

Abstract
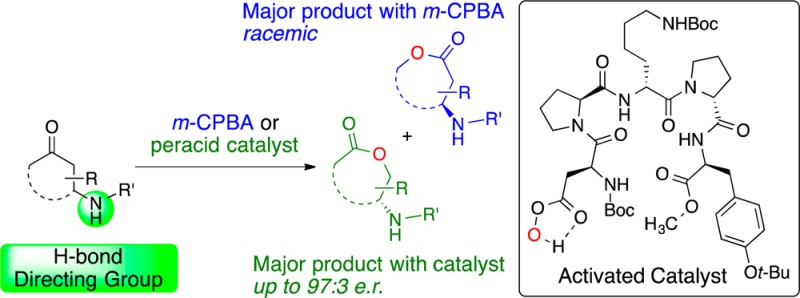
We report a peptide-based catalyst that can strongly influence the regio- and enantioselectivity of the Baeyer–Villiger (BV) oxidation of cyclic ketones bearing amide, urea, or sulfonamide functional groups. Both types of selectivity are thought to arise from a catalyst–substrate hydrogen-bonding interaction. Furthermore, in selected cases, the reactions exhibit the hallmarks of parallel kinetic resolution. The capacity to use catalysis to select between BV products during an asymmetric process may have broad utility for both the synthesis and diversification of complex molecules, including natural products.
The oxidation of ketones to esters, known as the Baeyer–Villiger (BV) reaction,1 has long been regarded as a powerful tool for organic synthesis. As such, there have been many attempts to render the process catalytic and enantioselective, which have seen the use of both enzymes2 and small-molecule catalysts.3 However, despite this storied history, the reaction still presents numerous challenges, such as the issue of regioselectivity, which can be difficult to predict, let alone control. For cyclic ketones, the effects of ring strain are an added complication; if the reaction forms a more strained lactone from a less strained ketone, then overcoming the energy barrier to C–C bond migration can be difficult without substantial activation. Finally, if complex molecule substrates are to be used, then the catalyst must also be sufficiently chemoselective to tolerate functionality in addition to the reacting ketone. Ideal catalysts must therefore be able to oxidize relatively unstrained ketones to deliver products with both regio- and enantioselectivity, while also tolerating the presence of additional functional groups.
We have been studying a catalytic cycle for oxidation in which the side-chain of a peptide-embedded aspartic acid shuttles between the carboxylic acid and the corresponding peracid in situ.4 This system, which has proven particularly effective for electrophilic oxidation, is well suited to a variety of substrate and reaction types, such as the oxidation of allylic carbamate 1 to its epoxide (2, Figure 1a). The selectivity of the reaction is controlled by the peptide sequence, which may be tuned for the regio- and enantioselective oxidation of various substrates.5 Notably, this approach may also be applied to cases where the reversal of a substrate’s intrinsic reactivity is desired. For example, indole 3 has a strong proclivity to form diastereomer 4 when treated with various electrophilic oxidants; yet, it is converted to 5 in high yield in the presence of peptide catalyst 6 (Figure 1b).6 In the context of the BV oxidation, a peracid functions as a nucleophile, not as an electrophile (Figure 1c). We thus wondered whether the aspartic acid system might be portable to the BV reaction, meeting the analogous requirements for selective oxidation in functional group-rich molecular environments, despite the fundamentally different role of the peracid intermediate.
Figure 1.
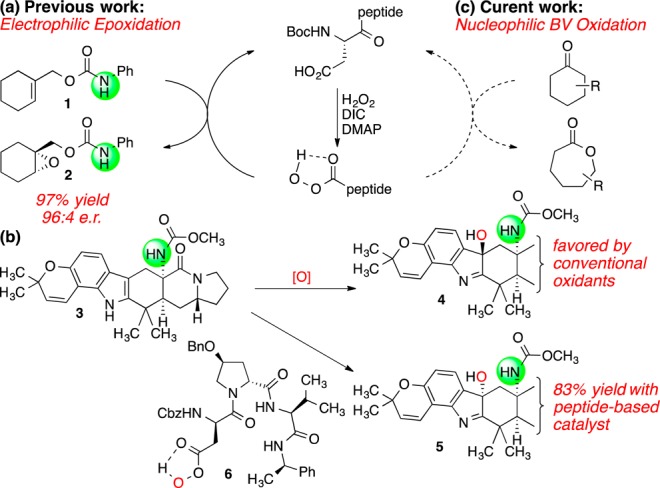
(a) Previous electrophilic epoxidation of alkenes. (b) Diastereoselective oxidation of an advanced intermediate. (c) Proposed nucleophilic oxidation of ketones. Cbz = benzyloxycarbonyl.
In earlier work, we demonstrated the viability of the acid/peracid catalytic cycle for the BV reaction.7 However, despite exploration of many peptide sequences that had given high enantioselectivity for epoxidation reactions in the intervening years, we were unable to achieve comparable levels of selectivity for the fundamentally different BV reaction. We therefore turned our attention to combinatorial screening.8
For our initial screen, we synthesized an on-bead library of catalysts, as depicted in Figure 2a. We evaluated each catalyst in the kinetic resolution (KR)9 of ketone 7 via oxidation to lactone 8 (Figure 2b), due to this reaction’s facile analysis. After screening 245 beads, we identified several “hit” catalysts that exhibited a modicum of selectivity (e.g., catalyst 9a, krel = 1.7; see SI). Intriguingly, the “Pro-dXaa-Pro” motif was conserved in the i + 1 to i + 3 positions, suggesting that this sequence might be especially suited to the BV reaction manifold.
Figure 2.
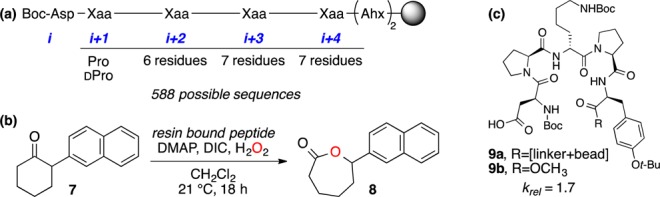
Combinatorial screening of BV catalysts. (a) Library composition. (b) Test reaction for on-bead screening. (c) Best performing catalyst from screen.
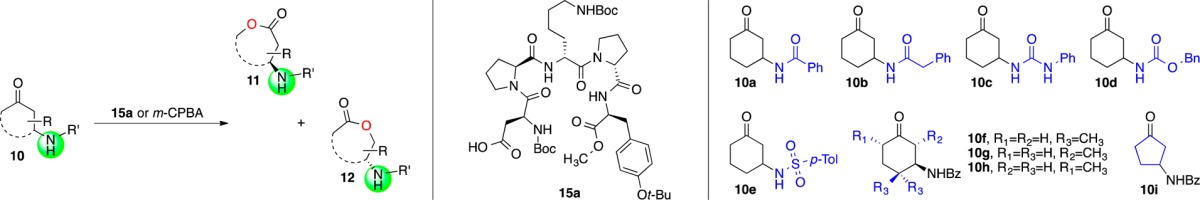
While the initial threshold of selectivity for these hits was low, our experience dictated that stronger catalyst control could be achieved if the substrates possessed additional functional groups that could engage the peptide through H-bonding.10 We thus turned our attention to the application of catalyst 9b (the soluble methyl ester analog of 9a) to the BV oxidation of 10a (Figure 3a). Ketone 10a presents an excellent test substrate in that both isomeric lactones 11a and 12a are formed upon exposure to 3-chloroperoxybenzoic acid (m-CPBA). When this stoichiometric oxidant is used, lactone 11a dominates, in a ratio of 7.9 to 1. However, with catalyst 9b (10 mol %), we observed notable catalyst control, including reversal of intrinsic migratory aptitude, with lactone 12a being favored 3.4 to 1 at 36% conversion. Moreover, all three species (unreacted substrate and both lactones) were enantioenriched (Table 1, entry 1), with 12a being formed in 91:9 enantiomeric ratio (er).
Figure 3.
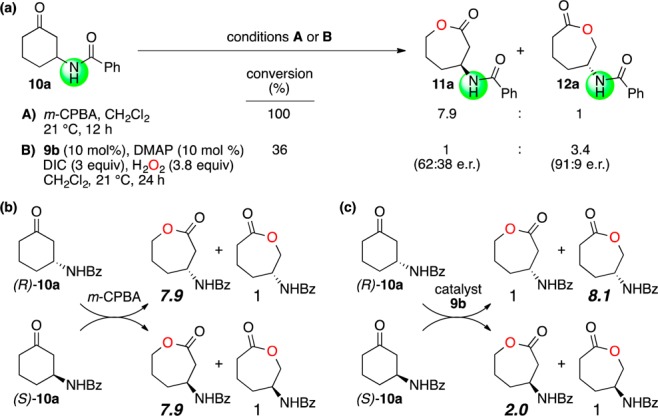
(a) BV oxidation of 10a. (b) Tracking the regiochemistry of O atom insertion for each enantiomer of 10a with m-CPBA, and (c) catalyst 9b. See SI for absolute stereochemical assignment.
Table 1. Effect of Peptide Sequence on Selectivitya.
| erc |
||||||
|---|---|---|---|---|---|---|
| entry | catalyst | conv. (%)b | ratio of 11:12c | 10a | 11a | 12a |
| 1 | 9b | 36 | 1:3.4 | 66:34 | 62:38 | 91:9 |
| 2 | 13 | 12 | 1.6:1 | 51:49 | 58:42 | 72:28 |
| 3 | 14 | 31 | 1:3.7 | 64:36 | 61:39 | 92:8 |
| 4 | 15a | 32 | 1:3.1 | 64:36 | 67:33 | 94:6 |
| 5 | 15b | 27 | 1:2.8 | 61:39 | 64:36 | 94:6 |
| 6 | 15c | 19 | 1:1.6 | 55:45 | 61:39 | 90:10 |
Reaction conditions depicted in Figure 3, conditions B.
Conversion = ee10/(ee10 + eepdt), where eepdt is the aggregate enantiomeric excess (ee) of both lactone products.
Determined by HPLC.
This reaction exhibits characteristics of a parallel kinetic resolution (PKR),11 in that each enantiomer of substrate is converted preferentially to a different product. However, unlike an archetypical PKR, the enantiomers exhibit an ∼5-fold rate difference.12 This situation creates the opportunity to track the degree to which catalyst-controlled reversal of intrinsic migratory aptitude occurs. Of course, with the achiral oxidant, each enantiomer of ketone 10a delivers an equivalent ratio of lactones (11a/12a, 7.9:1; Figure 3b). Yet, with the chiral catalyst 9b, one sees that the regioselectivity is more pronounced for (R)-10a (11a/12a, 1:8.1; Figure 3c) than for (S)-10a, which forms a less selective mixture of lactone isomers (11a/12a, 2.0:1). Thus, catalyst 9b exhibits the hallmarks of a stereochemically “matched” case for reaction with (R)-10a; catalyst 9b may be considered “mismatched” in reactions with (S)-10a.13
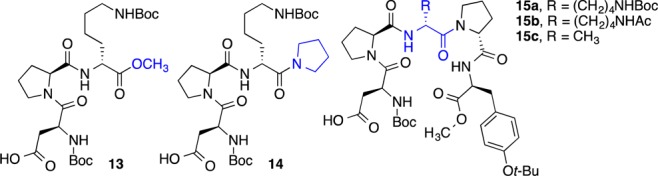
Fascinated by this catalyst-controlled regioselectivity, as well as the encouraging enantioselectivity, we wished to explore the role of the peptide sequence, with an eye toward optimization and identification of mechanistic roles for the different regions of the catalyst (Chart 1). One hypothesis emerging from the initial on-bead catalyst screen was that the Pro-dXaa-Pro motif is a crucial sequence element. Indeed, truncated peptide 13, which lacked the i + 3 Pro, led to a plummet in both conversion and selectivity, with “normal” lactone 11a being favored (11a/12a, 1.6:1; residual 10a nearly racemic; Table 1, entry 2). Conversely, when the two C-terminal residues were replaced with a simple pyrrolidine (14), the conversion and selectivity for the “abnormal” lactone were restored to levels comparable to catalyst 9b (11a/12a, 1:3.7; 12a with 92:8 er; Table 1, entry 3). Given the performance of catalyst 14, the role of the i + 4 position is unclear, but a brief survey revealed dPro-Tyr(t-Bu)-OCH3 as an optimal C-terminal sequence (15a), leading to lactone 12a with as high as 94:6 er (11a/12a, 1:3.1; Table 1, entry 4). The role of the dLys(Boc), where Boc = tert-butyloxycarbonyl, side-chain in the i + 2 position is not definitively clear. Replacement of the Boc carbamate with an acetamide (15b) produces a minimal change in behavior (11a/12a, 1:2.8; 12a with 94:6 er; Table 1, entry 5). However, substitution with dAla in this position (15c) leads to a drop in both conversion and selectivity (11a/12a, 1:1.6; 12a with 90:10 er; Table 1, entry 6). We thus chose 15a as our catalyst for further study.
Chart 1. Sample Peptides from Sequence Screen.
A brief survey of reaction solvents showed halogenated solvents to be superior, with chloroform giving the best results in terms of both conversion and selectivity (11a/12a, 1:3.7; 12a with 96:4 er; Table 2, entry 1). We next wished to test our hypothesis that the benzamide was acting mainly as an H-bonding handle, but that modifications that preserved this capability should be tolerated. Indeed, we find that when a CH2 is inserted between the phenyl ring and the amide, as in 10b, the selectivity not only remains but in fact improves, with 12b forming in as high as 97:3 er at similar conversion (11b/12b, 1:4.5; Table 2, entry 2). Intriguingly, when the CH2 is replaced with a NH (urea 10c), the reaction shows a pronounced increase in conversion, seeming to reflect an accelerated reaction rate for the “mismatched” enantiomer. Importantly, the rate acceleration in the mismatched series does not come at the expense of enantioselectivity. Instead, this reaction is more similar to an ideal PKR, with 11c and 12c forming in 93:7 and 92:8 er, respectively (11c/12c, 1:1.8; Table 2, entry 3). On the other hand, the reaction performs poorly with substrate 10d, wherein the directing group is a carbamate. Decreases in both conversion and selectivity (11d/12d, 1:3.1; 12d with 95:5 er; Table 2, entry 4) are observed in this case, possibly indicating that, in contrast to 10c, the introduction of an oxygen, which is a nominal H-bond acceptor, creates Coulombic repulsion that weakens the catalyst–substrate interaction. Finally, tosylamide 10e is also a competent substrate, exhibiting similar trends in both enantio- and regioselectivity (11e/12e, 1:1.8; 12e with 91:9 er; Table 2, entry 5). Taken together, these data suggest an H-bonding interaction between the NH of the substrate and an as yet unidentified H-bond acceptor on the catalyst.
Table 2. Effect of Substituents and Ring Size on Selectivitya.
| reversal
of regioselectivity: regioisomer ratio
(11:12)b |
erb |
|||||||
|---|---|---|---|---|---|---|---|---|
| entry | substrate | conv. (%)c | m-CPBAd | (15a, total) | 15a, matchede | 10 | 11 | 12 |
| 1 | 10a | 41 | 7.9:1 | (1:3.7) | 1:12 | 73:27 | 70:30 | 96:4 |
| 2 | 10b | 45 | 2.5:1 | (1:4.5) | 1:28 | 76:24 | 85:15 | 97:3 |
| 3 | 10c | 64 | 1.4:1 | (1:1.8) | 1:24 | 71:29 | 93:7 | 92:8 |
| 4 | 10d | 20f | 3.0:1 | (1:3.1) | 1:8.2 | NDg | 64:36 | 95:5 |
| 5 | 10e | 20 | 6.1:1h | (1:1.8) | 1:4.4 | 56:44 | 64:36 | 91:9 |
| 6 | 10f | 44 | 5.2:1 | (1:7.3) | 1:23 | 80:20 | 71:29 | 93:7 |
| 7i | 10g | 51 | 1:2.3 | (1:38) | <1:100 | 85:15 | 78:22 | 85:15 |
| 8 | 10h | 52f | >100:1 | (5.2:1) | 2.3:1 | 54:46 | 56:44 | 99:1 |
| 9 | 10i | 44f | 3.3:1 | (1.2:1) | 1:4.3 | 53:47 | 83:17 | 88:12 |
Conditions: 15a (10 mol %), 4-dimethylaminopyridine (DMAP, 10 mol %), N,N′-diisopropylcarbodiimide (DIC, 3.0 equiv), H2O2 (3.8 equiv), CHCl3 (0.1 M), 21 °C, 24 h.
Determined by HPLC.
See Table 1, footnote b.
m-CPBA (1.1 equiv), CHCl3 (0.1 M), 21 °C, 12 h.
Product ratio for the enantiomer that reacts more rapidly with the catalyst.
Conversion approximated from comparison of HPLC peak integrations of the products to the substrate.
ND = not determined.
m-CPBA (2.0 equiv) used.
DIC (1.5 equiv), H2O2 (1.9 equiv) used.
Having evaluated the catalyst’s performance with a variety of H-bonding handles, we next wished to investigate the effect of modification to the ring. The catalyst tolerates substitution adjacent to the directing group, with 4,4-dimethyl substrate 10f giving selectivity comparable to parent benzamide 10a (11f/12f, 1:7.3; 12f with 93:7 er; Table 2, entry 6). The introduction of a 2-methyl group adjacent to the amide (10g) creates a slight preference for the formation of lactone 12g when m-CPBA is used (11g/12g, 1:2.3; Table 2, entry 7). This is strongly amplified by catalyst 15a, leading to an almost undetectable level of 11g (11g/12g, 1:38; Table 2, entry 7). This reaction, which is now similar to a classical KR, proceeds with good enantioselectivity, corresponding to a krel of 11. Conversely, isomeric substrate 10h exhibits a strong preference for lactone 11h (11h/12h, >100:1 with m-CPBA; Table 2, entry 8). Even so, catalyst 15a forms an appreciable quantity of 12h in almost enantiopure form (11h/12h, 5.2:1; 12h with 99:1 er). Finally, we find that cyclopentanone 10i also undergoes clean oxidation with “PKR-like” behavior, in which products 11i and 12i are formed in near equal quantities, with similar levels of enantioenrichment (11i/12i, 1.2:1; 12i with 88:12 er, Table 2, entry 9).
Since the catalyst mediates a range of substrate-dependent reaction paradigms, from classical KR to PKR, we selected three substrates that exemplify distinct modes of reactivity and subjected them to minimally optimized BV conditions in order to demonstrate the isolation of enantioenriched products in each of these scenarios. Ketone 10g, which undergoes a KR with catalyst 15a, can be oxidized at −8 °C, to give 38% yield of 12g in 93:7 er and 50% recovery of 10g with 86:14 er (krel of 27, Scheme 1a). The oxidation of 10b, which represents an “in between” form of KR/PKR since each substrate enantiomer leads to a different regioisomer but at different rates, allows for the isolation of “m-CPBA-disfavored” lactone 12b in 24% yield (>58:1, 12b/11b) with 97:3 er (Scheme 1b). Finally, ketone 10c, which leads to a more “PKR-like” reaction, may be converted to lactones 11c and 12c in 56% combined yield, with 96:4 and 92:8 er respectively (Scheme 1c).
Scheme 1.

In summary, we have described catalysts that influence both the regio- and enantioselectivity of BV reactions involving functionalized ketones. The catalysts show the capacity to overturn the regioselectivity exhibited by m-CPBA, suggesting more generally that intrinsically disfavored BV products may be accessed with peptide catalysis. Furthermore, the mechanism underlying the selectivity, which likely stems from interactions between the peptide and H-bonding functionality of the substrate, constitutes a distinct approach to selective BV oxidation, relative to known small molecule catalysts. Further interrogation of this mechanism as well as applications to both enantioselective synthesis and natural product modification are ongoing objectives in our laboratory.
Acknowledgments
This work was supported by National Institutes of Health (NIH R01-GM096403). We also thank Dr. Brandon Q. Mercado for X-ray crystallographic analyses.
Supporting Information Available
Additional figures, experimental details and characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Baeyer A.; Villiger V. Ber. Dtsch. Chem. Ges. 1899, 32, 3625. [Google Scholar]; b Krow G. R. Org. React. 1993, 43, 251. [Google Scholar]; c Ito K. Comprehensive Chirality; Carreira E. M., Yamamoto H.; Elsevier: Amsterdam, 201251 [Google Scholar]; d Uyanik M.; Ishihara K. ACS Catal. 2013, 3, 513. [Google Scholar]
- Leisch H.; Morley K.; Lau P. C. Chem. Rev. 2011, 111, 4165. [DOI] [PubMed] [Google Scholar]
- a Bolm C.; Schlingloff G.; Weickhardt K. Angew. Chem., Int. Ed. 1994, 33, 1848. [Google Scholar]; b Bolm C.; Schlingloff G. J. Chem. Soc., Chem. Commun. 1995, 1247. [Google Scholar]; c Paneghetti C.; Gavagnin R.; Pinna F.; Strukul G. Organometallics 1999, 18, 5057. [Google Scholar]; d Murahashi S.; Ono S.; Imada Y. Angew. Chem., Int. Ed. 2002, 41, 2366. [DOI] [PubMed] [Google Scholar]; e Watanabe A.; Uchida T.; Irie R.; Katsuki T. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5737. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Xu S.; Wang Z.; Zhang X.; Zhang X.; Ding K. Angew. Chem., Int. Ed. 2008, 47, 2840. [DOI] [PubMed] [Google Scholar]; g Xu S.; Wang Z.; Li Y.; Zhang X.; Wang H.; Ding K. Chem.—Eur. J. 2010, 16, 3021. [DOI] [PubMed] [Google Scholar]; h Zhou L.; Liu X.; Ji J.; Zhang Y.; Hu X.; Lin L.; Feng X. J. Am. Chem. Soc. 2012, 134, 17023. [DOI] [PubMed] [Google Scholar]
- Peris G.; Jakobsche C. E.; Miller S. J. J. Am. Chem. Soc. 2007, 129, 8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kolundzic F.; Noshi M. N.; Tjandra M.; Movassaghi M.; Miller S. J. J. Am. Chem. Soc. 2011, 133, 9104. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lichtor P. A.; Miller S. J. Nat. Chem. 2012, 4, 990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado-Marin E. V.; Garcia-Reynaga P.; Romminger S.; Pimenta E. F.; Romney D. K.; Lodewyk M. W.; Williams D. E.; Andersen R. J.; Miller S. J.; Tantillo D. J.; Berlinck R. G. S.; Sarpong R. Nature 2014, 509, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris G.; Miller S. J. Org. Lett. 2008, 10, 3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtor P. A.; Miller S. J. ACS Comb. Sci. 2011, 13, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan H. B.; Fiaud J. C. In Topics in Stereochemistry; John Wiley & Sons, Inc.: New York, 1988; p 249. [Google Scholar]
- Miller S. J.; Copeland G. T.; Papaioannou N.; Horstmann T. E.; Ruel E. M. J. Am. Chem. Soc. 1998, 120, 1629. [Google Scholar]
- a Vedejs E.; Chen X. J. Am. Chem. Soc. 1997, 119, 2584. [Google Scholar]; b Dehli J. R.; Gotor V. Chem. Soc. Rev. 2002, 31, 365. [DOI] [PubMed] [Google Scholar]
- As conversion increases, the er of starting material and “mismatched” lactone should increase, whereas the er of the “matched” lactone should decrease. The ratio of matched to mismatched lactone should also decrease (see SI for details).
- The situation is reminiscent of double diastereoselection, as codified previously, but pertains to regio- and enantioselectivity in these cases. See:Masamune S.; Choy W.; Petersen J. S.; Sita L. R. Angew. Chem., Int. Ed. 1985, 24, 1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.