization to release a phenol, though this approach could likely also be applied to the direct uncaging of biologically active secondary amines. The cleavage kinetics achieved here are similar to existing single-photon photosensitizer-based uncaging methods, despite using much lower intensity light.7a Moreover, unlike these methods, the compounds used here display minimal intrinsic phototoxicity, which will allow for biological responses to be confidently assigned to drug release. We note that it is plausible that alteration of the cyanine scaffold will yield significant improvements, and such efforts are underway.
The findings presented here represent key steps toward a general near-IR photocaging method. The broad use of heptamethine cyanines, particularly for in vivo fluorescence imaging, and their biocompatibility suggest a number of avenues for future application. More generally, we have shown that chemistry associated with the photochemical decomposition of long wavelength fluorophores can serve as inspiration for the discovery of biologically useful reactions. Our ongoing efforts are focused on advanced uncaging protocols and the development of near-IR light-targeted drug delivery approaches.
Acknowledgments
We thank members of the Chemical Biology Laboratory for helpful comments and suggestions. We thank Dr. Joseph Barchi for NMR assistance and Dr. James Kelley for mass spectrometric analysis. We also gratefully acknowledge Dr. Hisataka Kobayashi for advice and assistance with the LED light source, Dr. Stephen Lockett, Ms. Kim Peifley, and the Optical Microscopy and Analysis Laboratory (Advanced Technology Program, Frederick National Laboratory for Cancer Research) for their invaluable expertise and assistance in obtaining confocal fluorescence images, and Ms. Kathleen Noer, Ms. Roberta Matthai, and the Frederick CCR Flow Cytometry Core (Cancer and Inflammation Program, NCI-Frederick) for help with flow cytometry analysis. This work was supported by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, and the National Cancer Institute, National Institutes of Health.
Supporting Information Available
Figures providing additional data, experimental details, and copies of 1H and 13C NMR spectra of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Lee H. M.; Larson D. R.; Lawrence D. S. ACS Chem. Biol. 2009, 4, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Deiters A. ChemBioChem 2010, 11, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Klan P.; Solomek T.; Bochet C. G.; Blanc A.; Givens R.; Rubina M.; Popik V.; Kostikov A.; Wirz J. Chem. Rev. 2013, 113, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ellis-Davies G. C. R. Nat. Methods 2007, 4, 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissleder R. Nat. Biotechnol. 2001, 19, 316. [DOI] [PubMed] [Google Scholar]
- Warther D.; Gug S.; Specht A.; Bolze F.; Nicoud J. F.; Mourot A.; Goeldner M. Bioorg. Med. Chem. 2010, 18, 7753. [DOI] [PubMed] [Google Scholar]
- Dore T. D.; Wilson H. C.. Chromophores for the Delivery of Bioactive Molecules with Two-Photon Excitation. In Photosensitive Molecules for Controlling Biological Function; Chambers J. J., Kramer R. H., Eds.; Springer Science: New York, 2011; Vol. 55, pp 57–92. [Google Scholar]
- Yang Y. M.; Shao Q.; Deng R. R.; Wang C.; Teng X.; Cheng K.; Cheng Z.; Huang L.; Liu Z.; Liu X. G.; Xing B. G. Angew. Chem., Int. Ed. 2012, 51, 3125. [DOI] [PubMed] [Google Scholar]
- a Ruebner A.; Yang Z. W.; Leung D.; Breslow R. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 14692. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rotaru A.; Mokhir A. Angew. Chem., Int. Ed. 2007, 46, 6180. [DOI] [PubMed] [Google Scholar]; c Jiang M. Y.; Dolphin D. J. Am. Chem. Soc. 2008, 130, 4236. [DOI] [PubMed] [Google Scholar]
- a Bio M.; Nkepang G.; You Y. Chem. Commun. 2012, 48, 6517. [DOI] [PubMed] [Google Scholar]; b Bio M.; Rajaputra P.; Nkepang G.; Awuah S. G.; Hossion A. M. L.; You Y. J. Med. Chem. 2013, 56, 3936. [DOI] [PubMed] [Google Scholar]
- a Shell T. A.; Shell J. R.; Rodgers Z. L.; Lawrence D. S. Angew. Chem., Int. Ed. 2014, 53, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Smith W. J.; Oien N. P.; Hughes R. M.; Marvin C. M.; Rodgers Z. L.; Lee J.; Lawrence D. S. Angew. Chem., Int. Ed. 2014, 10.1002/anie.201406216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q.; Juette M. F.; Jockusch S.; Wasserman M. R.; Zhou Z.; Altman R. B.; Blanchard S. C. Chem. Soc. Rev. 2014, 43, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangioni J. V. Curr. Opin. Chem. Biol. 2003, 7, 626. [DOI] [PubMed] [Google Scholar]
- Alford R.; Simpson H. M.; Duberman J.; Hill G. C.; Ogawa M.; Regino C.; Kobayashi H.; Choyke P. L. Mol. Imaging 2009, 8, 341. [PubMed] [Google Scholar]
- Henary M.; Mojzych M.. Stability and Reactivity of Polymethine Dyes. In Solution In Heterocyclic Polymethine Dyes: Synthesis, Properties and Applications; Strekowski L., Ed.; Topics in Heterocyclic Chemistry 14; Springer-Verlag: Berlin, 2008; pp 221–238. [Google Scholar]
- Byers G. W.; Gross S.; Henrichs P. M. Photochem. Photobiol. 1976, 23, 37. [DOI] [PubMed] [Google Scholar]
- Lepaja S.; Strub H.; Lougnot D. J. Z. Naturforsch., A 1983, 38, 56. [Google Scholar]
- Chen P.; Li J.; Qian Z. G.; Zheng D. S.; Okasaki T.; Hayami M. Dyes Pigments 1998, 37, 213. [Google Scholar]
- Renikuntla B. R.; Rose H. C.; Eldo J.; Waggoner A. S.; Armitage B. A. Org. Lett. 2004, 6, 909. [DOI] [PubMed] [Google Scholar]
- Toutchkine A.; Kraynov V.; Hahn K. J. Am. Chem. Soc. 2003, 125, 4132. [DOI] [PubMed] [Google Scholar]
- Chen X. Y.; Peng X. J.; Cui A. J.; Wang B. S.; Wang L.; Zhang R. J. Photochem. Photobiol., A 2006, 181, 79. [Google Scholar]
- Toutchkine A.; Nguyen D. V.; Hahn K. M. Org. Lett. 2007, 9, 2775. [DOI] [PubMed] [Google Scholar]
- Engel E.; Schraml R.; Maisch T.; Kobuch K.; Koenig B.; Szeimies R. M.; Hillenkamp J.; Baumler W.; Vasold R. Invest. Ophthalmol. Visual Sci. 2008, 49, 1777. [DOI] [PubMed] [Google Scholar]
- Samanta A.; Vendrell M.; Das R.; Chang Y. T. Chem. Commun. 2010, 46, 7406. [DOI] [PubMed] [Google Scholar]
- Strekowski L.; Lipowska M.; Patonay G. J. Org. Chem. 1992, 57, 4578. [Google Scholar]
- Saari W. S.; Schwering J. E.; Lyle P. A.; Smith S. J.; Engelhardt E. L. J. Med. Chem. 1990, 33, 97. [DOI] [PubMed] [Google Scholar]
- a Kassab K. J. Photochem. Photobiol., B 2002, 68, 15. [DOI] [PubMed] [Google Scholar]; b Conceição D. S.; Ferreira D. P.; Ferreira L. F. V. Int. J. Mol. Sci. 2013, 14, 18557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K.; Rajfur Z.; Vitriol E.; Hahn K. Trends Cell Biol. 2008, 18, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voitenko Z.; Mazieres M. R.; Sanchez M.; Wolf J. G. Tetrahedron 2001, 57, 1059. [Google Scholar]
- Bacher E.; Gompper R.; Mertz R.; Wagner H. Z. Naturforsch., B 1989, 44, 839. [Google Scholar]
- Sinha D. K.; Neveu P.; Gagey N.; Aujard I.; Benbrahim-Bouzidi C.; Le Saux T.; Rampon C.; Gauron C.; Goetz B.; Dubruille S.; Baaden M.; Volovitch M.; Bensimon D.; Vriz S.; Jullien L. ChemBioChem 2010, 11, 653. [DOI] [PubMed] [Google Scholar]
- Desta Z.; Ward B. A.; Soukhova N. V.; Flockhart D. A. J. Pharmacol. Exp. Ther. 2004, 310, 1062. [DOI] [PubMed] [Google Scholar]
- a Deiters A. Curr. Opin. Chem. Biol. 2009, 13, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Brieke C.; Rohrbach F.; Gottschalk A.; Mayer G.; Heckel A. Angew. Chem., Int. Ed. 2012, 51, 8446. [DOI] [PubMed] [Google Scholar]; c Liu Q. Y.; Deiters A. Acc. Chem. Res. 2014, 47, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cruz F. G.; Koh J. T.; Link K. H. J. Am. Chem. Soc. 2000, 122, 8777. [Google Scholar]; b Lin W. Y.; Albanese C.; Pestell R. G.; Lawrence D. S. Chem. Biol. 2002, 9, 1347. [DOI] [PubMed] [Google Scholar]
- Link K. H.; Shi Y. H.; Koh J. T. J. Am. Chem. Soc. 2005, 127, 13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Agasti S. S.; Vinegoni C.; Waterman P.; DePinho R. A.; Weissleder R. Bioconjugate Chem. 2012, 23, 1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inlay M. A.; Choe V.; Bharathi S.; Fernhoff N. B.; Baker J. R.; Weissman I. L.; Choi S. K. Chem. Commun. 2013, 49, 4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar M. D.; Tasic B.; Miyamichi K.; Li L.; Luo L. Genesis 2007, 45, 593. [DOI] [PubMed] [Google Scholar]
- Badea T. C.; Wang Y.; Nathans J. J. Neurosci. 2003, 23, 2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M. C.; Freire V.; Asumendi A.; Araiz J.; Herrera I.; Castiella G.; Corcostegui I.; Corcostegui G. Invest. Ophthalmol. Visual Sci. 2010, 51, 6018. [DOI] [PubMed] [Google Scholar]
- Yang X.; Shi C.; Tong R.; Qian W.; Zhau H. E.; Wang R.; Zhu G.; Cheng J.; Yang V. W.; Cheng T.; Henary M.; Strekowski L.; Chung L. W. Clin. Cancer. Res. 2010, 16, 2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.