

Abstract

The concise, enantioselective total syntheses of (−)-citrinadin A and (+)-citrinadin B in a total of only 20 and 21 steps, respectively, from commercially available starting materials are described. Our strategy, which minimizes refunctionalization and protection/deprotection operations, features the highly diastereoselective, vinylogous Mannich addition of a dienolate to a chiral pyridinium salt to set the first chiral center. The absolute stereochemistry of this key center was then relayed by a sequence of substrate-controlled reactions, including a highly stereoselective epoxidation/ring opening sequence and an oxidative rearrangement of an indole to furnish a spirooxindole to establish the remaining stereocenters in the pentacyclic core of the citrinadins. An early stage intermediate in the synthesis of (−)-citrinadin A was deoxygenated to generate a dehydroxy compound that was elaborated into (+)-citrinadin B by a sequence of reaction identical to those used to prepare (−)-citrinadin A. These concise syntheses of (−)-citrinadin A and (+)-citrinadin B led to a revision of their stereochemical structures.
Introduction
Secondary metabolites of fungi have a long and rich history as an important reservoir of biologically active molecules having unusual structures.1 The marine-derived fungus Penicillium citrinum has been a source of a number different classes of natural products,2 and in 2004 and 2005 Kobayashi and co-workers reported the isolation of the two related spirooxindole alkaloids (−)-citrinadin A (1) and (+)-citrinadin B (2) (Figure 1), which exhibited notable activity against murine leukemia L1210 (1, IC50 6.2 μg/mL; 2, 10 μg/mL) and human epidermoid carcinoma KB cells (1, IC50 10 μg/mL).3 The absolute and relative stereochemistry of the pentacyclic cores of the citrinadins were assigned based upon a combination of 1D and 2D NMR experiments and electronic circular dichroism (ECD) spectra. Specifically, ROESY correlations between protons on the N,N-dimethylvalinyl moiety and protons on the E ring of a chlorohydrin derivative of citrinadin A were used to assign the absolute stereochemical relationships in the E ring relative to (S)-valine. Use of the negative first Cotton effect observed in the ECD spectrum of this compound to assign the absolute stereochemistry of the spirocenter as being S was consistent with ROESY data observed for (−)-citrinadin A.3b The interpretation of the ROESY data presumed a preferred orientation of the N,N-dimethylvalinyl side chain relative to the E-ring. The absolute stereochemistry of the epoxide moiety of citrinadin A was assigned as being (S) by comparing its vibrational circular dichroism (VCD) spectrum with those of model α-keto epoxides of known absolute stereochemistry.
Figure 1.

Originally proposed structures for citrinadins A (1) and B (2), PF1270A–C (3–5), and revised structures of citrinadins A (6) and B (7).
A similar group of alkaloids designated as PF1270 A–C (3–5, Figure 1) were subsequently isolated from Penicillium waksmanii strain PF1270 by Kushida in 2007; PF1270 A (3), B (4) and C (5) exhibit submicromolar affinities for human H3 histamine receptor (Ki 0.047, 0.12, and 0.22 μM, respectively).4 Although the absolute stereochemistry of 3–5 was not assigned, the relative stereochemical relationships between the pentacyclic core and the epoxy ketone side chain were established by X-ray crystallographic analysis of 3. A comparison of the structures of 1–5 reveals that the relative stereochemical relationships between the pentacyclic core and the epoxy ketone moieties are opposite.
Interest in the citrinadins by members of the synthetic community has been driven by a combination of their unusual molecular architecture coupled with their biological activity. The pentacyclic framework comprises a spirooxindole that is connected by a cyclopentane ring to a highly substituted quinolizidine ring. The pentacyclic ring system of citrinadin A is punctuated with six stereocenters that include two functionalized tertiary carbon atoms, two contiguous quaternary carbon atoms, and there is an α–epoxy ketone moiety appended to the aromatic ring. Several approaches to the citrinadins have been published,5 and the Wood group recently completed the first total synthesis of (+)-citrinadin B using a convergent strategy wherein the spirooxindole ring system and the piperidine ring were joined via a [3 + 2] cycloaddition.6 Simultaneous with the report of Wood and co-workers, we disclosed the first total synthesis of (−)-citrinadin A7 by a linear approach, in which the first chiral center was created by an enantioselective vinylogous Mannich addition, and the remaining stereocenters on the pentacyclic core were set by substrate based control.8 It is notable that these two total syntheses led to revisions of the stereochemical structures of citrinadin A and citrinadin B as being 6 and 7, respectively, thereby demonstrating that the relative stereochemical relationships of the citrinadins in fact correspond to those reported for PF1270A–C (3–5). Herein we provide the details of our synthetic efforts toward the citrinadins that culminated in the enantioselective total syntheses of (−)-citrinadin A (6) and its congener (+)-citrinadin B (7).
Results and Discussion
First Generation Approach
Our initial retrosynthetic plan toward citrinadins A (6) and B (7) was a convergent one and is outlined in Scheme 1. Because these two alkaloids share a closely related pentacyclic skeleton, we envisioned a unified route for their synthesis that employed the spirooxindole 9 as a common intermediate. For example, coupling 9 with the organozinc reagent of an appropriately substituted piperidine 11 (R1 = H, OR) would give adducts that could be transformed into citrinadin A or citrinadin B via the pentacyclic intermediates 8. Specifically, the 1,2-amino alcohol motif in 6 and 7 would be established by selective epoxidation of 8 from the more accessible face of the carbon–carbon double bond followed by ring-opening, and the epoxy ketone could be introduced via a cross-coupling reaction. In early work directed toward the citrinadins, we developed a novel, oxidation/rearrangement approach for the enantioselective synthesis of spirooxindoles that served as precursors of compounds related to 9,5a but we found that organozinc reagents related to 11 (R1 = H) were unstable, and attempts to induce their desired coupling with 9 were unsuccessful. Accordingly, we adopted an alternate strategy to generate a precursor of 8. This plan featured a vinylogous Mannich reaction9 involving a dienolate generated from 10 and the pyridinium salt 12, the selection of which was inspired by related work of Comins.10 Implicit in this approach was the hope that the spiro stereocenter in the dienolate derived from 9 would either direct the diastereofacial outcome in the addition to an achiral variant of 12 or that this stereocenter would not interfere with directive effects of 12 bearing a chiral carbamate auxiliary.
Scheme 1. First Generation Retrosynthesis of Citrinadins.

To evaluate the diastereoselectivity in the addition of a dienolate such as 10 to an achiral pyridinium salt, we synthesized the functionalized spirooxindole 18 from 15 (Scheme 2), which was readily prepared in 64% overall yield from commercially available 2,2-dimethylcyclohexane-1,3-dione (14) in four straightforward steps that involved protection,11 crossed Claisen condensation, enol triflate formation, and methylation. Hydrolysis of the ketal moiety in 15, followed by phenylhydrazone formation and Fischer indole synthesis, produced the tricyclic indole 16, which was protected to give the Boc carbamate 17. Treatment of 17 with dimethyldioxirane (DMDO) gave an intermediate epoxide that underwent a facile semipinacol rearrangement upon exposure to SiO2 to provide the spirooxindole 18.5a,12 Several preliminary experiments to generate the dienolate from 18 suggested that the imide moiety might be unstable to these reaction conditions, so the oxindole was reduced, and the intermediate N,O-hemiacetal was converted to the more robust N,O-acetal 19. Deprotonation of 19 with lithium diisopropylamide (LDA), followed by transmetalation with ZnCl2 and reaction of the enolate thus formed with TIPS-pyridinium salt 12,10b,10c gave the adduct 21, which was treated with K2CO3 in MeOH to induce cyclization. A single diastereoisomeric pentacyclic product 22 was isolated in ∼10% yield, but a NOESY experiment suggested the relative stereochemistry at C(16) was opposite that required. It is perhaps noteworthy that addition of 20 to the methoxypyridinium salt 13, which lacks the TIPS group, gave a mixture (∼1:1) of diastereomeric adducts. Based upon these preliminary experiments, it appeared that substrate-control enforced by the stereochemistry at the spirocenter C(3) of 19 was opposite that found in the citrinadins.
Scheme 2. Addition of a Spirooxindole Dienolate to a Substituted Pyridinium Salt.

Second Generation Approach
To address the undesired stereochemical outcome in the vinylogous Mannich reaction of 19, we developed an alternate plan in which we envisioned stereochemistry at C(16) of 23 might direct the stereoselectivity of the oxidation/rearrangement to establish the requisite stereochemistry at the C(3) spirocenter of 8 (Scheme 3). We also knew from early work that the stereochemistry of spirooxindole formation could be controlled using a chiral auxiliary on the indole nitrogen atom of 23,5a which would be accessible from 24 via a Fisher indole reaction. Synthesis of the tricyclic intermediate 24 from its precursor 25 simply required N-deprotection and cyclization. A key step in this new approach involves formation of 25 by a stereoselective, vinylogous Mannich reaction between the dienolate derived from 15 and either an iminium ion such as 26 or a pyridinium salt 27 bearing a chiral auxiliary, Xc. Mindful of precedent that additions of small nucleophiles to piperidine-derived iminium salts like 26 proceed to give cis-2,6-disubstituted products as a consequence of the combined effects of A1,2-strain and stereoelectronic control,13 we hypothesized that a more bulky nucleophile, such as a dienolate derived from 15, might add to 26 via a boat-like transition state to give a trans-2,6-disubstituted piperidine. If this outcome was not observed, we would then use 27 as the electrophilic partner in the vinylogous Mannich reaction in accord with the work of Comins.10
Scheme 3. Second Generation Approaches to the Citrinadins.

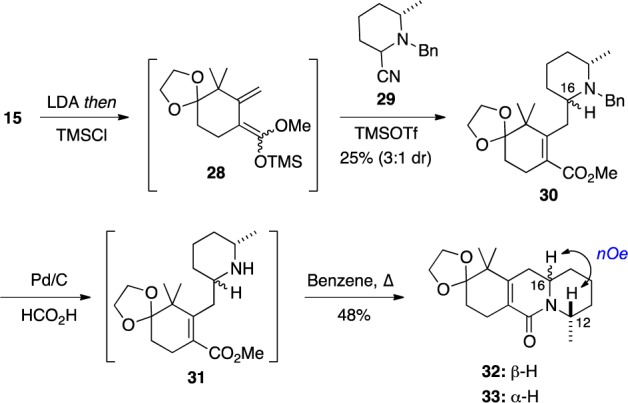
Toward probing the diastereoselectivity of vinylogous Mannich reactions with substituted iminium ions 26, the dienol ether 28 and racemic 29, which was prepared by a procedure reported by Rychnovsky,14 were allowed to react in the presence of TMSOTf to give a mixture (3:1) of isomeric adducts 30 (Scheme 4). Because it was impossible to assign the structure of the major isomer, this mixture was subjected to N-debenzylation using transfer hydrogenation, and the resulting amines 31 were subjected to cyclization by heating in benzene to form lactams 32 and 33. The major diastereomer of this mixture was purified by chromatography. Unfortunately, a NOESY experiment revealed a nOe between the protons at C(12) and C(16) of this compound, suggesting that the major product was 32. The vinylogous Mannich reaction had thus proceeded preferentially with the undesired diastereofacial selectivity.
Scheme 4. Vinylogous Mannich Addition to a Substituted 1,2-Dehydropiperidine.
Inspired by the seminal work of Comins and Sahn,10d we then examined the stereoselectivity of the addition of the zinc dienolate 34 to the chiral pyridinium salt 35, which was generated in situ by the reaction of 3-TIPS-4-methoxypyridine and the chloroformate derivative of (+)-trans-2-(α-cumyl)cyclohexanol [(+)-TCC] (Scheme 5). This reaction proceeded successfully to give 36 with high diastereoselectivity (dr = 92:8), and the absolute stereochemistry at the newly created stereocenter at C(16) was initially assigned by analogy with the extensive findings of Comins.10 This preliminary assignment would later be verified by X-ray crystallography of a derived intermediate (cf. Figure 2).
Scheme 5. Vinylogous Mannich Addition of a Dienolate to a Chiral Pyridinium Salt.

Figure 2.

X-ray structure of 56.
Having at long last discovered a reliable and efficient strategy to establish the requisite absolute stereochemistry at C(16) of the citrinadins, we turned our attention to forming a suitable CDE tricyclic intermediate (Scheme 6). In the event, exposure of 36 to Cs2CO3 in methanol induced removal of the chiral auxiliary, and spontaneous cyclization ensued to provide the lactam 37 in 84% ee together with about 70% recovered (+)-TCC. Gratifyingly, we found that the optical purity of 37 could be easily improved to 98% ee (chiral HPLC, see Supporting Information) after a recrystallization. Desilylation of the lactam 37 was most efficiently effected employing excess TBAF and microwave heating to afford enone 38.
Scheme 6. Preparation of a Key Tricyclic Intermediate.

The stereoselective 1,4-addition of a methyl group to 38 proved to be more problematic than anticipated, presumably owing to the relatively planar nature of the tricyclic ring system. Additions of simple methyl-derived cuprates and related reagents proceeded with low (<2:1) diastereoselectivity. Similarly, the 1,4-additions of organocopper reagents derived from metalated bis(phenylthio)methane15 and tris(phenylthio)methane16 were rather unselective and also low yielding. After extensive experimentation, we eventually discovered that the organocopper reagent generated from (dimethylphenylsilyl)methylmagnesium chloride17 and CuBr·DMS added smoothly to 38 in the presence of BF3·OEt2 to give a mixture of ketones that were directly reduced with high stereoselectivity using l-selectride to give 39 in 71% yield over two steps; 19% of the C(12) epimer of 39 was also isolated. We resisted the temptation to protect the secondary hydroxyl group and heated 39 with TBAF in a microwave oven to obtain the unsaturated lactam 40.18
At this juncture, the challenging tasks associated with stereoselective elaboration of the spirooxindole ring and introduction of the amino alcohol moiety were at hand. We first wanted to ascertain whether the stereocenter at C(16) in a pentacyclic intermediate such as 41 might direct the stereochemical outcome of an oxidation/rearrangement sequence to generate a spirooxindole. Because we wished to conserve the more valuable, enantiomerically pure intermediates in the planned exploratory experiments, we prepared the racemic ketal rac-40 from the product of the vinylogous Mannich reaction of 13 and 34 in accord with the transformations outlined in Scheme 6. When rac-40 was heated with o-chlorophenylhydrazine hydrochloride in refluxing sulfuric acid (5% v/v), rac-41 was isolated in 69% yield (Scheme 7).19 Toward converting rac-41 into the spirooxindole rac-43, a number of standard oxidants, including OsO4, NBS, and Pb(OAc)4 were evaluated,20 but mixtures of products were invariably obtained. However, we found that treating rac-41 with tert-BuOCl, followed by exposure of the intermediate chloroindolenines rac-42 to aqueous acid, provided a mixture (1.5:1 by 1H NMR) of diastereomeric spirooxindoles rac-43 as an inseparable mixture.21 This result suggested that the resident chirality in rac-41 is too remote to influence the diastereofacial selectivity of the chlorination of rac-41 to preferentially give the epimer of rac-42 that is required for the stereoselective formation of rac-43.
Scheme 7. Preliminary Experiment To Induce Stereoselective Formation of a Spirooxindole.

The finding that the oxidation/rearrangement of 41 was not very selective clearly mandated a change in strategy for the end game of the synthesis. We considered the possibility of introducing a chiral carbamate on the indole nitrogen atom because we had previously shown that reactions of such compounds with DMDO followed by rearrangement of the intermediate epoxide could be highly diasteroselective.5a However, such an approach suffered from the need to use a second chiral auxiliary as a stoichiometric reagent, so we considered that plan to be stereochemically inefficient relative to exploiting substrate control to establish relative stereochemistry following the diastereoselective vinylogous Mannich reaction. We were thus intrigued by an alternative approach in which the amino alcohol moiety in 45 was envisioned to direct the stereochemical course of an oxidation/rearrangement sequence to furnish 44 (Scheme 8).
Scheme 8. Revised Endgame for the Synthesis of the Citrinadins.

The transformation of enantiomerically pure 40 into 45 was surprisingly straightforward. Epoxidation of 40 with peroxytrifluoroacetic acid in the presence of sodium carbonate was highly diastereoselective, proceeding from the more accessible, slightly convex face to furnish a single epoxide 47 (Scheme 9).22 Although this epoxidation could also be performed in unbuffered media, the ketal moiety suffered hydrolysis under those conditions. The reaction of epoxide 47 with aqueous methylamine in a sealed tube also occurred with high diastereoselectivity to deliver the requisite amino alcohol 46 in 95% yield. Heating 46 with o-bromophenylhydrazine hydrochloride in aqueous sulfuric acid provided the indole 45 in 81% yield; trace amounts of the debromo derivative of 45 were observed by LCMS.23
Scheme 9. Preparation of Pentacyclic Indole Core 45.

The stage was now set to test the feasibility of creating the critical spirocenter at C(3) by a substrate-controlled oxidation/rearrangement of 45. Anticipating that some of the reactions might be troublesome, we again decided to perform our exploratory experiments using rac-45, which was conveniently prepared from rac-40 following the sequence of reactions depicted in Scheme 9. As noted previously, creation of spirooxindoles from indoles by oxidation followed by skeletal rearrangement is well documented.19,20 However, inducing such a transformation on indole rac-45 proved to be much more challenging than anticipated, and many established protocols did not give the desired spirocyclic product. The functionality present in rac-45 appeared to conspire against us, but we were reluctant to indulge in a series of unpredictable protecting group maneuvers.
In early studies with model systems, we had examined the possible use of Davis’ oxaziridine 48 to induce the oxidation and rearrangement of simple indoles to generate spirooxindoles, but these efforts were unsuccessful. Driven by desperation and inspired by several reports of Williams, who recently used oxaziridines in elegant syntheses of spirooxindole alkaloids related to the notoamides,24 we treated rac-45 with an excess of 48 and isolated a moderately stable epoxide that was exposed acetic acid to initiate a facile semipinacol rearrangement (Scheme 10). Gratifyingly, the structure of the product was established by X-ray crystallography to be the desired spirooxindole rac-50. The stereochemical outcome of this sequence is consistent with a hydroxyl group-directed delivery of 48 to the bottom face of rac-45 to give rac-49.
Scheme 10. Synthesis of Racemic Pentacyclic Core of Citrinadin A: The Initial Plan.

We had thus successfully prepared the pentacyclic core characteristic of citrinadins A and B. The euphoria associated with achieving this milestone was short-lived, however, because all efforts to selectively reduce the tertiary δ-lactam moiety in rac-50 in the presence of the secondary γ-lactam to cleanly give rac-44 using a variety of hydride reducing agents were uniformly unsuccessful.25 Alane was found to be the best reagent, giving rac-44 along with the products from reduction of the γ-lactam and both lactams in variable yields. Attempts to optimize the reduction by varying reaction temperature and amount of alane failed to improve the chemoselectivity.
To avoid the problems associated with the unselective lactam reduction, we decided to reverse the steps and reduce the lactam in rac-45 prior to the oxidation/rearrangement sequence (Scheme 11). In the event, reduction of rac-45 with the complex of alane and dimethylethylamine (DMEA)20b,25b delivered rac-51 in excellent yield. We found it was necessary to protonate the basic amino groups in rac-51 to protect them from oxidation under the conditions of the next step. Accordingly, sequential treatment of rac-51 with trifluoroacetic acid (TFA) and then an excess of Davis’ oxaziridine 48 directly delivered the spirooxindole rac-53 as a single diastereoisomer, presumably via the intermediate epoxide rac-52. To our dismay, however, X-ray crystallographic analysis revealed that the product was rac-53, wherein the relative stereochemistry at C(3) is opposite that present in the citrinadins. The aberrant stereochemical outcome of this oxidation relative to that of rac-45 can be rationalized if the protonated, axial N-methylammonium group in rac-51 directs the oxidation by Davis’ oxaziridine 48 from the top face to give rac-52, which then undergoes rearrangement leading to the formation of the undesired spirooxindole rac-53.
Scheme 11. Successful Synthesis of Racemic Pentacyclic Core of Citrinadin A.

This analysis suggested that a bulky Lewis or Brønsted acid might associate with the axial secondary amine group, thereby blocking the top face of rac-51 and enforcing approach of Davis’ oxaziridine 48 from the bottom face. Indeed, we had utilized this tactic to direct the stereochemistry of chlorination of an indole ring in our successful syntheses of the Strychnos alkaloids akuammicine and strychnine.26 A variety of Brønsted acids (e.g., AcOH, camphorsulfonic acid, and TsOH) and Lewis acids (e.g., AlEt3, SnCl4, ZnCl2 and BF3·OEt2) were screened, but none of these led to the desired spirooxindole rac-44. We eventually discovered that treating rac-51 sequentially with pyridinium p-toluenesulfonate (PPTS), excess of Davis’ oxaziridine 48, and then acetic acid provided the spirooxindole rac-44 as a single stereoisomer, the structure of which was unambiguously determined by X-ray analysis. The apparently unique role that PPTS plays in controlling the stereochemical outcome of this oxidation is not understood, but we tentatively surmise that ion-pairing effects may be involved that render oxidation from the top face of rac-51 sterically unfavorable.
Having thus validated our approach to the stereoselective synthesis of the pentacyclic core of the citrinadins, it was necessary to prepare 44 in enantiomerically pure form and to install the dimethyl valine side chain on the hydroxyl group at C(14) and the epoxy ketone side chain on the aromatic ring to complete the synthesis of citrinadin A (6). In the event, following the procedures outlined in Scheme 11, enantiomerically pure 45 was readily transformed into enantiomerically pure 44 (Scheme 12). After unsuccessfully investigating the possibility of converting the aryl bromide group in 44 directly into an α,β-unsaturated ketone using a carbonylative cross-coupling reaction we had previously developed,27 we turned to a stepwise procedure that commenced with the Sonogashira coupling between 44 and 3-methylbut-1-yne to furnish the alkyne 55.28O-Acylation of the hydroxyl group at C(14) with N,N-dimethyl-l-valine in the presence of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDCI) and 4-N,N-dimethylaminopyridine (DMAP) provided 56 in excellent yield.29 The absolute and relative stereochemistry of 56 was established unequivocally by X-ray crystallography (Figure 2). The gold-promoted oxidation of 56 using 2-bromopyridine N-oxide according to a method reported by Zhang gave the enone 57.30 Finally, diastereoselective epoxidation of 57 to deliver a mixture (5:1) of 6 and 58, respectively, was achieved by applying an Enders protocol for the enantioselective synthesis of (S)-epoxides from α,β-unsaturated ketones using (S,S)-N-methylpseudoephedrine as the chiral ligand.31
Scheme 12. First Total Synthesis of (−)-Citrinadin A (6).

The CD spectrum of the synthetic 6 thus obtained as the free base is identical with that reported by Kobayashi for (−)-citrinadin A,3b whereas the CD spectrum for 58 is very different (Figure 3). The 1H and 13C NMR data of the free base forms of 6 and 58 are wholly consistent with their assigned structures, and the 1H and 13C NMR data of 6 as its putative bis-hydrochloride salt, which was formed upon standing in CDCl3, are also in good agreement with those reported for a bis-salt of (−)-citrinadin A (see Supporting Information).3 Because we were unable to obtain an authentic sample of (−)-citrinadin A or its bis-salt, a direct comparison of natural material with the synthetic sample was not possible. Nevertheless, the CD spectrum of synthetic 1 coupled with the crystallographic data for 56 strongly suggests that the correct stereochemical structure of (−)-citrinadin A is 6, so the absolute stereochemistry of the pentacyclic core of the citrinadins is opposite that assigned by Kobayashi.3 Notably, this revised structure is in agreement with the findings of Wood and co-workers, who completed the first total synthesis of (+)-citrinadin B.6
Figure 3.

(a) CD Spectra of 6 and 58. (b) CD spectra of citrinadins A and B. Adapted with permission from (3b). Copyright 2005 American Chemical Society.
Further corroboration of the correctness of the stereochemical assignment of (−)-citrinadin A as being that shown in 6 was obtained by the independent synthesis of 1, the compound that Kobayashi had assigned as being (−)-citrinadin A (Scheme 13).3 In the event, the vinylogous Mannich reaction of the zinc dienolate of 15 with the chiral pyridinium salt ent-35, which has (−)-TCC as the chiral auxiliary, provided the adduct ent-36 in 61% yield (dr = 94:6). Following the same sequence of reactions described for the conversion of 36 into 57 (Schemes 6, 9, and 12), ent-36 was transformed into 59 in 4.2% overall yield over 14 steps. It is significant that en route to 59, the absolute configuration of ent-44 was unambiguously established by X-ray crystallography (Figure 4). Diastereoselective epoxidation of enone 59 with (S,S)-N-methylpseudoephedrine according to the Enders protocol afforded a separable mixture (2:1) of 60 and 1.31 The CD spectra of neither the major isomer 60 nor 1 matches the CD spectrum for (−)-citrinadin A reported by Kobayashi (Figure 5).3b On the contrary, the CD spectrum of 60 is clearly opposite to that of naturally occurring (−)-citrinadin A. This observation is consistent with the absolute stereochemistry of the pentacyclic core of 60 being enantiomeric to that of (−)-citrinadin A, which must therefore have the stereochemical structure depicted in 6.
Scheme 13. Synthesis of (−)-Citrinadin A Isomer 1.

Figure 4.

X-ray structure of ent-44. A molecule of CHCl2 and AcOH was removed for clarity.
Figure 5.

CD Spectra of 60, 1, and 6.
Total Synthesis of (+)-Citrinadin B
The difference between citrinadin A (6) and citrinadin B (7) is the presence of a dimethyl valine ester at C(14). Accordingly, one may envisage that deoxygenation at C(14) of a late stage intermediate in the synthesis of citrinadin A would give a precursor of citrinadin B. In several exploratory experiments, we examined the feasibility of deoxygenating rac-50 and rac-44 using procedures reported by Barton32 and Myers,33 but because mixtures of products were obtained, we turned to the removal of the C(14) hydroxyl group an earlier stage. Thus, Barton deoxygenation of 40 afforded 61 in 73% yield (Scheme 14). Epoxidation of 61 using buffered peroxytrifluoroacetic acid gave epoxide 62 (77% yield) as a single stereoisomer, and when 62 was heated with aqueous methylamine, the desired amino alcohol 63 was isolated in 94% yield. The Fisher indole reaction of 63 with o-bromophenylhydrazine hydrochloride furnished the pentacyclic indole 64, which was then converted into the spirooxindole 65 in 34% overall yield by sequential hydride reduction of the tertiary lactam, indole oxidation with Davis’ oxaziridine 48, and acid-catalyzed rearrangement of the intermediate epoxide. The aryl bromide moiety of 65 was then elaborated via a Sonogashira coupling with 3-methylbut-1-yne to furnish the alkyne 66 that was processed to the enone 67 in 68% overall yield from 65 by reaction with 2-bromopyridine N-oxide in the presence of gold.30 Finally, epoxidation of enone 67 using the Enders procedure31 afforded a separable mixture of 7 and 68 (dr = 2.5:1). The CD spectrum of major isomer 7 was in agreement with CD data obtained for natural (+)-citrinadin (B), and the spectral data (1H and 13C NMR) of the synthetic 7 thus isolated were consistent with those reported by Wood.6
Scheme 14. Total Synthesis of (+)-Citrinadin B.

Summary and Conclusions
In summary, we completed the enantioselective total syntheses of (−)-citrinadin A (6) and (+)-citrinadin B (7) by concise sequences of reactions involving only 20 and 21 steps, respectively, from commercially available starting materials. The brevity of the approach was enabled by minimizing protecting group operations and unproductive refunctionalization. The syntheses of 6 and 7 feature a highly diastereoselective, vinylogous Mannich reaction of a dienolate with a chiral pyridinium salt to establish the first stereogenic center. The stereochemical efficiency of the syntheses was then possible because the chirality of this carbon atom was exploited to introduce the remaining stereocenters of the pentacyclic core in a linear sequence of reactions that were subject to substrate-control. Notable stereoselective transformations include an epoxidation/ring opening sequence and an oxidation/rearrangement of an indole to furnish a spirooxindole. Our completion of the syntheses of 6 and 7 coupled with the corroborative findings of Wood6 led to the revision of the stereochemical structures from those that were originally proposed as being 1 and 2, respectively, by Kobayashi,3b who relied upon ROESY and ECD data to assign absolute and relative stereochemical relationships in the pentacyclic cores of the citrinadins. The misassignment of the absolute stereochemistry of the citrinadin structures by Kobayashi serves as a reminder of the potential pitfalls associated with using ROESY, as well as ECD, when assigning stereochemistry, especially when other stereoisomers are not available for comparison.
Acknowledgments
We thank the National Institutes of Health (GM 25439) and the Robert A. Welch Foundation (F-0652) for generous support of this research. We are grateful to Dr. Vincent Lynch (The University of Texas, Austin) for X-ray crystallography and Professor John Wood (Baylor University) for suggestions for introducing the epoxy ketone moiety and for discussions regarding the structural assignments of the citrinadins. We also thank Professor Daniel Comins (North Carolina State University) for a generous gift of (−)-TCC, Dr. James Sahn (The University of Texas, Austin) for helpful suggestions, Dr. Eun Jeong Cho (The University of Texas, Austin) for technical assistance, and Mr. Shawn Blumberg (The University of Texas, Austin) for valuable technical support.
Supporting Information Available
Complete experimental procedures, full characterization of new compounds, X-ray crystallographic data for ent-36, ent-46, ent-47, rac-50, rac-53, rac-44, ent-44, 56, comparison of CD spectra of 7 and 68 compared with those published for citrinadins A and B, comparison of 1H and 13C NMR data for 6 as its free base and as a bis-salt with those published for a bis-salt of (−)-citrinadin A, and a comparison of 1H and 13C NMR data for 7 with those obtained by Wood. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Department of Chemistry, Hendrix College, 1600 Washington Ave., Conway, AR 72032.
Author Present Address
‡ Pfizer Worldwide Research & Development, 610 Main St., Cambridge MA, 02139.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Bugni T. S.; Ireland C. M. Nat. Prod. Rep. 2004, 21, 143–163. [DOI] [PubMed] [Google Scholar]; b Saleem M.; Ali M. S.; Hussain S.; Jabbar A.; Ashraf M.; Lee Y. S. Nat. Prod. Rep. 2007, 24, 1142–1152. [DOI] [PubMed] [Google Scholar]; c Ratebab M. E.; Ebel R. Nat. Prod. Rep. 2011, 28, 290–344. [DOI] [PubMed] [Google Scholar]
- a Tsuda M.; Sasaki M.; Mugishima T.; Komatsu K.; Sone T.; Tanaka M.; Mikami Y.; Kobayashi J. J. Nat. Prod. 2005, 68, 273–276. [DOI] [PubMed] [Google Scholar]; b Lu Z.-Y; Lin Z.-J.; Wang W.-L.; Du L.; Zhu T.-J.; Fang Y.-C; Gu Q.-Q.; Zhu W.-M. J. Nat. Prod. 2008, 71, 543–546. [DOI] [PubMed] [Google Scholar]; c Ueda J.; Hashimoto J.; Inaba S.; Takagi M.; Shin-ya K. J. Antibiot. 2010, 63, 203–205. [DOI] [PubMed] [Google Scholar]; d Khamthong N.; Rukachaisirikul V.; Phongpaichit S.; Preedanon S.; Sakayaroj J. Tetrahedron 2012, 68, 8245–8250. [Google Scholar]; e El-Neketi M.; Ebrahim W.; Lin W.; Gedara S.; Badria F.; Saad H.-E. A.; Lai D.; Proksch P. J. Nat. Prod. 2013, 76, 1099–1104. [DOI] [PubMed] [Google Scholar]
- a Tsuda M.; Kasai Y.; Komatsu K.; Sone T.; Tanaka M.; Mikami Y.; Kobayashi J. Org. Lett. 2004, 6, 3087–3089. [DOI] [PubMed] [Google Scholar]; b Mugishima T.; Tsuda M.; Kasai Y.; Ishiyama H.; Fukushi E.; Kawabata J.; Watanabe M.; Akao K.; Kobayashi J. J. Org. Chem. 2005, 70, 9430–9435. [DOI] [PubMed] [Google Scholar]
- Kushida N.; Watanabe N.; Okuda T.; Yokoyama F.; Gyobu Y.; Yaguchi T. J. Antibiot. 2007, 60, 667–673. [DOI] [PubMed] [Google Scholar]
- a Pettersson M.; Knueppel D.; Martin S. F. Org. Lett. 2007, 9, 4623–4626. [DOI] [PubMed] [Google Scholar]; b McIver A. L.; Deiters A. Org. Lett. 2010, 12, 1288–1291. [DOI] [PubMed] [Google Scholar]; c Chandler B. D.; Roland J. T.; Li Y.; Sorensen E. J. Org. Lett. 2010, 12, 2746–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Guerrero C. A.; Sorensen E. J. Org. Lett. 2011, 13, 5164–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Mundal D. A.; Sarpong R. Org. Lett. 2013, 15, 4952–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Matsumaru T.; McCallum M. E.; Enquist J. A.; Smith G. M.; Kong K.; Wood J. L. Tetrahedron 2014, 70, 4089–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong K.; Enquist J. A. Jr.; McCallum M. E.; Smith G. M.; Matsumaru T.; Menhaji-Klotz E.; Wood J. L. J. Am. Chem. Soc. 2013, 135, 10890–10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Z.; Marvin C. C.; Martin S. F. J. Am. Chem. Soc. 2013, 135, 10886–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an early example highlighting the advantages of a stereochemically linear synthesis, see:; a Smith A. B. III; Fukui M.; Vaccaro H. A.; Empfield J. R. J. Am. Chem. Soc. 1991, 113, 2071–2092. [Google Scholar]; b Smith A. B. III; Empfield J. R. Chem. Pharm. Bull. 1999, 47, 1671–1678. [Google Scholar]
- For reviews of the vinylogous Mannich reaction, see:; a Martin S. F. Acc. Chem. Res. 2002, 35, 895–904. [DOI] [PubMed] [Google Scholar]; b Casiraghi G.; Battistini L.; Curti C.; Rassu G.; Zanardi F. Chem. Rev. 2011, 111, 3076–3154. [DOI] [PubMed] [Google Scholar]
- a Comins D. L.; Hong H. J. Am. Chem. Soc. 1993, 115, 8851–8852. [Google Scholar]; b Comins D. L.; Goehring R. R.; Joseph S. P.; O’Connor S. J. Org. Chem. 1990, 55, 2574–2576. [Google Scholar]; c Comins D. L.; Joseph S. P.; Goehring R. R. J. Am. Chem. Soc. 1994, 116, 4719–4728. [Google Scholar]; d Comins D. L.; Sahn J. J. Org. Lett. 2005, 7, 5227–5228. [DOI] [PubMed] [Google Scholar]
- For ketalization of 14, see:Shibuya S.; Isobe M. Tetrahedron 1998, 54, 6677–6698. [Google Scholar]
- a Zhang X.; Foote C. S. J. Am. Chem. Soc. 1993, 115, 8867–8868. [Google Scholar]; b Adam W.; Ahrweiler M.; Peters K.; Schmiedeskamp B. J. Org. Chem. 1994, 59, 2733–2739. [Google Scholar]
- a Stevens R. V. Acc. Chem. Res. 1984, 17, 289–296. [Google Scholar]; b Neipp C. E.; Martin S. F. J. Org. Chem. 2003, 68, 8867–8878. [DOI] [PubMed] [Google Scholar]; c Roulland E.; Cecchin F.; Husson H.-P. J. Org. Chem. 2005, 70, 4474–4477. [DOI] [PubMed] [Google Scholar]; d Miller K. A.; Shanahan C. S.; Martin S. F. Tetrahedron 2008, 64, 6884–6900. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Amorde S. M.; Jewett I. T.; Martin S. F. Tetrahedron 2009, 65, 3222–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolckenhauer S. A.; Rychnovsky S. D. Tetrahedron 2005, 61, 3371–3381. [Google Scholar]
- a Mukaiyama T.; Narasaka K.; Furusato M. J. Am. Chem. Soc. 1972, 94, 8641–8642. [Google Scholar]; b Ager D. J.; East M. B. J. Org. Chem. 1986, 51, 3983–3992. [Google Scholar]
- a Manas A.-R. B.; Smith R. A. J. Chem. Soc., Chem. Commun. 1975, 216–217. [Google Scholar]; b Cohen T.; Nolan S. M. Tetrahedron Lett. 1978, 19, 3533–3536. [Google Scholar]
- Comins D. L.; Al-awar R. S. J. Org. Chem. 1995, 60, 711–716. [Google Scholar]
- Heitzman C. L.; Lambert W. T.; Mertz E.; Shotwell J. B.; Tinsley J. M.; Va P.; Roush W. R. Org. Lett. 2005, 7, 2405–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews, see:; a Robinson B. Chem. Rev. 1963, 63, 373–401. [Google Scholar]; b Robinson B. Chem. Rev. 1969, 69, 227–250. [DOI] [PubMed] [Google Scholar]; c Roberson C. W.; Woerpel K. A. J. Am. Chem. Soc. 2002, 124, 11342–11348. [DOI] [PubMed] [Google Scholar]
- For a review, see:; a Marti C.; Carreira E. M. Eur. J. Org. Chem. 2003, 2209–2219. [Google Scholar]; See also:; b Peterson A. C.; Cook J. M. J. Org. Chem. 1995, 60, 120–129. [Google Scholar]; c Cushing T. D.; Sanz-Cervera J. F.; Williams R. M. J. Am. Chem. Soc. 1996, 118, 557–579. [Google Scholar]; d Wearing X. Z.; Cook J. M. Org. Lett. 2002, 4, 4237–4240. [DOI] [PubMed] [Google Scholar]
- a Martin S. F.; Mortimore M. Tetrahedron Lett. 1990, 31, 4557–4560. [Google Scholar]; b Martin S. F.; Benage B.; Geraci L. S.; Hunter J. E.; Mortimore M. J. Am. Chem. Soc. 1991, 113, 6161–6171. [Google Scholar]
- Fehr C. Angew. Chem., Int. Ed. 1998, 37, 2407–2409. [DOI] [PubMed] [Google Scholar]
- For examples of the loss of halogen atoms under the conditions of the Fischer indole synthesis, see:; a Carlin R. B.; Larson G. W. J. Am. Chem. Soc. 1957, 79, 934–941. [Google Scholar]; b Chen J.; Chen W.; Hu Y. Synlett 2008, 77–82. [Google Scholar]
- a Greshock T. J.; Grubbs A. W.; Tsukamoto S.; Williams R. M. Angew. Chem., Int. Ed. 2007, 46, 2262–2265. [DOI] [PubMed] [Google Scholar]; b Greshock T. J.; Grubbs A. W.; Williams R. M. Tetrahedron 2007, 63, 6124–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Greshock T. J.; Grubbs A. W.; Jiao P.; Wicklow D. T.; Gloer J. B.; Williams R. M. Angew. Chem., Int. Ed. 2008, 47, 3573–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For some examples:; a Dutton J. K.; Steel R. W.; Tasker A. S.; Popsavin V.; Johnson A. P. J. Chem. Soc., Chem. Commun. 1994, 765–766. [Google Scholar]; b Cushing T. D.; Sanz-Cervera J. F.; Williams R. M. J. Am. Chem. Soc. 1996, 118, 557–579. [Google Scholar]
- Martin S. F.; Rüeger H.; Williamson S. A.; Grzejszczak S. J. Am. Chem. Soc. 1987, 109, 6124–6134. [Google Scholar]
- a O’Keefe B. M.; Simmons N.; Martin S. F. Org. Lett. 2008, 10, 5301–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]; b O’Keefe B. M.; Simmons N.; Martin S. F. Tetrahedron 2011, 67, 4344–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sonogashira K.; Tohda Y.; Hagihara N. Tetrahedron Lett. 1975, 4467–4470. [Google Scholar]; b Sonogashira K. In Contemporary Organic Synthesis; Trost B. M., Fleming I., Eds.; Pergamon: Oxford, 1991; Vol. 3, pp 521–549. [Google Scholar]
- Neises B.; Steglich W. Angew. Chem., Int. Ed. 1978, 17, 522–524. [Google Scholar]
- Lu B.; Li C.; Zhang L. J. Am. Chem. Soc. 2010, 132, 14070–14072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enders D.; Zhu J.; Raabe G. Angew. Chem., Int. Ed. 1996, 35, 1725–1728. [Google Scholar]
- For a review of procedures to effect deoxygenations of alcohols, see:; a Hartwig W. Tetrahedron 1983, 39, 2609–2645. [Google Scholar]; See also:; b Barton D. H. R.; McCombie S. W. J. Chem. Soc., Perkin Trans. 1 1975, 1574–1585. [Google Scholar]; c Pettus T. R. R.; Inoue M.; Chen X.-T.; Danishefsky S. J. J. Am. Chem. Soc. 2000, 122, 6160–6168. [Google Scholar]
- Myers A. G.; Movassaghi M.; Zheng B. J. Am. Chem. Soc. 1997, 119, 8572–8573. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.