

Abstract
Mutations in ADNP were recently identified as a frequent cause of syndromic autism, characterized by deficits in social communication and interaction and restricted, repetitive behavioral patterns. Based on its functional domains, ADNP is a presumed transcription factor. The gene interacts closely with the SWI/SNF complex by direct and experimentally verified binding of its C-terminus to three of its core components. A detailed and systematic clinical assessment of the symptoms observed in our patients allows a detailed comparison with the symptoms observed in other SWI/SNF disorders. While the mutational mechanism of the first 10 patients identified suggested a gain of function mechanism, an 11th patient reported here is predicted haploinsufficient. The latter observation may raise hope for therapy, as addition of NAP, a neuroprotective octapeptide named after the first three amino acids of the sequence NAPVSPIQ, has been reported by others to ameliorate some of the cognitive abnormalities observed in a knockout mouse model. It is concluded that detailed clinical and molecular studies on larger cohorts of patients are necessary to establish a better insight in the genotype phenotype correlation and in the mutational mechanism.
Keywords: autism, SWI/SNF, BAF complexes, ADNP
INTRODUCTION
Autism is a neurodevelopmental disorder characterized by limitations in social interaction and communication in combination with stereotypic, repetitive behavior and restricted interest [APA, 2013]. The symptoms usually emerge in early childhood, before the age of three. The prevalence of the disorder appears to be on the rise over the last decades, with as many as 1 in 68 individuals affected according to the most recent estimates [Surveillance, 2014]. Consistent among all prevalence estimates, more boys than girls are affected. All population studies indicate a significant contribution of genetic components underlying the disorder. While the heritability—the proportion of phenotypic variation explained by genetic factors—was once estimated to be as high as 90% in the first reported twin studies [Folstein and Rutter, 1977; Steffenburg et al., 1989], there is now a consensus that these initial studies may have overestimated the genetic contribution of the disorder. The largest population based study so far reports a heritability of 50% with an increased risk of recurrence of about 10-fold for a first degree relative and of about 2-fold for cousins [Sandin et al., 2014].
While the genetic causes of non-syndromic autism remain elusive, searches for a genetic cause have been successful to some extent in syndromic forms of autism. Syndromic autism is defined as autism in combination with additional clinical features. Co-morbidities often observed include intellectual disability, epilepsy, and psychiatric disorders. Frequent monogenic causes of syndromic autism include the fragile X syndrome and Rett syndrome [Amir et al., 1999; Rooms and Kooy, 2011]. Specific genomic disorders—submicroscopic chromosomal deletions or duplications at fixed positions in the genome—presenting with autism, include the duplication of the Prader–Willi/Angelman region at 15q11-13 and recurrent copy number variants (CNV) at 16p11.2 [Sanders et al., 2011]. A microscopically visible chromosomal abnormality associated with autism is an additional supernumerary isodicentric chromosome 15 [Mendelsohn and Schaefer, 2008]. Apart from recurrent genomic disorders, an excess of non-recurrent de novo CNVs as compared to healthy siblings and control subjects is also evident in patients with Autistic Spectrum Disorder (ASD) [Pinto et al., 2010].
Most recently, whole exome—consisting of all protein coding regions in the genome—sequencing (WES) has been applied to identify the genes involved in autism. This approach was inspired by the successful introduction of WES to identify genetic causes of neurodevelopmental disorders [Veltman and Brunner, 2012]. After a proof of principle study comparing the exomes of a pilot cohort of 10 ID patients with those of the parents, likely causative mutations were identified in six cases [Vissers et al., 2010]. Subsequent studies using the same so called trio approach in larger cohorts of up to 250 patients showed a diagnostic yield in the range of up to 50% [de Ligt et al., 2012; Rauch et al., 2012; Yang et al., 2013]. In autism, it was discovered that de novo potentially deleterious (e.g., amino acid changing) SNPs were significantly more prevalent in patients than in unaffected relatives or controls. Based on the trio and quartet approach, consisting of patient, parents, and unaffected sibling, the most frequently mutated genes identified so far include CDH8, SCN2A, DYRK1A, and CTNNB1 [O’Roak et al., 2012a; Krumm et al., 2014]. Many genes implicated in autism were previously identified as causative for ID, epilepsy, schizophrenia or bipolar disorder, suggesting an overlap in the underlying etiology of neurodevelopmental disorders.
Collectively, the genetic abnormalities identified in autism to date explain maximally 15% of cases [Mendelsohn and Schaefer, 2008; Carter and Scherer, 2013]. While no single gene is mutated in more than 1% of patients, autism genes functionally converge to commonly affected cellular pathways and protein-protein interaction networks [O’Roak et al., 2012b; Krumm et al., 2014; Pinto et al., 2014]. The most commonly affected networks are the developmental wnt signaling pathway, the pathway involving synaptic function, and the chromatin remodeling pathway.
The Identification of ADNP Mutations in Autistic Patients
In an attempt to identify novel genes responsible for autism, a first de novo p.Lys408Valfs*31 mutation in the activity-dependent neuroprotective protein (ADNP) gene was identified in a large cohort of autistic patients [O’Roak et al., 2012b]. As sequencing of the 209 families in this cohort did not reveal a second mutation in ADNP nor hardly in any other candidate gene, a large scale resequencing study of the most promising candidates, including ADNP, was initiated using the molecular inversion probe (MIP) sequencing technology [O’Roak et al., 2012a]. In this study of 2,446 probands, an additional p. Tyr719*de novo ADNP mutation was identified. However, a single p.Q361* mutation in a patient of the NHLBI GO Exome Sequencing Project (ESP) cohort, consisting of patients with non-neurological disorders affecting heart, lung, and blood was present, and it was concluded that statistical evidence was not sufficient to prove causality of the mutations [ESP]. Confusingly, a p. Gly1094Profs*5 mutation, inherited from an unaffected parent, was reported in this same large scale targeted resequencing study. While this finding seemed to argue against causality at first sight, this mutation is located close to the C-terminus of the encoded protein, beyond the last known functional domain. Typically variations that close to the end of a protein are unlikely to affect protein function.
In a trial of optimizing the diagnostic workflow for the introduction of WES in a diagnostic setting, we discovered another ADNP mutation in a small cohort of ID/autism patients [Helsmoortel et al., 2014a]. Targeted screening of a cohort of 148 autistic patients revealed yet another mutation. By combining the data from WES and targeted resequencing studies initiated in multiple centers, we identified a total of 10 patients with mutations in ADNP, including the cases described above [Helsmoortel et al., 2014b]. We calculated that the frequency of truncating de novo mutations in ADNP is significantly higher (p: 0.001852, odds ratio 13.24668, one-sided Fisher’s exact test) in patients compared to the ESP cohort and additional controls from the Simons Siblings. In addition to the case-control analysis, we calculated a locus specific enrichment for truncating variation using a probabilistic model as described [O’Roak et al., 2012a]. The probability of detecting eight or more de novo truncating events in ADNP within our cohort by chance was estimated to be P = 2.65e-18 (binomial test) under a de novo rate of 1.2 non-synonymous coding variants per individual. To further delineate the clinical characteristics of this novel disorder, an online portal was set up to collect phenotypic information of additional patients in a collaborative effort (Fig. 1). One additional patient with a de novo c.118C > T (p.Q40*) mutation was already submitted to the system.
Figure 1.

http://adnpgene.com forms a portal of a collaborative research project to further characterize the phenotype and future development of patients with ADNP mutations.
The ADNP gene
The ADNP gene was first identified in murine P19 carcinoma cells as a vasoactive intestinal peptide (VIP) responsive gene showing increased expression after VIP treatment [Bassan et al., 1999; Pinhasov et al., 2003]. VIP is a neuro-protective peptide that is active during embryonic development, especially during the time of neuronal tube closure, and protects damaged nerve cells from cell death by inducing glia-derived, survival promoting substances [Said, 1996]. The human orthologue, ADNP, spans about 40 kb of genomic DNA and consists of five exons and four introns with alternative splicing of an untranslated second intron [Zamostiano et al., 2001]. Human and murine mRNA are 90% identical and the region is highly conserved between vertebrates. The encoded protein contains nine zinc fingers and a homeobox domain with a strong homology to that found in hox genes, suggesting a firm role in embryonic development (Fig. 2). Bioinformatic analysis also identified PxVxL as a potential heterochromatin protein 1α (HP1α) binding motif, together with an ARKS motif in the homeobox domain [Mosch et al., 2011]. Indeed, HP1α is found in co-immunoprecipitates from P19 nuclear protein extracts with ADNP antibodies and vice versa. Additional proteins in the precipitates with ADNP antibodies were BRG1, BAF250A, and BAF170, all members of the mating-type switching/sucrose non-fermenting (SWI/SNF) remodeling complex. Despite the presence of a bipartite nuclear localization signal (NLS), the protein is predominantly, but not exclusively, cytoplasmatic in neuronal cells, though in non-neural cells it is mostly located in the nucleus [Gennet et al., 2008; Mandel et al., 2008]. The protein also contains signals involved in cellular secretion and uptake and ADNP has been found in the extracellular space of VIP stimulated astrocytes [Furman et al., 2004]. Finally, ADNP exhibits a strong neuroprotective function that can be attributed in its entirety to an octapeptide Asn-Ala-Pro-Val-Ser-Ile-Pro-Alaor NAPVSPIQ domain called NAP [Bassan et al., 1999; Magen and Gozes, 2014]. The mechanism of action is believed to involve P53, a key regulator of cellular apoptosis, as silencing of ADNP results in an increase of p53 [Zamostiano et al., 2001]. Subsequently, it was shown that addition of NAP to PC12 cells subjected to oxidative stress protected the cells against elevated p53 levels, normally caused by this treatment [Gozes et al., 2004]. ADNP2 is a sister protein to ADNP which is 33% identical at the protein level, shares the zinc finger and homeobox domains, but lacks the NAP motif [Zamostiano et al., 2001]. Expression levels of ADNP and ADNP2 appear to be correlated [Dresner et al., 2011; Helsmoortel et al., 2014b].
Figure 2.

a: Schematic representation of the ADNP protein. Symbols:  previous cases,
previous cases,  new case,
new case,  controls and
controls and  cases for which expression analysis was performed. b: Amino acid positions of the different domains.
cases for which expression analysis was performed. b: Amino acid positions of the different domains.
ADNP Knockout Mouse Model
A knockout mouse model has been generated by targeted replacement of the protein coding exons III-V with a neomycin cassette [Pinhasov et al., 2003]. Homozygous mice are not viable and die prenatally at E8.5-9 due to a failure of cranial neural tube closure. Heterozygous mice develop normally, be it with a slight developmental delays. A detailed differential expression analysis of E9 full knockout, heterozygous and control mice in parallel with the same analysis in P19 cells showed a significant upregulation of genes involved in lipid transport, lytic vacuoles, and coagulation. Downregulated genes clustered in pathways involving regulation of transcription, organogenesis, and neurogenesis [Pinhasov et al., 2003; Mandel et al., 2007]. Typical examples in the first category include the apolipoproteins A1 and E, metalotionine 1 and neurogenin with overexpression in the range of 5–30 fold. For the second category, examples include myosin light chain 2 and neurogenin 1 with a 12-fold and 5-fold underexpression, respectively. The heterozygous knockout mouse model shows tauopathy and cognitive abnormalities as demonstrated in the passive avoidance and Morris water maze tests [Vulih-Shultzman et al., 2007; Gozes et al., 2014]. Interestingly, treatment with NAP or its derivative isoNAP was able to, at least partially, ameliorate the abnormalities in the behavioral tests.
CLINICAL PRESENTATION
All patients had autism, co-morbid in each case with mild to severe intellectual disability. Dysmorphic features as described in the first cohort of 10 patients included a prominent forehead, high hairline, eversion or notch of the eyelid, broad nasal bridge, and thin upper lip [Helsmoortel et al., 2014b]. In the meantime an 11th patient has been identified. His phenotype shares many of the characteristics of the first 10 patients (Tables I and II).
TABLE I.
Comparison of Clinical Features of Patients Caused by Mutations in ADNP or Coffin-Siris genes (Growth, Craniofacial Features, Skeletal Features, Complications, Neurology, Development and Behavior)
| Genes | ADNP | Coffin-siris genes caused by mutations in SMARCB1, SMARCA4, SMARCE1, ARID1A |
|---|---|---|
| Growth and feeding | ||
| Prenatal growth | ||
| Birth weight | −0,17 (n=8) | −1.3 (n=31) |
| Birth length | +0,34 (n=7) | −1.7 (n=17) |
| Birth OFC | +0,7 (n=4) | −0.9 (n=15) |
| Postnatal growth at last observation | ||
| Weight (mean SD score) | +0,5 (n=10) | −2.3 (n=28) |
| Height (mean SD score) | −1,0 (n=10) | −3.3 (n=33) |
| OFC (mean SD score) | +0,2 (n=9) | −2.6 (n=28) |
| Sucking/feeding difficulty | 64% (7/11) | 99% (32/33) |
| Tube feeding | 0% (0/9) | 95% (19/20) |
| Weaned off tube feeding | 0% (0/9) | 38% (6/16) |
| Craniofacial features | ||
| Sparse scalp hair | 0% (0/8) | 65% (20/31) |
| Hypertrichosis | 0% (0/7) | 91% (29/32) |
| Thick eyebrows | 0% (0/8) | 82% (28/34) |
| Long eyelashes | 0% (0/8) | 90% (28/31) |
| Ptosis | 33% (3/9) | 58% (19/33) |
| Nasal bridge | ||
| Wide | 66% (6/9) | 33% (10/30) |
| Flat | 11% (1/9) | 27% (8/30) |
| Normal | 33% (3/9) | 27% (8/30) |
| Narrow | 0% (0/9) | 13% (4/30) |
| Philtrum | ||
| Long | 13% (1/8) | 30% (9/30) |
| Short | 13% (1/8) | 27% (8/30) |
| Normal | 75% (6/8) | 17% (5/30) |
| Broad/long | 13% (1/8) | 17% (5/30) |
| Broad | 0% (0/8) | 7% (2/30) |
| Broad/short | 0% (0/8) | 3% (1/30) |
| Upper lip vermilion | ||
| Thin | 90% (9/10) | 53% (16/30) |
| Normal | 10% (1/10) | 30% (9/30) |
| Everted | 10% (1/10) | 13% (4/30) |
| Thick | 0% (0/10) | 3% (1/30) |
| Lower lip vermilion | ||
| Thick | 25% (2/8) | 82% (27/33) |
| Normal | 75% (6/8) | 18% (6/33) |
| Palatal abnormalities | ||
| Cleft palate | 0% (0/9) | 30% (9/30) |
| Skeletal-limb features | ||
| Hypoplastic 5th fingers or toes | 14%(1/7) | 88% (29/33) |
| Hypoplastic 5th fingernails or toenails | 0% (0/7) | 97% (33/34) |
| Hypoplastic other fingernails and toenails | 14%(1/7) | 65% (20/31) |
| Prominent interphalangeal joints | 14%(1/7) | 33% (9/27) |
| Prominent distal phalanges | 14%(1/7) | 50% (13/26) |
| Scoliosis | 33% (3/9) | 39% (11/28) |
| Internal complications | ||
| Cardiovascular | 27% (3/11) | 15/34 (44%) |
| Gastrointestinal | 66% (6/9) | 65% (20/31) |
| Genitourinary | 11% (1/9) | 32% (10/31) |
| Hernia | 0% (0/7) | 56% (14/25) |
| Hearing and vision | ||
| Hearing impairment | 22% (2/9) | 46% (13/28) |
| Visual impairment | 73% (8/11) | 56% (14/25) |
| Immunology | ||
| Frequent infection | 64% (7/11) | 72% (21/29) |
| Neurology | ||
| Hypotonia | 73% (8/11) | 73% (24/33) |
| Seizures | 18% (2/11) | 44% (14/32) |
| Structural CNS abnormalities | 50% (5/10) | 92% (24/26) |
| Development and intelligence | ||
| Developmental delay and ID | ||
| Severe | 55% (6/11) | 56% (19/32) |
| Moderate to severe | 0% (0/11) | 6% (2/32) |
| Moderate | 18% (2/11) | 22% (7/32) |
| Mild | 27% (3/11) | 13% (4/32) |
| Speech impairment | ||
| No words | 22% (2/9) | 63% (19/30) |
| Several words | 66% (6/9) | 10% (3/30) |
| Sentences | 11% (1/9) | 27% (8/30) |
| Behavior | ||
| Behavioral abnormalities | 78% (7/9) | 65% (15/23) |
SD, standard deviation; CNS, central nervous system; ID, intellectual disability.
TABLE II.
Additional Features of ADNP Patients
| Clinical features | Percentage affected |
|---|---|
| Craniofacial features | |
| High hairline | 63% (5/8) |
| Prominent forehead | 70% (7/10) |
| Malformed ears | 66% (6/9) |
| Obesity | 57% (4/7) |
| Visual impairment | |
| Strabismus | 27% (3/11) |
| Hypermetropia | 55% (6/11) |
| Cardiovascular complications | |
| Atrial Septal Defect | 18% (2/11) |
| Bladder training delay | 100% (5/5) |
| Sleep problems | 45% (5/11) |
| Autism | 100% (11/11) |
| ADHD | 18% (2/11) |
Family history was unremarkable in each case. At the time of birth, the parental ages were in the normal range. None of the couples were consanguineous and no affected siblings have been observed. All children were born at term with birth weight, length, and occipitofrontal circumference in the normal range. Developmental milestones were delayed in all patients. They could sit between 7.5 months and 12 months and walked independently between 19 months and 4.5 years old. All of them have speech problems including one patient that has never developed speech and is now 8.5 years old. Seven of 11 patients have had feeding difficulties, including decreased sucking or chewing and abnormally increased appetite. Bladder training was delayed in five patients. In infancy, eight children have had hypotonia and two have had seizures, but without abnormalities on EEG. More than half of the children have eye defects, mostly hypermetropia or strabismus, but these abnormalities did not always result in visual impairment. A high anterior hairline is seen in five of eight patients of whom sufficiently detailed clinical information is available and one patient has a low hairline (Fig. 3). Seven of 10 patients have a prominent forehead. Other craniofacial features include ptosis, abnormal slant of palpebral fissures, wide nasal bridge, upturned nasal tip, and a thin upper lip. Six of nine patients have ear abnormalities, including small low-set ears, protruding cup shaped ears, and bilateral helical indentation.
Figure 3.

a–f: Patients 1 (a), 2 (b), 4 (c), 5 (d), 6 (e) and 8 (f) at young ages. Note the clinical similarities, including a prominent forehead, a thin upper lip and a broad nasal bridge. Reproduced with permission from Helsmoortel et al., 2004.
Joint hyperlaxity is noted in six of nine children. Almost all children have hand abnormalities, including clinodactyly, polydactyly, small fifth fingers, fetal finger pads, prominent interphalangeal joints, and distal phalanges. Three of eleven patients have cardiac defects, namely atrial septal defect and mitral valve prolapse. Six of nine patients have had gastro-intestinal problems, including esophageal reflux disease, frequent vomiting, and constipation. Most of the children have recurrent infections, including upper respiratory tract and urinary tract infections. Sleep problems are present in five of eleven patients, with good response on melatonin. Several patients are obese. Eight patients have had brain imaging by MRI. Five of them showed abnormalities, including atypical white matter lesions, wide ventricles, and choroid cysts. Behavioral problems are common. Two children were reported to have anxiety problems, three have an obsessive compulsive disorder and two have affectional problems. One of them shows aggressive behavior.
ADNP mutation patients share several clinical features with Coffin-Siris patients, namely feeding difficulties, gastrointestinal problems, visual impairment, frequent infections, hypotonia, structural central nervous system abnormalities, speech impairment, intellectual disability, developmental delay, and behavioral problems (Table I). The ADNP patients identified so far do not have a coarse face with sparse scalp hair, hypertrichosis, thick eyebrows or long eyelashes as observed in patients with Coffin-Siris syndrome, nor have intrauterine or postnatal growth retardation been reported (Tables I and II).
ADNP IN nBAF (mSWI/SNF) COMPLEXES
In vertebrates, ATP-dependent chromatin remodeling is mediated by BAF complexes, which are the functional equivalents of the SWI/SNF complexes in yeast. The human BAF complex consists of 15 subunits, where one of both homologue ATPase core subunits, SMARCA4 or SMARCA2, is always present [Ronan et al., 2013]. Additional components vary with developmental stage and tissue, with theoretically several hundred possible configurations. The exact subunit composition of the complex determines the final functional characteristics and tissue specificity. With regard to neuronal development, a switch of three subunits in the complex composition predisposes the neuronal progenitor cells to a post-mitotic state and initiates activity-dependent dendritic outgrowth and axonal development. This transition occurs in all neurons, and illustrates the fundamental role of BAF complexes in development [Lessard et al., 2007]. Defects hampering the global function of the complex can result in multiple defects in an organism [Ho and Crabtree, 2010]. Illustrative for these global effects of deregulating BAF-mediated chromatin remodeling is that mutations in six components of the neuronal BAF (nBAF) complex (SMARCB1, SMARCA4, SMARCA2, SMARCE1, ARID1A, and ARID1B) have been reported to cause distinct, albeit overlapping, syndromic ID disorders [Santen et al., 2013]. These conditions are now commonly referred to as the “SWI/SNF-related ID syndromes” [Kosho et al., 2013].
Ample evidence exists that ADNP is of key importance for proper functioning of the nBAF complex. This functional relation between ADNP and the nBAF complex is mediated by direct protein-protein interaction of ADNP with several of the BAF subunits [Mandel and Gozes, 2007]. ADNP directly binds to SMARCA2, SMARCA4, and SMARCC2, through its C-terminal end (Fig. 4). Furthermore, reciprocal interactions have been demonstrated between ADNP, the heterochromatin-enriched protein HP1α, and BAF complexes. ADNP is recruited to the pericentromeric heterochromatin through binding of the P*V*L and ARKs motif to HP1α. It can be hypothesized that ADNP by its zinc fingers and homeobox domain functions as an anchoring protein that binds to specific positions in the DNA to guide the protein remodeling complex to its target regions. This way, dysfunction of ADNP results in deregulation of BAF-mediated chromatin remodeling.
Figure 4.
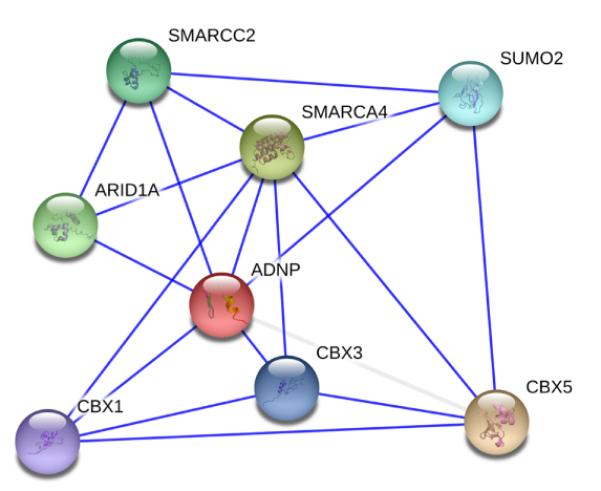
First-level interaction partners of ADNP. Data were retrieved from the STRING database and are based on experimental data only [von Mering et al., 2007].
Mutational Mechanism
The first reported patients all had stop mutations in the fifth and last exon of ADNP. Consistent with predicted escape from nonsense mediated decay for mRNA mutations in the last exon, ADNP mRNA levels were not down-regulated in the four available cell lines of patients with stop mutations [Kervestin and Jacobson, 2012; Helsmoortel et al., 2014b]. Unexpectedly, instead of downregulation, overexpression of the ADNP transcripts was observed in these cell lines. Because three of the mutations clustered within basepairs of each other, we were able to discriminate between expression of the mutant and wild type mRNA. Despite the presence of only one copy of the ADNP gene in the genome, this analysis showed unaltered mRNA levels of the wild-type transcript and the excess level of mRNA in these patients corresponds to the amount of mutated mRNA. It would be interesting to see whether the mutated RNA is translated into protein, but Western blots have so far not been reported for these patients. Recently, the expression of the ADNP gene was reported to be auto-regulated by a negative feedback loop mechanism [Oz et al., 2012]. Hence, a plausible explanation for the observed overexpression in the cell lines of the patients is the inability of the mutant protein to bind to its own promoter region, resulting in a homeostatic correction by up regulation of transcription.
These observations are compatible with a dominant negative mutational mechanism. A dominant negative model is also in line with a discordance in over- or underexpression in our patients versus the results in mice/P19 cells for several genes, but p53 and ADNP2 [Vulih-Shultzman et al., 2007; Helsmoortel et al., 2014b]. However, since the initial report on 10 patients, an 11th patient has been identified with a stop mutation in the fourth exon, which is unlikely to escape nonsense mediated decay. Since the clinical presentation of this patient is not different from that of patients with a mutation in exon five, it challenges the dominant negative hypothesis as the only mutational mechanism. It would be interesting to investigate whether the homeostatic correction of ADNP mRNA levels observed in the initial patients with mutations in exon five, escaping nonsense mediated decay, is present in this patient, where mRNA degradation is predicted. Full homeostatic correction in the presence of NMD would favor the dominant-negative model, but leaves the clinical presentation of Patient 11 unexplained. A reduced ADNP expression in haploinsufficient samples would indicate that the mutant protein actively deregulates the feedback loop. Deregulation might be a consequence of the inability to harvest additional cofactors to the promoter region, while at the same time preventing wild-type protein to bind to the promoter binding site. Similarly, mutant ADNP might occupy alternative target sequences, while being unable to bind to HP1α or BAF. As such, despite the presence of functional BAF complexes, chromatin remodeling is hampered, leading to downstream alterations in gene expression patterns. To test this hypothesis, ChIP-seq and RNA-seq analysis of cell lines of the available patients could be useful.
Therapeutic Potential of NAP
The octapeptide NAP has femtomolar activity and is able to restore some of the anomalies caused by haploinsufficiency of the entire ADNP protein [Bassan et al., 1999; Zamostiano et al., 2001]. In cellular models, it is able to protect the cells from chemical, electrical or stress induced damage [reviewed in Gozes et al., 2005]. The presumed mode of action is NAPs ability to bind to tubulin, facilitating microtubule assembly and increasing cellular survival. Davunetide, the drug name for NAP, is a candidate for the treatment of multiple selected neurological disorders [Gozes, 2011]. Intranasal and intravenous formulations of the drug exist and both have been shown to cross the blood-brain barrier [Gozes et al., 2000a; Leker et al., 2002]. The drug has been in Phase II and even in Phase III clinical trials and appears to be well tolerated without significant side effects [Magen and Gozes, 2014]. As stated above, specific cognitive abnormalities were also ameliorated by NAP in an ADNP heterozygous knockout mouse model. While these observations may raise hope for treatment in patients with ADNP mutations, it has to be noted that the mouse model has not been evaluated for autistic traits so far. Consequently, it is not known whether davunetide is able to interfere with the autistic traits. A second point of interest is that, generally speaking, drug testing in a mouse model needs independent confirmation because of the intrinsic variance associated with this type of experiments and because environmental conditions might interfere with the test result [Crabbe et al., 1999; Wahlsten et al., 2003].
EPILOGUE
The data presented show that mutations in the ADNP gene cause syndromic autism. These findings are in line with the observations by others that genes involved in the chromatin remodeling pathway are over represented in autism/ID. In contrast to related disorders caused by mutations in the SWI/SNF genes SMARCB1, SMARCA4, SMARCA2, SMARCE1, ARID1A, and ARID1B, ADNP is not part of the core nBAF complex, suggesting that a potentially much broader range of SWI/SNF related disorders might exist. All patients reported so far suffer from a combination of clinical characteristics that show some consistency. It should be realized though that all are in their childhood (5–12 years old) and we do not know the clinical presentation of older patients. At present the clinical heterogeneity appears too large to enable identification of additional patients on the basis of clinical selection criteria only. ADNP-related syndromic ASD seems to be more clinically heterogeneous than existing monogenic syndromes. However, this greater heterogeneity between patients with ADNP mutations might be caused by applying a more unbiased gene identification strategy. Until recently, patients were selected for screening a specific gene based on clinical similarity with an existing disorder. This biased selection procedure increases the chance of detecting a mutation in the target gene in two patients with convincing clinical similarity. In contrast, WES is applied on very large cohorts without phenotypic bias, except for a small set of broad inclusion criteria, such as ASD/ID. The clinical heterogeneity of these cohorts diminishes the chances that identical mutations are found in two or more patients with a strong clinical resemblance. If this hypothesis is correct, the phenotypic spectrum of many existing, clinically well delineated rare disorders is likely to expand in the post-exomic era [Yu et al., 2013].
It remains to be seen whether specific ADNP mutations may be associated with non-syndromic autism. The phenotypes of the patients reported here, range from severe autism, co-morbid with ID to less severely affected patients. It is very well possible that the phenotype in larger cohorts of patients with truncating or less damaging ADNP mutations will expand into a much milder range. Seemingly, support of this hypothesis comes from the presence of a likely pathogenic ADNP mutation in the ESP cohort. However, care should be taken in treating ESP as a healthy control cohort with regard to neurological disorders. This is illustrated by multiple likely pathogenic variants in several other genes involved in SWI/SNF related syndromes (Table III), we identified in the ESP database by in silico analysis [Tsurusaki et al., 2012; Kircher et al., 2014]. Unfortunately, detailed phenotypic information is not available on the individuals included in these control databases. The detailed molecular and phenotypical characterization of larger patient cohorts extending in all age groups is mandatory for a better understanding of the role of ADNP mutations in human disease.
TABLE III.
Likely Pathogenic Variants in Coffin-Siris Genes, Present in the Exome Sequencing Project
| Gene | Variant type | Protein change | ESP frequency | Phred (CADD) |
|---|---|---|---|---|
| ARID1A | Truncating | p.(M940Hfs*67) | 0.00399361 | 33.0 |
| Truncating | p.(L2259Vfs*19) | 0.003035144 | 37.0 | |
| ARID1B | Truncating | p.(V2005Wfs*16) | 0.000159898 | 44.0 |
| SMARCA2 | Missense | p.(D82H) | 0.000153775 | 27.6 |
| Missense | p.(P109A) | 0.000153775 | 32.0 | |
| Missense | p.(K862R) | 0.000153775 | 26.9 | |
| Missense | p.(V1567L) | 0.001999077 | 26.9 | |
| Missense | p.(D1573N) | 0.002460403 | 28.5 | |
| SMARCA4 | Missense | p.(R425Q) | 0.000153775 | 34.0 |
| Missense | p.(R1119H) | 0.000153775 | 31.0 | |
| Missense | p.(S1209N) | 0.000153775 | 24.0 | |
| Missense | p.(R1463H) | 0.000153846 | 34.0 | |
| Missense | p.(V1501M) | 0.000153775 | 24.5 | |
| SMARCE1 | Missense | p.(G360D) | 0.003229279 | 25.8 |
| Missense | p.(E336K) | 0.000153775 | 27.5 | |
| Missense | p.(R313H) | 0.000461326 | 32.0 | |
| Missense | p.(R229W) | 0.000153775 | 23.6 | |
| Missense | p.(R148C) | 0.000153775 | 34.0 |
Genes were selected as causative for Coffin-Siris from Tsurusaki et al. [2012]. ESP frequency is combined frequency for all populations. Phred (CADD) is the Phred-scaled relative rank of CADD score for this variant [Kircher et al., 2014]. Scaled scores higher than 23 represent the 0.5% most likely pathogenic variants amongst all 8.6 billion possible genomic variants.
In an attempt to identify novel genes responsible for autism, a first de novo p. Lys408Valfs*31 mutation in the activity-dependent neuroprotective protein (ADNP)gene was identified in a large cohort of autistic patients.
The ADNP gene was first identified in murine P19 carcinoma cells as a vasoactive intestinal peptide (VIP) responsive gene showing increased expression after VIP treatment. VIP is a neuroprotective peptide that is active during embryonic development, especially during the time of neuronal tube closure, and protects damaged nerve cells from cell death by inducing glia-derived, survival promoting substances.
ADNP mutation patients share several clinical features with Coffin-Siris patients, namely feeding difficulties, gastrointestinal problems, visual impairment, frequent infections, hypotonia, structural central nervous system abnormalities, speech impairment, intellectual disability, developmental delay and behavioral problems (Table I).
Ample evidence exists that ADNP is of key importance for proper functioning of the nBAF complex. This functional relation between ADNP and the nBAF complex is mediated by direct protein-protein interaction of ADNP with several of the BAF subunits. ADNP directly binds to SMARCA2, SMARCA4 and SMARCC2, through its C-terminal end.
These observations are compatible with a dominant negative mutational mechanism. A dominant negative model is also in line with a discordance in over- or underexpression in our patients versus the results in mice/P19 cells forseveral genes, but p53 and ADNP2.
ACKNOWLEDGMENTS
This work was funded by the Belgian National Fund for Scientific Research-Flanders (FWO) to GV and RFK, the Special Research Fund of the University of Antwerp (Bijzonder Onderzoeksfonds (BOF-IWT)) to CH, by grants from the Dutch Organization for Health Research and Development (917-86-319 and 40-00812-98-12109 to BBAdV and 907-00-365 to TK), the EU-funded GENCODYS project (EU-7th-2010-241995 to ATVvS, BBAdV, and TK), Simons Foundation Autism Research Initiative award (SFARI191889EE to EEE) and NIH (MH101221 to EEE). EEE is an investigator of the Howard Hughes Medical Institute.
Grant sponsor: Dutch Organization for Health Research and Development; Grant number: 917-86-319, 40-00812-98-12109, 907-00-365; Grant sponsor: EU; Grant number: EU-7th-2010-241995; Grant sponsor: Simons Foundation Autism Research Initiative award; Grant number: SFARI191889EE; Grant sponsor: NIH; Grant number: MH101221.
Footnotes
Competing financial interests: EEE is on the scientific advisory boards for Pacific Biosciences, Inc., SynapDx Corp., and DNAnexus, Inc.
Contributor Information
Geert Vandeweyer, Department of Medical Genetics of the University of Antwerp, Belgium..
Céline Helsmoortel, Department of Medical Genetics of the University of Antwerp, Belgium..
Anke Van Dijck, Department of Medical Genetics of the University and Universtiy Hospital Antwerp, Belgium..
Anneke T. Vulto-van Silfhout, Department of Human Genetics in the Nijmegen Centre for Molecular Life Sciences from the Radboud University Medical Center in Nijmegen, The Netherlands..
Bradley P. Coe, Departement of Genome Sciences of the University of Washington, USA..
Raphael Bernier, Department of Psychiatry of the University of Washington, USA..
Jennifer Gerdts, Department of Psychiatry of the University of Washington, USA..
Liesbeth Rooms, Departement of Medical Genetics of the Antwerp University Hospital..
Madhura Bakshi, Children’s Hospital of Westmead, USA..
Jenneke van den Ende, Departement of Medical Genetics of the University and University Hospital of Antwerp..
Meredith Wilson, Department of Clinical Genetics at the Children’s Hospital of Westmead, USA..
Ann Nordgren, Department of Molecular Medicine and Surgery of the Karolinska Institutet, Sweden..
Laura G. Hendon, University of Mississippi Medical Center of Jackson, USA..
Omar A. Abdulrahman, University of Mississippi Medical Center of Jackson, USA..
Corrado Romano, I.R.C.C.S. Associazione Oasi Maria Santissima, Troina, Italy..
Bert B.A. de Vries, Department and at the Human Genetics in the Nijmegen Centre for Molecular Life Sciences, both from the Radboud University Medical Center in Nijmegen, The Netherlands..
Tjitske Kleefstra, Department and at the Human Genetics in the Nijmegen Centre for Molecular Life Sciences, both from the Radboud University Medical Center in Nijmegen, The Netherlands..
Evan E. Eichler, University of Washington, USA and an investigator of the Howard Hughes Medical Institute..
Nathalie Van der Aa, Departement of Medical Genetics of the Antwerp University and University Hospital..
R. Frank Kooy, Department of Medical Genetics of the University of Antwerp, Belgium..
REFERENCES
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- APA . American Psychiatric Association: Diagnostic and statistical manual of mental disorders DSM-V. 5th edition American Psychiatric Association; Text revision Washington DC: 2013. [Google Scholar]
- Bassan M, Zamostiano R, Davidson A, Pinhasov A, Giladi E, Perl O, Bassan H, Blat C, Gibney G, Glazner G, Brenneman DE, Gozes I. Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J Neurochem. 1999;72:1283–1293. doi: 10.1046/j.1471-4159.1999.0721283.x. [DOI] [PubMed] [Google Scholar]
- Carter MT, Scherer SW. Autism spectrum disorder in the genetics clinic: A review. Clin Genet. 2013;83:399–407. doi: 10.1111/cge.12101. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Wahlsten D, Dudek BC. Genetics of mouse behavior: Interactions with laboratory environment. Science (New York, NY) 1999;284:1670–1672. doi: 10.1126/science.284.5420.1670. [DOI] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, del Rosario M, Hoischen A, Scheffer H, de Vries BB, Brunner HG, Veltman JA, Vissers LE. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- Dresner E, Agam G, Gozes I. Activity-dependent neuroprotective protein (ADNP) expression level is correlated with the expression of the sister protein ADN P2: Deregulation in schizophrenia. Eur Neuropsychopharmacol. 2011;21:355–361. doi: 10.1016/j.euroneuro.2010.06.004. [DOI] [PubMed] [Google Scholar]
- ESP. NGESP Exome Variant Server.
- Folstein S, Rutter M. Genetic influences and infantile autism. Nature. 1977;265:726–728. doi: 10.1038/265726a0. [DOI] [PubMed] [Google Scholar]
- Furman S, Steingart RA, Mandel S, Hauser JM, Brenneman DE, Gozes I. Subcellular localization and secretion of activity-dependent neuroprotective protein in astrocytes. Neuron Glia Biol. 2004;1:193–199. doi: 10.1017/S1740925X05000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennet N, Herden C, Bubb VJ, Quinn JP, Kipar A. Expression of activity-dependent neuroprotective protein in the brain of adult rats. Histol Histopathol. 2008;23:309–317. doi: 10.14670/HH-23.309. [DOI] [PubMed] [Google Scholar]
- Gozes I. Microtubules, schizophrenia and cognitive behavior: Preclinical development of davunetide (NAP) as a peptide-drug candidate. Peptides. 2011;32:428–431. doi: 10.1016/j.peptides.2010.10.030. [DOI] [PubMed] [Google Scholar]
- Gozes I, Giladi E, Pinhasov A, Bardea A, Brenneman DE. Activity-dependent neurotrophic factor: Intranasal administration of femtomolar-acting peptides improve performance in a water maze. J Pharmacol Exp Ther. 2000a;293:1091–1098. [PubMed] [Google Scholar]
- Gozes I, Morimoto BH, Tiong J, Fox A, Sutherland K, Dangoor D, Holser Cochav M, Vered K, Newton P, Aisen PS, Matsuoka Y, van Dyck CH, Thal L. NAP: Research and development of a peptide derived from activity-dependent neuroprotective protein (ADNP) CNS Drug Rev. 2005;11:353–368. doi: 10.1111/j.1527-3458.2005.tb00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozes I, Schirer Y, Idan Feldman A, David M, Furman Assaf S. NAP Alpha-Amino-isobutyric Acid (IsoNAP) J Mol Neurosci. 2014;52:1–9. doi: 10.1007/s12031-013-0103-8. [DOI] [PubMed] [Google Scholar]
- Gozes I, Steingart RA, Spier AD. NAP mechanisms of neuroprotection. J Mol Neurosci. 2004;24:67–72. doi: 10.1385/JMN:24:1:067. [DOI] [PubMed] [Google Scholar]
- Gozes I, Zamostiano R, Pinhasov A, Bassan M, Giladi E, Steingart RA, Brenneman DE. A novel VIP responsive gene. Activity dependent neuroprotective protein. Ann N Y Acad Sci. 2000b;921:115–118. doi: 10.1111/j.1749-6632.2000.tb06957.x. [DOI] [PubMed] [Google Scholar]
- Helsmoortel C, Vandeweyer G, Ordoukhanian P, Van Nieuwerburgh F, Van der Aa N, Kooy RF. Challenges and opportunities in the investigation of unexplained intellectual disability using family based whole exome sequencing. Clinical genetics:Clin Genet. 2014a Aug; doi: 10.1111/cge.12470. 2014. 2011. doi: 2010. 1111/cge.12470. [DOI] [PubMed] [Google Scholar]
- Helsmoortel C, Vulto-van Silfhout AT, Coe BP, Vandeweyer G, Rooms L, van den Ende J, Schuurs-Hoeijmakers JH, Marcelis CL, Willemsen MH, Vissers LE, Yntema HG, Bakshi M, Wilson M, Witherspoon KT, Malmgren H, Nordgren A, Anneren G, Fichera M, Bosco P, Romano C, de Vries BB, Kleefstra T, Kooy RF, Eichler EE, Van der Aa N. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat Genet. 2014b;46:380–384. doi: 10.1038/ng.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervestin S, Jacobson A. NMD: A multifaceted response to premature translational termination. Nat Rev Mol Cell Biol. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosho T, Okamoto N, Ohashi H, Tsurusaki Y, Imai Y, Hibi-Ko Y, Kawame H, Homma T, Tanabe S, Kato M, Hiraki Y, Yamagata T, Yano S, Sakazume S, Ishii T, Nagai T, Ohta T, Niikawa N, Mizuno S, Kaname T, Naritomi K, Narumi Y, Wakui K, Fukushima Y, Miyatake S, Mizuguchi T, Saitsu H, Miyake N, Matsumoto N. Clinical correlations of mutations affecting six components of the SWI/SNF complex: Detailed description of 21 patients and a review of the literature. Am J Med Genet. 2013;161A:1221–1237. doi: 10.1002/ajmg.a.35933. [DOI] [PubMed] [Google Scholar]
- Krumm N, O’Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014;37:95–105. doi: 10.1016/j.tins.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leker RR, Teichner A, Grigoriadis N, Ovadia H, Brenneman DE, Fridkin M, Giladi E, Romano J, Gozes I. NAP, a femtomolar-acting peptide, protects the brain against ischemic injury by reducing apoptotic death. Stroke. 2002;33:1085–1092. doi: 10.1161/01.str.0000014207.05597.d7. [DOI] [PubMed] [Google Scholar]
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 2007;55:201–215. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magen I, Gozes I. Davunetide: Peptide therapeutic in neurological disorders. Curr Med Chem. 2014;21:2591–2598. doi: 10.2174/0929867321666140217124945. [DOI] [PubMed] [Google Scholar]
- Mandel S, Gozes I. Activity-dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J Biol Chem. 2007;282:34448–34456. doi: 10.1074/jbc.M704756200. [DOI] [PubMed] [Google Scholar]
- Mandel S, Rechavi G, Gozes I. Activity- dependent neuroprotective protein (ADNP) differentially interacts with chromatin to regulate genes essential for embryogenesis. Dev Biol. 2007;303:814–824. doi: 10.1016/j.ydbio.2006.11.039. [DOI] [PubMed] [Google Scholar]
- Mandel S, Spivak-Pohis I, Gozes I. ADNP differential nucleus/cytoplasm localization in neurons suggests multiple roles in neuronal differentiation and maintenance. J Mol Neurosci. 2008;35:127–141. doi: 10.1007/s12031-007-9013-y. [DOI] [PubMed] [Google Scholar]
- Mendelsohn NJ, Schaefer GB. Genetic evaluation of autism. Semin Pediatr Neurol. 2008;15:27–31. doi: 10.1016/j.spen.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Mosch K, Franz H, Soeroes S, Singh PB, Fischle W. HP1 recruits activity-dependent neuroprotective protein to H3K9me3 marked pericentromeric heterochromatin for silencing of major satellite repeats. PloS One. 2011;6:e15894. doi: 10.1371/journal.pone.0015894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, Munson J, Hiatt JB, Turner EH, Levy R, O’Day DR, Krumm N, Coe BP, Martin BK, Borenstein E, Nickerson DA, Mefford HC, Doherty D, Akey JM, Bernier R, Eichler EE, Shendure J. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science (New York, NY) 2012a;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012b;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz S, Ivashko-Pachima Y, Gozes I. The ADNP derived peptide, NAP modulates the tubulin pool: Implication for neurotrophic and neuroprotective activities. PloS One. 2012;7:e51458. doi: 10.1371/journal.pone.0051458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinhasov A, Mandel S, Torchinsky A, Giladi E, Pittel Z, Goldsweig AM, Servoss SJ, Brenneman DE, Gozes I. Activity-dependent neuroprotective protein: A novel gene essential for brain formation. Brain Res Dev Brain Res. 2003;144:83–90. doi: 10.1016/s0165-3806(03)00162-7. [DOI] [PubMed] [Google Scholar]
- Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z, Vorstman JA, Thompson A, Regan R, Pilorge M, Pellecchia G, Pagnamenta AT, Oliveira B, Marshall CR, Magalhaes TR, Lowe JK, Howe JL, Griswold AJ, Gilbert J, Duketis E, Dombroski BA, De Jonge MV, Cuccaro M, Crawford EL, Correia CT, Conroy J, Conceicao IC, Chiocchetti AG, Casey JP, Cai G, Cabrol C, Bolshakova N, Bacchelli E, Anney R, Gallinger S, Cotterchio M, Casey G, Zwaigenbaum L, Wittemeyer K, Wing K, Wallace S, van Engeland H, Tryfon A, Thomson S, Soorya L, Roge B, Roberts W, Poustka F, Mouga S, Minshew N, McInnes LA, McGrew SG, Lord C, Leboyer M, Le Couteur AS, Kolevzon A, Jimenez Gonzalez P, Jacob S, Holt R, Guter S, Green J, Green A, Gillberg C, Fernandez BA, Duque F, Delorme R, Dawson G, Chaste P, Cafe C, Brennan S, Bourgeron T, Bolton PF, Bolte S, Bernier R, Baird G, Bailey AJ, Anagnostou E, Almeida J, Wijsman EM, Vieland VJ, Vicente AM, Schellenberg GD, Pericak-Vance M, Paterson AD, Parr JR, Oliveira G, Nurnberger JI, Monaco AP, Maestrini E, Klauck SM, Hakonarson H, Haines JL, Geschwind DH, Freitag CM, Folstein SE, Ennis S, Coon H, Battaglia A, Szatmari P, Sutcliffe JS, Hallmayer J, Gill M, Cook EH, Buxbaum JD, Devlin B, Gallagher L, Betancur C, Scherer SW. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94:677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BH, Cochrane L, Corsello C, Crawford EL, Crossett A, Cytrynbaum C, Dawson G, de Jonge M, Delorme R, Drmic I, Duketis E, Duque F, Estes A, Farrar P, Fernandez BA, Folstein SE, Fombonne E, Freitag CM, Gilbert J, Gillberg C, Glessner JT, Goldberg J, Green A, Green J, Guter SJ, Hakonarson H, Heron EA, Hill M, Holt R, Howe JL, Hughes G, Hus V, Igliozzi R, Kim C, Klauck SM, Kolevzon A, Korvatska O, Kustanovich V, Lajonchere CM, Lamb JA, Laskawiec M, Leboyer M, Le Couteur A, Leventhal BL, Lionel AC, Liu XQ, Lord C, Lotspeich L, Lund SC, Maestrini E, Mahoney W, Mantoulan C, Marshall CR, McConachie H, McDougle CJ, McGrath J, McMahon WM, Merikangas A, Migita O, Minshew NJ, Mirza GK, Munson J, Nelson SF, Noakes C, Noor A, Nygren G, Oliveira G, Papanikolaou K, Parr JR, Parrini B, Paton T, Pickles A, Pilorge M, Piven J, Ponting CP, Posey DJ, Poustka A, Poustka F, Prasad A, Ragoussis J, Renshaw K, Rickaby J, Roberts W, Roeder K, Roge B, Rutter ML, Bierut LJ, Rice JP, Salt J, Sansom K, Sato D, Segurado R, Sequeira AF, Senman L, Shah N, Sheffield VC, Soorya L, Sousa I, Stein O, Sykes N, Stoppioni V, Strawbridge C, Tancredi R, Tansey K, Thiruvahindrapduram B, Thompson AP, Thomson S, Tryfon A, Tsiantis J, Van Engeland H, Vincent JB, Volkmar F, Wallace S, Wang K, Wang Z, Wassink TH, Webber C, Weksberg R, Wing K, Wittemeyer K, Wood S, Wu J, Yaspan BL, Zurawiecki D, Zwaigenbaum L, Buxbaum JD, Cantor RM, Cook EH, Coon H, Cuccaro ML, Devlin B, Ennis S, Gallagher L, Geschwind DH, Gill M, Haines JL, Hallmayer J, Miller J, Monaco AP, Nurnberger JI, Jr, Paterson AD, Pericak-Vance MA, Schellenberg GD, Szatmari P, Vicente AM, Vieland VJ, Wijsman EM, Scherer SW, Sutcliffe JS, Betancur C. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, Dufke A, Cremer K, Hempel M, Horn D, Hoyer J, Joset P, Ropke A, Moog U, Riess A, Thiel CT, Tzschach A, Wiesener A, Wohlleber E, Zweier C, Ekici AB, Zink AM, Rump A, Meisinger C, Grallert H, Sticht H, Schenck A, Engels H, Rappold G, Schrock E, Wieacker P, Riess O, Meitinger T, Reis A, Strom TM. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: An exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- Ronan JL, Wu W, Crabtree GR. From neural development to cognition: Unexpected roles for chromatin. Nat Rev Genet. 2013;14:347–359. doi: 10.1038/nrg3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooms L, Kooy RF. Advances in understanding fragile X syndrome and related disorders. Curr Opin Pediatr. 2011;23:601–606. doi: 10.1097/MOP.0b013e32834c7f1a. [DOI] [PubMed] [Google Scholar]
- Said SI. Molecules that protect: The defense of neurons and other cells. J Clin Invest. 1996;97:2163–2164. doi: 10.1172/JCI118655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Wright NR, Dhodapkar RM, DiCola M, DiLullo NM, Fernandez TV, Fielding-Singh V, Fishman DO, Frahm S, Garagaloyan R, Goh GS, Kammela S, Klei L, Lowe JK, Lund SC, McGrew AD, Meyer KA, Moffat WJ, Murdoch JD, O’Roak BJ, Ober GT, Pottenger RS, Raubeson MJ, Song Y, Wang Q, Yaspan BL, Yu TW, Yurkiewicz IR, Beaudet AL, Cantor RM, Curland M, Grice DE, Gunel M, Lifton RP, Mane SM, Martin DM, Shaw CA, Sheldon M, Tisch-field JA, Walsh CA, Morrow EM, Ledbetter DH, Fombonne E, Lord C, Martin CL, Brooks AI, Sutcliffe JS, Cook EH, Jr, Geschwind D, Roeder K, Devlin B, State MW. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. JAMA. 2014;311:1770–1777. doi: 10.1001/jama.2014.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen GW, Aten E, Vulto-van Silfhout AT, Pottinger C, van Bon BW, van Minderhout IJ, Snowdowne R, van der Lans CA, Boogaard M, Linssen MM, Vijfhuizen L, van der Wielen MJ, Vollebregt MJ, Breuning MH, Kriek M, van Haeringen A, den Dunnen JT, Hoischen A, Clayton-Smith J, de Vries BB, Hennekam RC, van Belzen MJ, Almureikhi M, Baban A, Barbosa M, Ben-Omran T, Berry K, Bigoni S, Boute O, Brueton L, van der Burgt I, Canham N, Chandler KE, Chrzanowska K, Collins AL, de Toni T, Dean J, den Hollander NS, Flore LA, Fryer A, Gardham A, Graham JM, JR, Harrison V, Horn D, Jongmans MC, Josifova D, Kant SG, Kapoor S, Kingston H, Kini U, Kleefstra T, Krajewska-Walasek M, Kramer N, Maas SM, Maciel P, Mancini GM, Maystadt I, McKee S, Milunsky JM, Nampoothiri S, Newbury-Ecob R, Nikkel SM, Parker MJ, Perez-Jurado LA, Robertson SP, Rooryck C, Shears D, Silengo M, Singh A, Smigiel R, Soares G, Splitt M, Stewart H, Sweeney E, Tassabehji M, Tuysuz B, van Eerde M, Vincent-Delorme C, Wilson LC, Yesil G. Coffin-Siris syndrome and the BAF complex: Genotype-phenotype study in 63 patients. Hum Mutat. 2013;34:1519–1528. doi: 10.1002/humu.22394. [DOI] [PubMed] [Google Scholar]
- Steffenburg S, Gillberg C, Hellgren L, Andersson L, Gillberg IC, Jakobsson G, Bohman M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry. 1989;30:405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- Surveillance DDMN. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63:1–21. [PubMed] [Google Scholar]
- Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, Fukushima Y, Homma T, Kato M, Hiraki Y, Yamagata T, Yano S, Mizuno S, Sakazume S, Ishii T, Nagai T, Shiina M, Ogata K, Ohta T, Niikawa N, Miyatake S, Okada I, Mizuguchi T, Doi H, Saitsu H, Miyake N, Matsumoto N. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet. 2012;44:376–378. doi: 10.1038/ng.2219. [DOI] [PubMed] [Google Scholar]
- Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- Vissers LELM, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, van Bon BW, Hoischen A, de Vries BB, Brunner HG, Veltman JA. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- von Mering C, Jensen LJ, Kuhn M, Chaffron S, Doerks T, Kruger B, Snel B, Bork P. STRING 7–recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007;35(Database issue):D358–362. doi: 10.1093/nar/gkl825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulih-Shultzman I, Pinhasov A, Mandel S, Grigoriadis N, Touloumi O, Pittel Z, Gozes I. Activity-dependent neuroprotective protein snippet NAP reduces tau hyper-phosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther. 2007;323:438–449. doi: 10.1124/jpet.107.129551. [DOI] [PubMed] [Google Scholar]
- Wahlsten D, Rustay NR, Metten P, Crabbe JC. In search of a better mouse test. Trends Neurosci. 2003;26:132–136. doi: 10.1016/S0166-2236(03)00033-X. [DOI] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, Hardison M, Person R, Bekheirnia MR, Leduc MS, Kirby A, Pham P, Scull J, Wang M, Ding Y, Plon SE, Lupski JR, Beaudet AL, Gibbs RA, Eng CM. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, Schmitz-Abe K, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–273. doi: 10.1016/j.neuron.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamostiano R, Pinhasov A, Gelber E, Steingart RA, Seroussi E, Giladi E, Bassan M, Wollman Y, Eyre HJ, Mulley JC, Brenneman DE, Gozes I. Cloning and characterization of the human activity-dependent neuro-protective protein. J Biol Chem. 2001;276:708–714. doi: 10.1074/jbc.M007416200. [DOI] [PubMed] [Google Scholar]