

Abstract
RNA-seq and especially differential RNA-seq-type transcriptomic analyses (dRNA-seq) are powerful analytical tools, as they not only provide insights into gene expression changes but also provide detailed information about all promoters active at a given moment, effectively giving a deep insight into the transcriptional landscape. Synechocystis sp. PCC 6803 (Synechocystis 6803) is a unicellular model cyanobacterium that is widely used in research fields from ecology, photophysiology to systems biology, modelling and biotechnology. Here, we analysed the response of the Synechocystis 6803 primary transcriptome to different, environmentally relevant stimuli. We established genome-wide maps of the transcriptional start sites active under 10 different conditions relevant for photosynthetic growth and identified 4,091 transcriptional units, which provide information about operons, 5′ and 3′ untranslated regions (UTRs). Based on a unique expression factor, we describe regulons and relevant promoter sequences at single-nucleotide resolution. Finally, we report several sRNAs with an intriguing expression pattern and therefore likely function, specific for carbon depletion (CsiR1), nitrogen depletion (NsiR4), phosphate depletion (PsiR1), iron stress (IsaR1) or photosynthesis (PsrR1). This dataset is accompanied by comprehensive information providing extensive visualization and data access to allow an easy-to-use approach for the design of experiments, the incorporation into modelling studies of the regulatory system and for comparative analyses.
Keywords: comparative transcriptome analysis, cyanobacteria, regulation of gene expression, sRNA, transcriptional unit
1. Introduction
1.1. Synechocystis sp. PCC 6803 as a model for fundamental and applied research in cyanobacteria
The production of biofuels based on oxygenic photosynthesis in metabolically engineered cyanobacteria is considered a major resource for future energy.1–8 Synechocystis sp. PCC 6803 (from here: Synechocystis 6803), a unicellular cyanobacterium naturally competent for DNA uptake and chromosomal integration by homologous recombination,9–12 is frequently used in these studies. However, Synechocystis 6803 is also widely used in fundamental research on ecological or photophysiological questions and to unravel the reactions and metabolites of photosynthetic primary metabolism.13–15
Based on global proteomic16,17 or microarray-based approaches,18–20 the response of Synechocystis 6803 to many different environmental conditions has been elucidated. A genome-wide mapping of its transcriptional start sites (TSSs) under standard growth conditions revealed 3,527 active TSSs,21 from which only one-third (1,165) was located within a distance of 100 nt upstream of an annotated gene or associated with pyrosequencing reads reaching into the coding sequence (gTSS), while another third (1,112) was on the reverse complementary strand of 866 genes (aTSS), suggesting massive antisense transcription. Last but not least, 429 seemingly orphan TSSs (nTSSs) located in intergenic regions led to the prediction of non-coding RNAs (ncRNAs). Thus, with ∼64% of all TSSs, the major transcriptional output was found to consist of ncRNA in a genome that is to 87% protein-coding.21 However, the existing genome-wide map of TSSs was inferred from cultures grown under standard growth conditions only. Therefore, the suite of promoters active under different conditions has largely remained unknown, rendering the systematic comparison of promoter sequences and putative regulatory elements impossible.
1.2. Comparative approaches to microbial transcriptomic analyses
RNA-seq and especially differential RNA-seq-type transcriptomic analyses (dRNA-seq22) are powerful analytical tools. Comparative transcriptomics has been used to study the differences in transcriptional regulation under different environmental conditions. Such an analysis compared the primary transcriptomes of the major human pathogen Helicobacter pylori under mid-logarithmic growth phase versus acid stress conditions, mimicking the host environment.22 Comparative analysis of the primary transcriptome of the cyanobacterium Anabaena sp. PCC 7120 revealed >10,000 TSSs active during the differentiation of N2-fixing heterocysts, from which >900 TSSs exhibited minimum fold changes of eight, suggesting a large number of hitherto unidentified regulators of cell differentiation and N2-fixation.23
Here, we describe a large-scale approach to analyse the response of the Synechocystis 6803 primary transcriptome to different, environmentally relevant stimuli. We established genome-wide maps of the 5,162 TSSs active under at least one of the 10 different growth conditions, which were employed to identify 4,091 transcriptional units (TUs) on the chromosome and the plasmids pSYSA, pSYSG, pSYSM and pSYSX. Based on a unique expression factor (UEF) for every TSS, we describe regulons and relevant promoter sequences for 10 conditions important for photosynthetic growth at single-nucleotide resolution.
2. Materials and methods
2.1. Biological material and growth conditions
We used the Synechocystis 6803 strain ‘PCC-M’24 throughout this work. Liquid cultures were grown in 100 ml Erlenmeyer flasks using 75 ml of BG11 medium25 at 30°C under continuous white light illumination of 50–80 µmol quanta m−2 s−1 and a continuous stream of air to the desired growth phase (OD750 = 0.6–0.8). For the transcriptome analyses, cells were harvested from exponentially growing cultures which were then transferred to nine different conditions: (i) cold stress, 15°C for 30 min; (ii) heat stress, 42°C for 30 min; (iii) Ci depletion, cells were washed three times with carbon-free BG11 and cultivated further for 20 h; (iv) dark, no light for 12 h; (v) Fe2+ limitation, addition of iron-specific chelator desferrioxamine B (DFB) and further cultivation for 24 h; (vi) high light, 470 µmol quanta m−2 s−1 for 30 min; (vii) N depletion, cells were washed three times with nitrogen-free BG11 and cultivated further for 12 h; (viii) P depletion, cells were washed three times with phosphate-free BG11 and further incubated for 12 h; (ix) stationary phase, cells were grown until an OD750 of 4.7 was reached.
2.2. RNA extraction, cDNA synthesis and sequencing
Synechocystis 6803 cultures were harvested by rapid filtration on hydrophilic polyethersulfone filters (Pall Supor 800 Filter, 0.8 µm). The filter covered with cells was immediately immersed in 1 ml of PGTX solution26 and frozen in liquid nitrogen. Total RNA was extracted and analysed by gel electrophoresis and Northern blotting as described.27 For each condition, total RNA from two independent cultures was pooled for the following analyses. For sequence analysis, cDNA libraries were constructed (vertis Biotechnologie AG, Germany) and analysed on an Illumina sequencer as previously described.23 The dRNA-seq protocol22 distinguishes treated and untreated libraries. For the treated libraries, total RNA was fragmented with ultrasound (four pulses of 30 s at 4°C) and RNA species that carry a 5′ mono-phosphate were degraded using Terminator™ 5′ phosphate-dependent exonuclease (TEX, Epicentre). The exonuclease-resistant mRNA (mainly primary transcripts with 5′-PPP) was poly(A)-tailed using poly(A) polymerase. Then, 5′-PPP RNA was cleaved enzymatically using tobacco acid pyrophosphatase (TAP), ligated to an RNA linker23 and first-strand cDNA synthesis initiated using an oligo(dT)-adapter primer and M-MLV reverse transcriptase. The second-strand cDNA synthesis was primed with a biotinylated antisense 5′-Solexa primer, after which cDNA fragments were bound to streptavidin beads. Bead-bound cDNA was blunted and 3′ ligated to a Solexa adapter. The cDNA fragments were amplified by 10–12 cycles of PCR. The resulting cDNA samples were double-stranded with a size of ∼150–700 bp. For the untreated libraries, we pooled total RNA from samples representing all 10 growth conditions and depleted rRNA using the MICROBExpress kit (Ambion). Except for the TEX treatment, the libraries were handled as described for the treated libraries.
2.3. Read mapping and data normalization
A total of 344,768,773 reads was obtained for the 10 different conditions. The data were deposited in the NCBI Sequence Read Archive under accession SRP032228. A total of 300,073,773 reads (56,611,278 non-ribosomal reads) were mapped to the genome with segemehl28 using default parameters. The data were normalized in two steps. First, the treated libraries were scaled to library sizes of 100 million reads. Positions with read starts ≥500 and with a ratio >0.5 between read starts and coverage were defined as true primary positions. In a second step, library-specific correction factors based on the fraction of counts at true primary positions were applied. For the two untreated libraries, we obtained 33,357,164 and 31,985,927 reads, of which 4,058,785 and 3,039,856 mapped to non-ribosomal regions. The counts for the two untreated libraries were scaled to counts per 100 million and averaged.
2.4. Prediction of TUs
Automated detection of TUs by RNAseg29 is based on start positions and coverage by reads from the treated libraries and the coverage by reads from the untreated libraries. We used a combined dataset of all treated libraries, which provides for each genomic position the maximum number of read starts from any library together with the coverage from the same library. For the TU prediction, RNAseg was applied with the following settings: ‘−R 500 −l 0.5 −a 100 −k 20,000 −u 200 −w 100’. Thus, a TU has to have a minimum of 500 reads from a treated library starting at its TSS, a ratio of at least 0.5 for reads starting from the treated library versus those covering the TSS, an average coverage of 100 reads from the untreated library and a maximum length of 20 kb. Non-TU regions must not exceed an average coverage of 200 from the untreated library. The chromosome was split into 35 parts of ∼100 kb length overlapping by 20,000 nt. For each part, RNAseg produced predictions with different numbers of TUs up to a maximum, which was imposed by the parameter settings. We extracted the maximal number of TUs for the individual parts and merged them. In the case of inconsistencies in the overlapping regions, the longer TU was kept. RNAseg cannot handle overlapping TUs, which originate from internal TSSs (iTSSs). As a result, a TU may end directly in front of an iTSS, while it likely extends to the same end as the transcript originating from this iTSS. Therefore, adjacent TUs were merged when their contact point was located within an annotated gene on the same strand. The contact points were stored as candidate iTSS. We termed a TU gTU when one or more annotated genes were covered, aTU when it was antisense to annotated genes or TUs (overlap ≥20 nt), iTU when the TU was located within an annotated gene and nTU when it was free-standing. The classification of a TU is frequently ambiguous, e.g. a TU covering annotated genes, extending into a region antisense to a downstream gene or TU on the opposite strand, as it is known for excludons.30 In such cases, we provided all possible notations with an order of g > a, which for gene internally located TSSs (iTSSs) was further complemented by an ‘i’. Thus, a TU starting at an iTSS, covering an annotated gene and extending into an antisense region, was termed gaiTU.
2.5. Leaderless transcript detection
gTSSs that were mapped to start codons, either the A of AUG or the preceding 10 nucleotides, were subjected to a closer inspection. If N-terminally shorter homologues in other bacteria were found, these gTSSs indicated mis-annotated start codons and the corresponding gene locations were updated.
2.6. Northern blot analysis
Selected transcripts were verified by Northern hybridization using single-stranded radioactively labelled RNA probes, which were generated by in vitro transcription as described.31 The oligonucleotides used to amplify the according probe templates are listed in Supplementary Table S3. For the analysis, 3 µg of total RNA were separated on 1.5% agarose gels, transferred to Hybond-N nylon membranes by capillary blotting and cross-linked by UV-illumination and hybridized, as described earlier.31 Signals were visualized by using a Personal Molecular Imager FX system with Quantity One software (Bio-Rad).
3. Results
3.1. High-resolution transcriptomes
We isolated total RNA from cells cultivated under 10 different conditions (darkness, high light, cold and heat stress, depletion of iron, phosphate, nitrogen or inorganic carbon, exponential and stationary growth phase) and prepared cDNA libraries enriched for primary transcripts22 (treated libraries). In addition, we prepared two untreated libraries for RNA pooled from all conditions. The combined data of the two untreated libraries provided sequencing reads distributed over the complete length of a transcript, based on which TUs were defined. Depending on the context, we term a TU gTU when it covers one or more annotated genes, aTU when it is antisense to another TU and nTU when it is free-standing (for details see Section 2). Each TU is associated with a TSS that is named accordingly, and the TSS's read count from the treated library defines the expression level of the TU. This and the complexity of TU definition are illustrated in Fig. 1. The flavoprotein (flv4-2) operon (TU156) comprises the genes, sll0217 (flv4), sll0218 and sll0219 (flv2), and several short asRNAs, of which As1_flv432 (TU158) is by far the most abundant one. As1_flv4 sets a threshold on the expression of the flv4-2 operon,32 thus preventing premature expression. Under standard conditions, the expression of this operon would be disadvantageous, because flavodiiron proteins encoded by this operon open up an alternative electron transfer route.33 The situation changes in high light or carbon limitation, when this photoprotective function is needed. Indeed, our data show strong induction of the flv4-2 gTSS under carbon limitation and high light (Fig. 1), consistent with previous observations that were obtained by other methods.32,34 Interestingly, we found an additional iTSS (within TU156) within the coding region of flv4.
Figure 1.

Annotation of TSSs and inference of TUs. Protein-encoding genes are shown in blue, non-coding elements in yellow and TUs in red. Arrows indicate TSSs. The colour-coded graphs represent the reads from treated libraries for all 10 conditions. The highlighted blocks at the beginning of the TUs visualize the read coverage from active TSSs. Their shape results from the enrichment of 5′-PPP carrying transcripts and the read length (101 nt) of the sequence analysis. Read coverage from the untreated libraries is shown in grey. For the definition of TUs, we distinguished between coding (gTU), non-coding (nTU) and antisense-oriented (aTU) TUs. If the TU matched more than one criterion, e.g. a gTU was also antisense to another TU, the descriptions were combined (e.g. gaTU). The locus around the flavoprotein-encoding genes sll0217 and sll0219 was chosen, since it illustrates the complexity of evaluating transcriptomic datasets.
Table 1 summarizes the numbers of different transcript types found for the different categories. The full list of all TUs is presented in Supplementary Table S1. A previous study21 reported 3,527 TSSs active at exponential growth conditions. When using the same threshold of two raw reads, our data for the exponential growth condition validated 3,282 (93%) of these. However, with the parallel analysis of nine further growth conditions and due to the higher sequencing depth, we found also many new TSSs. In summary, the 5,162 TSSs active at least in one of the tested conditions were employed to identify 4,091 TUs. Hence, one-fifth of the mapped TSSs are internal or alternative starts. The identified TUs provide information about the expression of 83% (3,101 of 3,725) of the annotated genes. Though our data provide a dramatically increased resolution, there are still 624 genes for which no transcript information was obtained.
Table 1.
Numbers of g, a, i and nTUs
| Number | |
|---|---|
| g(ai)TU | 2,012 |
| a(in)TU | 1,702 |
| iTU | 185 |
| nTU | 192 |
| Total TU | 4,091 |
3.2. Differential expression and regulation
Figure 2A shows the fractions of the TU types with a predicted TSS for each of the analysed conditions. We observed maximum numbers of active TUs in N-depleted and cold stress cultures, minimum numbers in stationary phase cultures and cultures kept for 12 h in darkness. We modelled the expression level of a TU by the read count from the treated libraries at the corresponding TSS. For the TU prediction, we set rather strict criteria, e.g. requiring a minimum number of 500 normalized reads from the treated library of at least one condition. While this is necessary for the specificity of the prediction, lower TSS read counts are still reasonable when assessing the expression level of a TU. The distribution of read counts at TSSs among all conditions is shown in Fig. 2B. The read counts can be modelled by a mixture of two gamma distributions, where the one for lower read counts (γ2 in Fig. 2B) resembles our background model for P-value calculation. Requiring a P-value of ≤0.05 results in a read count cut-off of 80. Following this criterion, we saw only 1,170 TUs (22%) present under all 10 conditions. However, most of the TUs were found in 9 of the 10 tested conditions (Fig. 2C).
Figure 2.
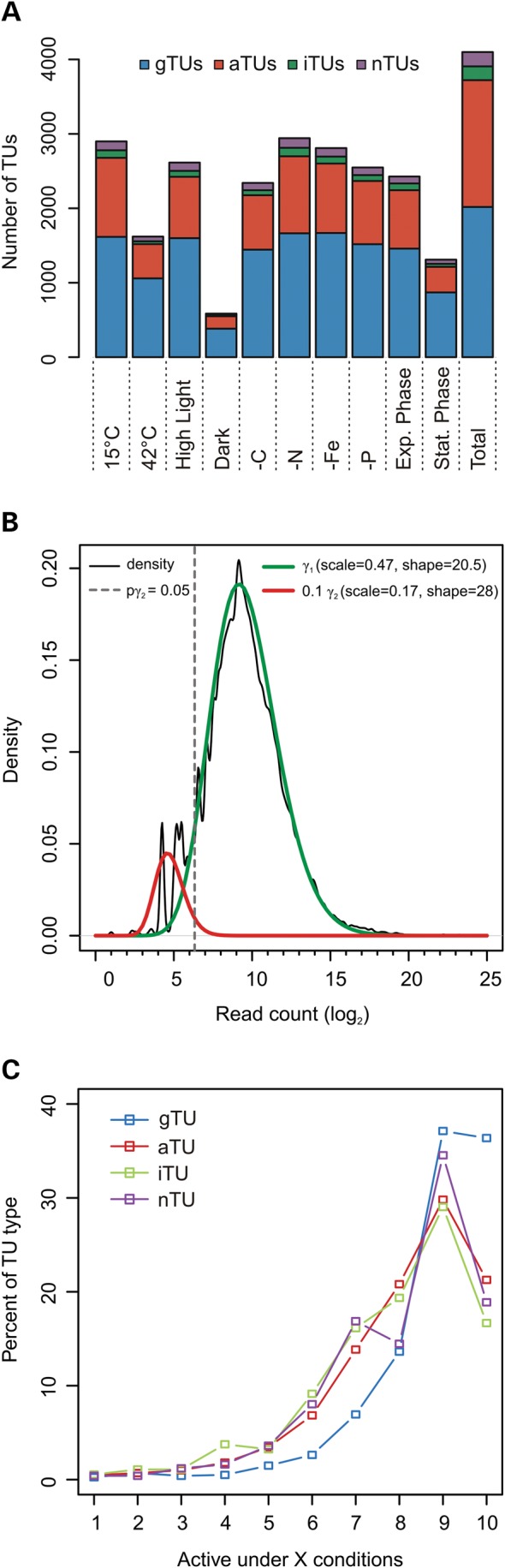
TU and TSS statistics. (A) Numbers of TU types defined for the different growth conditions. (B) Density of the TSS read counts among all conditions together with two gamma distributions (γ1 and γ2) as models for specific transcription (γ1) and unspecific low constitutive expression (γ2). The grey dashed line indicates a P-value of 0.05 for γ2, which corresponds to a read count of ∼80. Density was computed with the R density function using the Sheather–Jones bandwidth estimator. The distributions were fitted with the ‘fitdistr’ function from the MASS R package. For better visualization, γ2 is scaled with 0.1. (C) Occurrence of TUs under different numbers of conditions.
To identify genes whose expression is restricted to a single condition, we introduced the UEF, which for a single TU is the ratio of the read counts for the condition with the highest expression and the one with the second highest. Thus, TUs with a high UEF respond strongly to a particular stimulus. In Table 2, we show for each condition the TU associated with the highest number of reads and an UEF of >2.0. Interestingly, for many conditions we found sRNAs among the top-responding TUs.
Table 2.
List of TUs with highest absolute read numbers and UEF >2 for each of the 10 conditions
| TU ID | Type | Conditions | UEF | Genes | Annotation and comments |
|---|---|---|---|---|---|
| TU1715 | gaTU | Dark | 2.4 | ncr0700, ncr0710, ssr2227 | Ncr700 (cf. Fig. 5) | transposase |
| TU445 | gaTU | 42°C | 7.1 | sll1514 (hspA) | Chaperone |
| TU958 | gaTU | Exp. p. | 3.5 | slr1834 | Photosystem I P700 chlorophyll a apoprotein A1 |
| TU3332 | gaTU | HL | 2.9 | slr0040, slr0041, slr0042, slr0043, slr0044 | Bicarbonate transporter CmpA | bicarbonate transport system permease protein CmpB | probable porin | CmpC | CmpD |
| TU1322 | ga | −N | 3.5 | nsiR4, sll1698, sll1699 | Predicted in cluster 376 in ref.35; ncRNA ncl0540 or SyR12 in ref.21 see Fig. 5 | HP | oligopeptide-binding protein of the oligopeptide ABC transporter |
| TU1955 | nTU | −CO2 | 2.74 | NA | Possible ncRNA |
| TU2374 | nTU | Stat. p. | 25.7 | NA | Possible ncRNA |
| TU2304 | gTU | −Fe | 6.3 | sll1862 | HP |
| TU3627 | gaTU | −P | 58.7 | sll1552, sll0720 | HP | RTX toxin-activating protein homologue |
| TU5051 | gaTU | 15°C | 3.9 | sll5046, ssl5045, sll5044, sll5043 | HPs |
The TU ID and type are given, together with the condition with the maximum number of reads, the UEF, the respective gene IDs and the gene annotations. For further details see Supplementary Table S1.
Exp. p., exponential growth phase; Stat. p., stationary growth phase; RTX, repeats in toxin; NA, not applicable; HP, hypothetical protein.
3.2.1. The phosphate stress regulon
The phosphate stress regulon of Synechocystis 6803 depends on the regulatory protein PhoB and consists of 12 genes that responded in microarray analyses to phosphate depletion.36 To determine to what extent our dataset would cover this regulon, we ranked those TUs which had a maximum number of normalized read counts in the ‘−P’ condition according to their UEF (Table 3). This approach identified four TSSs strongly responding to phosphate stress that gives rise to four TUs that explain the expression of the 12 genes reported previously.36 Thus, our data fully explain the regulation of the known P stress regulon in Synechocystis 6803 and provide the exact TSSs, none of which had been known before (Table 3 and Supplementary Table S1). Moreover, our data identified seven additional TUs that were induced upon P stress, which cover additional 21 genes. Among these genes is sll0290, encoding a polyphosphate kinase, but surprisingly also two TUs seemingly unrelated to phosphate metabolism, covering the genes for phenoxybenzoate dioxygenase, a ferredoxin, the methyl-accepting chemotaxis-like protein PilJ and the regulator Hik39 as well as with chlLN the genes for two subunits of the protochlorophyllide reductase. Moreover, two of these TUs, PsiR1 (phosphate-stress-induced RNA 1, part of TU3627, Fig. 3) and TU79, are likely sRNAs or encode very short peptides. It should be noted, however, that our identification of TUs and the associated regulation is not always directly comparable with the results of microarray analyses that target the steady-state accumulation of an mRNA, while our measure of expression focuses on nascent transcripts. Furthermore, differences in sample preparation, signal readout and signal processing likely result in deviations between the methods.
Table 3.
TUs associated with the phosphate stress regulon
| TU ID | Type | −P | UEF | Genes | Annotation and comments |
|---|---|---|---|---|---|
| TU1428 | gaTU | 16,516 | 146.2 | slr1247, slr1248, slr1249, slr1250 | pstS2C2A2B2 phosphate ABC transporter; P stress regulon36 |
| TU3627 | gaTU | 179,694 | 58.7 | PsiR1, sll0720, sll1552 | Main accumulating transcript is an sRNA of ∼600 nt; new, see Fig. 3 |
| TU388 | gaTU | 3,047 | 38.1 | sll0654, ncl0120, sll0656 | Extracellular nuclease NucH | ncRNA | alkaline phosphatase PhoA; P stress regulon36 |
| TU679 | gTU | 8,559 | 32.4 | sll1294, sll1296, ssl2559, sll1297 | pilJ | hik39 | ferredoxin | pobA; new |
| TU79 | nTU | 4,910 | 26.1 | NA | sRNA; new |
| TU2789 | gTU | 8,248 | 22 | sll0679 | SphX protein ; P stress regulon36 |
| TU3169 | gaTU | 12,692 | 7.6 | sll0290 | Polyphosphate kinase; new |
| TU3594 | gaTU | 68,955 | 4.7 | slr0749, ssr1251, slr0750 | Light-independent protochlorophyllide reductase iron protein subunit ChlL | HP | light-independent protochlorophyllide reductase subunit ChlN |
| TU3107 | gTU | 8,054 | 3.7 | sll0594, sll0595 | Transcriptional regulator | HP |
| TU2264 | gaTU | 24,298 | 3.4 | slr0144, slr0145, slr0146, slr0147, slr0148, slr0149, slr0150, slr0151 | HP | HP |HP | HP | HP | HP | ferredoxin | petF-like protein | HP |
| TU2785 | gaTU | 40,135 | 3.3 | sll0680, sll0681, sll0682, sll0683, sll0684 | pstS1C1A1B1B2 phosphate ABC transporter; P stress regulon36 |
The identified TUs are given, together with the TU type, the normalized read count in the −P condition, the UEF, the gene IDs, the gene annotations and comments and sorted according to the UEF. The entries are sorted according to their UEF and filtered for a minimum read count of 3000.
HP, hypothetical protein.
Figure 3.

An sRNA as part of the P stress regulon in Synechocystis 6803. (A) Locus of the gene sll1552 for which a P-stress-activated TSS more than 600 nt upstream was detected. (B) Verification of the P-stress-specific expression of a short transcript upstream of sll1552, which we named phosphate-stress-induced RNA 1 (PsiR1). *The bar in (A) marks the part of the TU that is covered by the 32P-labelled probe. Since no additional TSS was detected closer to the coding region of sll1552, but TU3627 also covers this gene, PsiR1 probably results from post-transcriptional processing. The signal for the 5S rRNA was used as a loading control. (C) Comparison of putative Pho boxes (shaded repeats of PyTTAAPyPy(T/A) separated by 3 nt each35) within the upstream flanking regions of known phosphate-stress-responsive genes such as pstS2C2A2B2, phoA, but also for the newly discovered P-stress-related sRNA PsiR1. Putative −10 elements are underlined and an arrow highlights the first transcribed nucleotide. For the complete dataset on the P stress regulon, see also Table 3.
In previous work, the repetitive 8-bp Pho box consensus sequence PyTTAAPyPy(T/A) was described in the upstream regions of the phoA, sphX and pstS2 genes of Synechocystis 6803. This sequence was shown to be recognized by SphR, the PhoB homologue.36 Some of these elements are located more than 300 nt upstream of the respective genes; however, the exact locations of these elements within the respective promoter sequences were not investigated. Here, we found these elements to form tetrameric repeats within 61–182 nt upstream of the TSSs and identified very similar elements at the corresponding positions within the promoter of the newly identified P-stress-regulated PsiR1 transcript (Fig. 3C), providing further support to the idea that PsiR1 is a previously unknown member of the Pho regulon in Synechocystis 6803. Whereas our Northern experiment clearly indicated PsiR1 to accumulate in the form of a distinct ∼500 nt sRNA (Fig. 3B), we cannot exclude the possibility of its translation into one or two small peptides, as there are two short reading frames, 46 and 55 codons in length, present within PsiR1.
3.2.2. The iron stress response
As in our approach targeting the phosphate stress regulon, we ranked the iron-stress-specific TUs according to their maximum UEF values (Table 4). Known iron-stress-induced genes, such as that for the iron-stress-induced protein A (IsiA), were among the top induced TUs. Remarkably, again a putative sRNA, which we termed IsaR1 (iron stress-activated RNA 1), was among the top induced TSSs. In a previous microarray-based study, IsaR1 was identified as the sRNA whose expression responded most strongly on iron stress20 (called NC-181 there), but its TSS had not been mapped. The accumulation of IsaR1 under iron depletion was verified by Northern hybridization (Fig. 4).
Table 4.
TUs associated with the iron stress regulon
| TU ID | Type | −Fe | UEF | Genes | Annotation and comments |
|---|---|---|---|---|---|
| TU24 | ga | 151,423 | 1138.5 | sll1206 | Ferric aerobactin receptor |
| TU32 | ga | 106,482 | 86.3 | sll1404, sll1405, sll1406, sll1407, sll1408 | ExbB | ExbD | ferrichrome-iron receptor fhuA | probable methyltransferase | transcriptional regulator |
| TU1556 | ga | 160,751 | 76.11 | sll0247 | IsiA |
| TU3631 | ga | 4,683 | 40.7 | sll1549 | Salt-enhanced periplasmic protein |
| TU39 | ga | 10,769 | 29.5 | slr1489, slr1490 | Transcriptional regulator | ferrichrome-iron receptor |
| TU3327 | n | 89,915 | 23.4 | ncl1600 | IsaR1, potentially regulatory sRNA |
| TU37 | g | 7,871 | 19 | slr1484, slr1485, slr1488 | HP | putative phosphatidylinositol phosphate kinase | salt-induced periplasmic protein | multidrug resistance family ABC transporter |
| TU23 | ga | 6,258 | 17 | slr1318, slr1319 | Iron(III) dicitrate transport system ATP-binding protein | iron(III) dicitrate transport system substrate-binding protein |
| TU288 | ga | 26,880 | 8.4 | slr1295 | Iron transport protein |
| TU3030 | ga | 59,402 | 8.3 | slr0074, slr0075, slr0076, slr0077 | ABC transporter subunit | ABC transporter ATP-binding protein | HP | cysteine desulphurase |
| TU19 | g | 3,311 | 8.3 | slr1316 | Iron(III) dicitrate ABC transporter permease |
| TU3083 | ga | 5,331 | 6.7 | sll0477, sll0478, sll0479 | Putative biopolymer transport ExbB-like protein | HP | HP |
| TU2304 | g | 212,454 | 6.3 | sll1862 | HP |
| TU1071 | ga | 56,123 | 5.4 | ssl2250 | Glycoprotein |
| TU689 | ga | 7,378 | 4.4 | ssr2333, slr1392 | HP | ferrous iron transport protein B |
| TU1867 | ga | 174,398 | 3.8 | sll1878 | ABC transporter |
| TU3085 | ga | 23,007 | 3.6 | slr0513 | Periplasmic iron-binding protein |
| TU2548 | ga | 5,004 | 3.1 | sll0314 | Periplasmic protein | HP |
The identified TUs are given, together with the TU type, the normalized read count in the −Fe condition, the UEF, the gene IDs and the gene annotations. The entries are sorted according to their UEF and filtered for a minimum read count of 3000.
HP, hypothetical protein.
Figure 4.
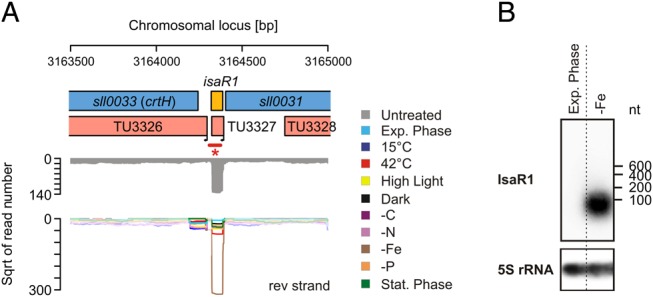
An sRNA as part of the iron stress regulon in Synechocystis 6803. (A) Transcriptomic organization of the locus in between the genes crtH and sll0031 indicate for an sRNA particularly accumulating under iron depletion and was therefore named iron-stress-activated RNA 1 (IsaR1). (B) Northern blot verifying the accumulation of IsaR1 in iron-limited cells. *The bar in (A) marks the part of the TU that is covered by the 32P-labelled probe. The signal for the 5S rRNA was used as a loading control for both strains.
3.2.3. ncRNAs relevant under conditions important for photosynthetic growth
In Fe and P stress conditions discussed above, sRNAs were among the most highly induced transcripts. Furthermore, the overall strongest TSS gives rise to the sRNA Ncr0700. This sRNA is highly expressed in darkness (>11 million normalized read counts), but also expressed at relatively high levels after heat shock or during stationary phase (4 and 4.8 million reads, respectively). We observed similar patterns also for mRNAs, for example, for the small protein Norf121 (Fig. 5), but in the following, we will focus on sRNAs, as these are the least-explored component of the regulatory apparatus.
Figure 5.

Differential expression of further transcripts. Genomic regions of PsrR1 (A), Ncr0700 (B), Norf1 (C), NsiR4 (D) as well as the verification of differential expression by Northern verification (E and F). In a separate hybridization, the membranes were also submitted to a probe specific for the 5S rRNA. Here, a representative reference signal is shown for the membranes used for PsrR1 (E) and NsiR4 (F) detection, respectively.
The sRNA PsrR1,37 which was previously described to be light-regulated21 (called SyR1 there), is one such example. As expected, PsrR1 was most highly expressed under high light and strongly decreased at darkness. Additionally, we found that it is relatively highly expressed under other conditions such as Fe or N depletion. Especially, a 10 times higher read number for cold compared with heat stress suggests a temperature-dependent regulation, which was confirmed by Northern hybridization (Fig. 5).
In addition to the light intensity, the availability of macronutrients, including nitrogen and inorganic carbon, is very important for photosynthetic growth. Commonly, in N-depleted cells especially genes encoding subunits of transport systems for nitrate [e.g. nrtABCD (sll1450-53)], urea [e.g. urtABC (slr0447, slr1200, slr1201)] and ammonium [e.g. amt1 (sll0108), amt2 (sll1017) and amt3 (sll0537)] become up-regulated.38–40 We observed a higher read number for the corresponding TSSs under N depletion, except for the nrt operon and amt1. However, as described above, TSS activity is not always directly comparable with steady-state levels measured by microarrays, which may explain the only partial agreement with existing microarray data. Beside uptake systems, also genes for enzymes involved in metabolizing nitrogen are regulated, e.g. glutamine synthetase (GS) type III (glnN, slr0288), which is responsible for the condensation of ammonium and glutamate, as well as isocitrate dehydrogenase (icd, slr1289), which provides 2-oxoglutarate for the GOGAT reaction. For both, an increased read number was detected in N-depleted cells which is consistent with previous reports.40 Also consistent with previous findings,40 the genes gifA (ssl1911) and gifB (sll1515), both of which encode inhibitory proteins for GS activity, are down-regulated in our data. Intriguingly, also under N-depletion, an sRNA (NsiR4 for nitrogen-stress-induced RNA 4, previously named SyR1221), which is encoded in between the genes sll1697 and sll1698, was found among the transcripts with the highest read numbers. Actually, NsiR4 was expressed in all conditions, but with a maximum under N depletion (UEF = 3.52, Table 2 and Fig. 5), which suggests a regulatory role in the metabolism of nitrogen.
A similar observation was made for carbon as an environmental factor. An sRNA (previously named SyR1421) located upstream of the gene slr1214, encoding a UV-dependent response regulator,41 was among the 20 most strongly expressed TUs for all conditions. Interestingly, its expression was completely shut down when cells were exposed to CO2 concentrations higher than the ambient concentration of 0.03% (Fig. 6). Thus, these data indicate a specific carbon-dependent regulation, and thus, we named this sRNA CsiR1 (carbon-stress-induced RNA 1).
Figure 6.
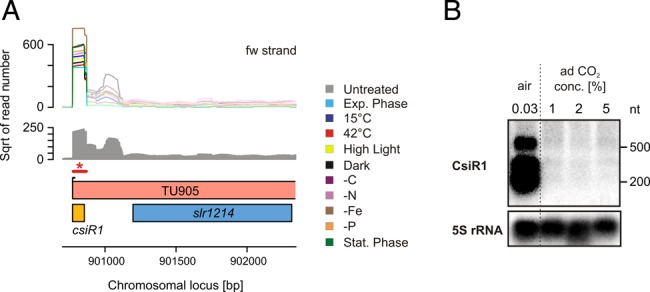
An sRNA specifically repressed by elevated CO2 levels. (A) Locus of the gene slr1214, for which a TSS more than 400 nt upstream was detected giving hint for a small RNA. The accumulation of small transcripts was also verified by Northern blots (B). *The bar in (A) marks the part that is covered by the labelled probe. Very likely, these transcripts are processing products of the large TU905. Under the tested conditions, the transcripts appeared constitutively expressed. However, when cells were bubbled with CO2-enriched air (1–5% CO2), these transcripts were not detectable indicating for carbon-regulated expression. Therefore, we named it carbon-stress-induced RNA 1 (CsiR1).
In summary, all these examples constitute likely important regulatory factors that have been overseen so far and show that sRNAs are often involved in regulatory responses.
3.3. Operons, 5′ and 3′ untranslated regions
We found 683 TUs that cover more than one gene and the longest TU comprised 22 genes (TU355; Supplementary Table S1). Its composition is rather diverse and contains mainly genes without an ortholog in any other cyanobacteria. It represents a major part of the rfb-glycosyltransferase gene cluster that has features of a genomic island and encodes several enzymes for the modification of cell surface structures.42 The finding that these genes are transcribed as a very long multicistronic operon is consistent with the idea that they form a gene cluster that was jointly transferred. The second-longest TU (TU837, 18 genes) and the adjacent third-longest TU, TU833 (9 genes; Supplementary Table S1), represent the S10/spc ribosomal protein operon.
The untranslated regions (UTRs) of mRNAs are functionally relevant elements, i.e. they can harbour regulatory features such as riboswitches or are targeted by regulatory sRNAs. Our TU-based approach intrinsically defines 5′ and 3′ UTRs and their length distribution is shown in Fig. 7. The median 5′ UTR length is 52 nt, which is somewhat longer than the preferred 5′ UTR length of 20–40 nt found in proteobacteria.43 The median 3′ UTR length is 118 nt.
Figure 7.
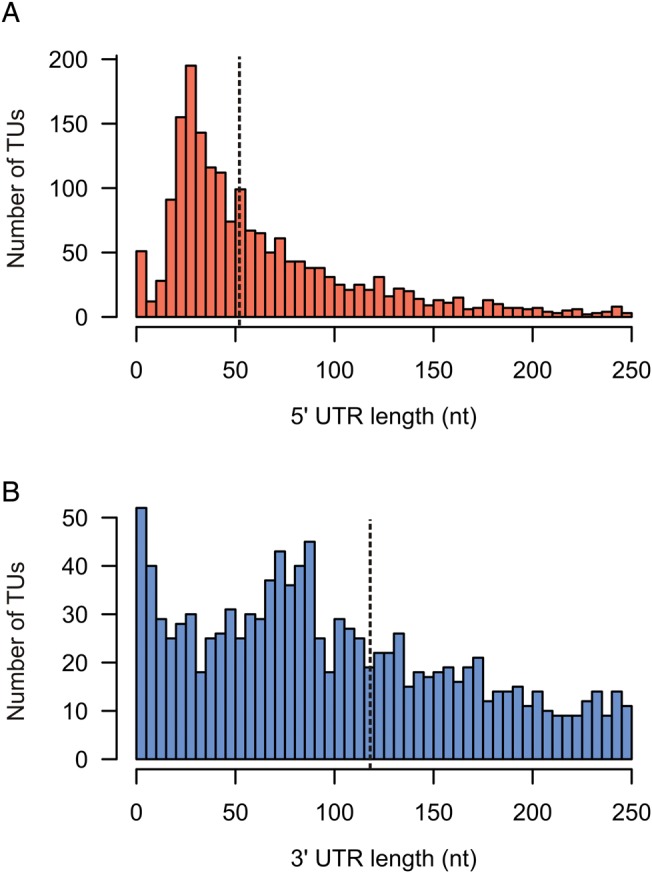
Distribution of UTR lengths. Histogram of the (A) 5′ UTR lengths and (B) 3′ UTR lengths of all TUs that contain annotated genes.
Leaderless mRNAs play a role in stress adaptation in E. coli44 and are more or less restricted to the respective condition. Our data provide evidence for 51 leaderless mRNAs in Synechocystis 6803, including the previously reported apcE (slr0335), slr1079 and slr0846 mRNAs.21 Interestingly, their expression was distributed over all conditions (Supplementary Table S2). In addition, based on iTSS locations shortly behind the annotated start codon and the conservation of reading frames, our data allowed the reannotation of the 5′ ends of 46 reading frames in Synechocystis 6803 (Supplementary Table S2).
4. Discussion
4.1. The TU approach
Traditional dRNA-seq-based studies so far were more focused on the identification of TSSs and often used fixed-length thresholds for assigning them to mRNA, sRNA or cis-asRNA transcripts. For example, in our previous analysis,21 a TSS was classified as gTSS if it was located 100 nt upstream of an annotated gene; in the work on Campylobacter jejuni, 300 nt were used; in the work on Anabaena sp. PCC 7120, this was 200 nt, unless specified otherwise due to additional information.23,45 These rather arbitrary assumptions need to be made for automatic annotation but are erroneous. For example, it is well known that the actual UTR lengths can differ greatly, e.g. the key transcription factor HetR in Anabaena sp. PCC 7120 is transcribed from four different gTSS, with the two most distal ones giving rise to 5′ UTRs of 696 and 728 nt.46–49 Here, we defined TUs, which directly provide information on TSSs, operons and UTRs, and lower the potential for false-positive TSS predictions, since a TSS needs to be followed by a region covered by reads from the untreated library.
4.2. Regulons inferred from dRNA-seq analysis
Our approach allows the definition of regulons and relevant promoter sequences at single-nucleotide resolution, demonstrated exemplarily for the Pho regulon. Based on the definition of the UEF, a statistical reliable quantitative data evaluation became possible. This approach allowed us to identify the suite of TSSs specifically linked to activation under phosphate stress, which fully explain the regulation of the known P-stress regulon in Synechocystis 680336 (Fig. 3 and Table 3). Moreover, we suggest seven additional TUs to belong to the P-stress regulon, two of which appear to encompass sRNAs.
4.3. Many growth conditions exhibit a strongly responding sRNA
For the majority of the conditions in our study, we identified an sRNA among the top expressed or induced transcripts. A change in the light conditions triggered the induction of PsrR1, which is probably a key player in the regulation of the photosynthetic apparatus (Georg et al. unpublished data). Interestingly, two other transcripts, the sRNA Ncr0700 and the short mRNA Norf1, behaved in completely the opposite way for the conditions tested, with a peak of expression in the dark and the minimum in high light (Fig. 5), suggesting a specific role under that condition.
In addition to the light intensity, the availability of macronutrients is very important for photosynthetic growth. Indeed, we identified several sRNAs with an according pattern of expression under iron stress (IsaR1), or which were induced specifically by the depletion of carbon (CsiR1), nitrogen (NsiR4) or phosphate (PsiR1). The phosphate-stress-induced sRNA PsiR1 is the second highest-induced transcript according to our data. Interestingly, PsiR1 seems to be part of the 5′ UTR of sll1552, from which it may originate by post-transcriptional cleavage. Alternatively, incomplete termination of PsiR1 transcription may allow read-through into sll1552. Thus, in both scenarios its TSS also drives the transcription of sll1552, such that they constitute an operon of an sRNA and a protein-coding gene. We found a similar organization for nsiR4, which forms an operon together with the protein-coding gene sll1698. Here, again, we validated NsiR4 as an individual transcript, which is highly induced upon N depletion.
Although the individual functions of the sRNAs remain to be elucidated at the molecular level, our findings are in line with the important role of sRNAs in many regulatory processes. Their identification in Synechocystis 6803, a cyanobacterial model organism with a well-studied physiology and genetics, is advantageous for their future detailed characterization.
5. Availability
Transcriptome data have been deposited in the short read archive (SRP032228). Genome-wide visualizations of all data are available at http://www.cyanolab.de/Supplementary.html.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft Focus program ‘Sensory and regulatory RNAs in Prokaryotes’ SPP1258, grant HE 2544/4-2 and by the Federal Ministry of Education and Research grant ‘e:bio RNAsys’ 0316165 (to W.R.H.).
Supplementary Material
Acknowledgement
We thank Martin Hagemann for critical reading of a previous manuscript version.
References
- 1.Georgianna D.R., Mayfield S.P. Exploiting diversity and synthetic biology for the production of algal biofuels. Nature. 2012;488:329–35. doi: 10.1038/nature11479. [DOI] [PubMed] [Google Scholar]
- 2.Peralta-Yahya P.P., Zhang F., del Cardayre S.B., Keasling J.D. Microbial engineering for the production of advanced biofuels. Nature. 2012;488:320–8. doi: 10.1038/nature11478. [DOI] [PubMed] [Google Scholar]
- 3.McKinlay J.B., Harwood C.S. Photobiological production of hydrogen gas as a biofuel. Curr. Opin. Biotechnol. 2010;21:244–51. doi: 10.1016/j.copbio.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 4.Deng M.-D., Coleman J.R. Ethanol synthesis by genetic engineering in cyanobacteria. Appl. Environ. Microbiol. 1999;65:523–8. doi: 10.1128/aem.65.2.523-528.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atsumi S., Higashide W., Liao J.C. Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde. Nat. Biotechnol. 2009;27:1177–80. doi: 10.1038/nbt.1586. [DOI] [PubMed] [Google Scholar]
- 6.Takahama K., Matsuoka M., Nagahama K., Ogawa T. Construction and analysis of a recombinant cyanobacterium expressing a chromosomally inserted gene for an ethylene-forming enzyme at the psbAI locus. J. Biosci. Bioeng. 2003;95:302–5. doi: 10.1016/s1389-1723(03)80034-8. [DOI] [PubMed] [Google Scholar]
- 7.Lindberg P., Park S., Melis A. Engineering a platform for photosynthetic isoprene production in cyanobacteria, using Synechocystis as the Model Organism. Metab. Eng. 2010;12:70–9. doi: 10.1016/j.ymben.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Schirmer A., Rude M.A., Li X., Popova E., del Cardayre S.B. Microbial biosynthesis of alkanes. Science. 2010;329:559–62. doi: 10.1126/science.1187936. [DOI] [PubMed] [Google Scholar]
- 9.Haselkorn R. Genetic systems in cyanobacteria. In: Jeffrey H.M., editor. Methods in Enzymology. Academic Press, Waltham, USA; 1991. pp. 418–30. [DOI] [PubMed] [Google Scholar]
- 10.Porter R.D. DNA transformation. In: Lester Packer A.N.G., editor. Methods in Enzymology. Academic Press, Waltham, USA; 1988. pp. 703–12. [DOI] [PubMed] [Google Scholar]
- 11.Williams J.G.K. Construction of specific mutations in photosystem II photosynthetic reaction center by genetic engineering methods in Synechocystis 6803. In: Lester Packer A.N.G., editor. Methods in Enzymology. Academic Press, Waltham, USA; 1988. pp. 766–78. [Google Scholar]
- 12.Grigorieva G., Shestakov S. Transformation in the cyanobacterium Synechocystis sp. 6803. FEMS Microbiol. Lett. 1982;13:367–70. [Google Scholar]
- 13.Kaniya Y., Kizawa A., Miyagi A., et al. Deletion of the transcriptional regulator cyAbrB2 deregulates primary carbon metabolism in Synechocystis sp. PCC 6803. Plant Physiol. 2013;162:1153–63. doi: 10.1104/pp.113.218784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knoop H., Zilliges Y., Lockau W., Steuer R. The metabolic network of Synechocystis sp. PCC 6803: systemic properties of autotrophic growth. Plant Physiol. 2010;154:410–22. doi: 10.1104/pp.110.157198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knoop H., Gründel M., Zilliges Y., et al. Flux balance analysis of cyanobacterial metabolism: the metabolic network of Synechocystis sp. PCC 6803. PLoS Comput Biol. 2013;9:e1003081. doi: 10.1371/journal.pcbi.1003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fulda S., Mikkat S., Huang F., et al. Proteome analysis of salt stress response in the cyanobacterium Synechocystis sp. strain PCC 6803. Proteomics. 2006;6:2733–45. doi: 10.1002/pmic.200500538. [DOI] [PubMed] [Google Scholar]
- 17.Battchikova N., Vainonen J.P., Vorontsova N., Keränen M., Carmel D., Aro E.-M. Dynamic changes in the proteome of Synechocystis 6803 in response to CO2 limitation revealed by quantitative proteomics. J. Proteome Res. 2010;9:5896–912. doi: 10.1021/pr100651w. [DOI] [PubMed] [Google Scholar]
- 18.Hihara Y., Kamei A., Kanehisa M., Kaplan A., Ikeuchi M. DNA microarray analysis of cyanobacterial gene expression during acclimation to high light. Plant Cell Online. 2001;13:793–806. doi: 10.1105/tpc.13.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Georg J., Voß B., Scholz I., Mitschke J., Wilde A., Hess W.R. Evidence for a major role of antisense RNAs in cyanobacterial gene regulation. Mol. Syst. Biol. 2009;5:305. doi: 10.1038/msb.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hernández-Prieto M.A., Schön V., Georg J., et al. Iron deprivation in Synechocystis: inference of pathways, non-coding RNAs, and regulatory elements from comprehensive expression profiling. G3 (Bethesda) 2012;2:1475–95. doi: 10.1534/g3.112.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitschke J., Georg J., Scholz I., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. USA. 2011;108:2124–9. doi: 10.1073/pnas.1015154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma C.M., Hoffmann S., Darfeuille F., et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–5. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 23.Mitschke J., Vioque A., Haas F., Hess W.R., Muro-Pastor A.M. Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp. PCC7120. Proc. Natl. Acad. Sci. USA. 2011;108:20130–5. doi: 10.1073/pnas.1112724108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trautmann D., Voß B., Wilde A., Al-Babili S., Hess W.R. Microevolution in cyanobacteria: re-sequencing a motile substrain of Synechocystis sp. PCC 6803. DNA Res. 2012;19:435–48. doi: 10.1093/dnares/dss024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rippka R., Deruelles J., Waterbury J.B., Herdman M., Stanier R.Y. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. Microbiology. 1979;111:1–61. [Google Scholar]
- 26.Pinto F., Thapper A., Sontheim W., Lindblad P. Analysis of current and alternative phenol based RNA extraction methodologies for cyanobacteria. BMC Mol. Biol. 2009;10:79. doi: 10.1186/1471-2199-10-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hein S., Scholz I., Voß B., Hess W.R. Adaptation and modification of three CRISPR loci in two closely related cyanobacteria. RNA Biol. 2013;10:852–64. doi: 10.4161/rna.24160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffmann S., Otto C., Kurtz S., et al. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009;5:e1000502. doi: 10.1371/journal.pcbi.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bischler T., Kopf M., Voß B. Transcript mapping based on dRNA-seq data. BMC Bioinformatics. 2014;15:122. doi: 10.1186/1471-2105-15-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sesto N., Wurtzel O., Archambaud C., Sorek R., Cossart P. The excludon: a new concept in bacterial antisense RNA-mediated gene regulation. Nat. Rev. Microbiol. 2013;11:75–82. doi: 10.1038/nrmicro2934. [DOI] [PubMed] [Google Scholar]
- 31.Steglich C., Futschik M.E., Lindell D., Voß B., Chisholm S.W., Hess W.R. The challenge of regulation in a minimal photoautotroph: non-coding RNAs in Prochlorococcus. PLoS Genet. 2008;4:e1000173. doi: 10.1371/journal.pgen.1000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eisenhut M., Georg J., Klähn S., et al. The antisense RNA As1_flv4 in the cyanobacterium Synechocystis sp. PCC 6803 prevents premature expression of the flv4-2 operon upon shift in inorganic carbon supply. J. Biol. Chem. 2012;287:33153–62. doi: 10.1074/jbc.M112.391755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bersanini L., Battchikova N., Jokel M., et al. Flavodiiron protein Flv2/Flv4-related photoprotective mechanism dissipates excitation pressure of PSII in cooperation with phycobilisomes in cyanobacteria. Plant Physiol. 2014;164:805–18. doi: 10.1104/pp.113.231969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang P., Allahverdiyeva Y., Eisenhut M., Aro E.-M. Flavodiiron proteins in oxygenic photosynthetic organisms: photoprotection of photosystem II by Flv2 and Flv4 in Synechocystis sp. PCC 6803. PLoS ONE. 2009;4:e5331. doi: 10.1371/journal.pone.0005331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voß B., Georg J., Schon V., Ude S., Hess W. Biocomputational prediction of non-coding RNAs in model cyanobacteria. BMC Genomics. 2009;10:123. doi: 10.1186/1471-2164-10-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki S., Ferjani A., Suzuki I., Murata N. The SphS-SphR two component system is the exclusive sensor for the induction of gene expression in response to phosphate limitation in Synechocystis. J. Biol. Chem. 2004;279:13234–40. doi: 10.1074/jbc.M313358200. [DOI] [PubMed] [Google Scholar]
- 37.Hess W.R., Berghoff B.A., Steglich C., Wilde A., Klug G. Riboregulators and the role of Hfq in photosynthetic prokaryotes. RNA Biol. 2014;11:1–14. doi: 10.4161/rna.28035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montesinos M.L., Muro-Pastor A.M., Herrero A., Flores E. Ammonium/methylammonium permeases of a cyanobacterium: identification and analysis of three nitrogen-regulated amt genes in Synechocystis sp. PCC 6803. J. Biol. Chem. 1998;273:31463–70. doi: 10.1074/jbc.273.47.31463. [DOI] [PubMed] [Google Scholar]
- 39.Wang H.-L., Postier B.L., Burnap R.L. Alterations in global patterns of gene expression in Synechocystis sp. PCC 6803 in response to inorganic carbon limitation and the inactivation of ndhR, a LysR family regulator. J. Biol. Chem. 2004;279:5739–51. doi: 10.1074/jbc.M311336200. [DOI] [PubMed] [Google Scholar]
- 40.Krasikov V., Aguirre von Wobeser E., Dekker H.L., Huisman J., Matthijs H.C.P. Time-series resolution of gradual nitrogen starvation and its impact on photosynthesis in the cyanobacterium Synechocystis PCC 6803. Physiol. Plant. 2012;145:426–39. doi: 10.1111/j.1399-3054.2012.01585.x. [DOI] [PubMed] [Google Scholar]
- 41.Song J.-Y., Cho H.S., Cho J.-I., Jeon J.-S., Lagarias J.C., Park Y.-I. Near-UV cyanobacteriochrome signaling system elicits negative phototaxis in the cyanobacterium Synechocystis sp. PCC 6803. Proc. Natl. Acad. Sci. U.S.A. 2011;108:10780–5. doi: 10.1073/pnas.1104242108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kopf M., Klähn S., Pade N., et al. Comparative genome analysis of the closely related Synechocystis strains PCC 6714 and PCC 6803. DNA Res. 2014;21:255–66. doi: 10.1093/dnares/dst055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim D., Hong J.S.-J., Qiu Y., et al. Comparative analysis of regulatory elements between Escherichia coli and Klebsiella pneumoniae by genome-wide transcription start site profiling. PLoS Genet. 2012;8:e1002867. doi: 10.1371/journal.pgen.1002867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vesper O., Amitai S., Belitsky M., et al. Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell. 2011;147:147–57. doi: 10.1016/j.cell.2011.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dugar G., Herbig A., Forstner K.U., et al. High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet. 2013;9:e1003495. doi: 10.1371/journal.pgen.1003495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buikema W.J., Haselkorn R. Expression of the Anabaena hetR gene from a copper-regulated promoter leads to heterocyst differentiation under repressing conditions. Proc. Natl. Acad. Sci. USA. 2001;98:2729–34. doi: 10.1073/pnas.051624898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ehira S., Ohmori M. NrrA directly regulates expression of hetR during heterocyst differentiation in the cyanobacterium Anabaena sp. strain PCC 7120. J. Bacteriol. 2006;188:8520–5. doi: 10.1128/JB.01314-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rajagopalan R., Callahan S.M. Temporal and spatial regulation of the four transcription start sites of hetR from Anabaena sp. strain PCC 7120. J. Bacteriol. 2010;192:1088–96. doi: 10.1128/JB.01297-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muro-Pastor A.M., Valladares A., Flores E., Herrero A. Mutual dependence of the expression of the cell differentiation regulatory protein HetR and the global nitrogen regulator NtcA during heterocyst development. Mol. Microbiol. 2002;44:1377–85. doi: 10.1046/j.1365-2958.2002.02970.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.