

Abstract
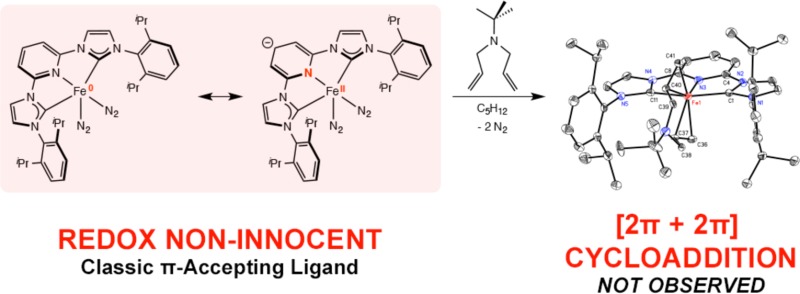
The electronic structures of pyridine N-heterocyclic dicarbene (iPrCNC) iron complexes have been studied by a combination of spectroscopic and computational methods. The goal of these studies was to determine if this chelate engages in radical chemistry in reduced base metal compounds. The iron dinitrogen example (iPrCNC)Fe(N2)2 and the related pyridine derivative (iPrCNC)Fe(DMAP)(N2) were studied by NMR, Mössbauer, and X-ray absorption spectroscopy and are best described as redox non-innocent compounds with the iPrCNC chelate functioning as a classical π acceptor and the iron being viewed as a hybrid between low-spin Fe(0) and Fe(II) oxidation states. This electronic description has been supported by spectroscopic data and DFT calculations. Addition of N,N-diallyl-tert-butylamine to (iPrCNC)Fe(N2)2 yielded the corresponding iron diene complex. Elucidation of the electronic structure again revealed the CNC chelate acting as a π acceptor with no evidence for ligand-centered radicals. This ground state is in contrast with the case for the analogous bis(imino)pyridine iron complexes and may account for the lack of catalytic [2π + 2π] cycloaddition reactivity.
Introduction
Iron dinitrogen complexes with meridionally coordinating nitrogen-based pincer-type ligands are of interest due to their activity and versatility in base-metal catalysis.1−3 In 2004, our laboratory reported the synthesis of (iPrPDI)Fe(N2)2 (iPrPDI = 2,6-(2,6-iPr2-C6H3-N=CMe)2C5H3N) and exploration of its activity in catalytic hydrogenation and hydrosilylation (Scheme 1).4,5 This iron dinitrogen compound has also proven to be an effective precatalyst for the [2π + 2π] cycloaddition of α,ω-dienes6 with ethylene and butadiene to form vinylcyclobutane7 and reductive cyclizations involving enynes and diynes.8 Modification of the bis(imino)pyridine chelate has resulted in iron dinitrogen complexes with improved catalytic performance. Reducing the size of the 2,6-aryl substituents from isopropyl to ethyl or methyl furnished the dimeric iron dinitrogen complexes [(RPDI)Fe(N2)]2(μ2-N2) (RPDI = 2,6-(2,6-R2-C6H3-N=CMe)2C5H3N; R = Me, Et), which exhibited dramatically improved activity for both olefin hydrogenation9 and hydrosilylation.10 The hydrosilylation chemistry produced no byproducts arising from olefin isomerization or dehydrogenative silylation, a distinct advantage over commercial precious-metal catalysts.
Scheme 1. Bis(imino)pyridine and Pyridine N-Heterocyclic Dicarbene Iron Dinitrogen Complexes That Are Active Precatalysts for Olefin Hydrogenation.
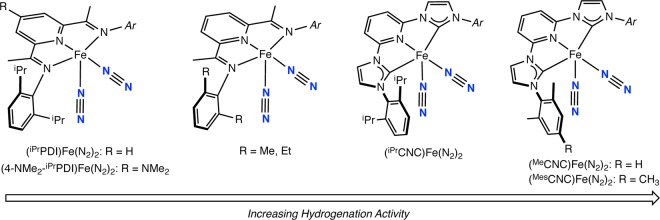
The dimers [(RPDI)Fe(N2)]2(μ2-N2) (R = Me, Et) are depicted as monomers for simplicity.
Electronic effects on the performance of aryl-substituted bis(imino)pyridine iron catalysts have also been recently evaluated. Introduction of electron-donating groups into the 4-position of the bis(imino)pyridine ligand, e.g. (4-Me2N-iPrPDI)Fe(N2)2,11 increased both hydrogenation12 and hydrosilylation activity.5 These findings prompted the study of even more electron rich iron bis(dinitrogen) complexes. Danopoulos and co-workers reported the synthesis of (iPrCNC)Fe(N2)2 (iPrCNC = 2,6-(2,6-iPr2-C6H3-imidazol-2-ylidene)2-C5H3N)13 and infrared stretching frequencies of the symmetric and asymmetric N2 stretching modes, which established an iron center more electron rich than that in the corresponding bis(imino)pyridine compounds. Accordingly, (iPrCNC)Fe(N2)2 and its less sterically protected variants (MeCNC)Fe(N2)2 and (MesCNC)Fe(N2)2 are extremely active hydrogenation precatalysts, with the latter compounds exhibiting rapid turnover for unactivated tri- and tetrasubstituted alkenes, which are challenging substrates for even the most active precious-metal catalysts.14
The observation of increased catalytic hydrogenation activity with more electron rich iron dinitrogen complexes has prompted additional studies aimed at understanding the origins of this effect. Before mechanistic studies can be meaningfully interpreted, the electronic structure of the various iron structural types must be established. Studies from our laboratory15,16 and others17−19 have established the redox activity and noninnocence20 of bis(imino)pyridine ligands in reduced iron,21 manganese,22 and cobalt23,24 complexes. Specifically, the five-coordinate bis(imino)pyridine iron bis(dinitrogen) complex (iPrPDI)Fe(N2)2 is best described as a hybrid structure with iron(0) and iron(II) canonical forms, where the chelating ligand serves as a classical π-acid with no evidence for radical character on either the ligand or the metal (Scheme 2). Because of the ambiguity in the oxidation state due to the high covalency in the molecule, we apply the term redox noninnocence in line with Jørgenson’s original definition.16,25 The electronic structure of the four-coordinate compound (iPrPDI)FeN2 is distinct from that of the five-coordinate species and is best described as an intermediate-spin iron(II) compound antiferromagnetically coupled to a triplet bis(imino)pyridine diradical dianion (Scheme 2).21 There is no ambiguity surrounding the oxidation state assignment of the metal, where the iron is undoubtedly intermediate spin Fe(II), and hence the bis(imino)pyridine diradical is termed “redox active”.16
Scheme 2. Difference in Electronic Structures between (iPrPDI)Fe(N2)2 and (iPrPDI)FeN2.
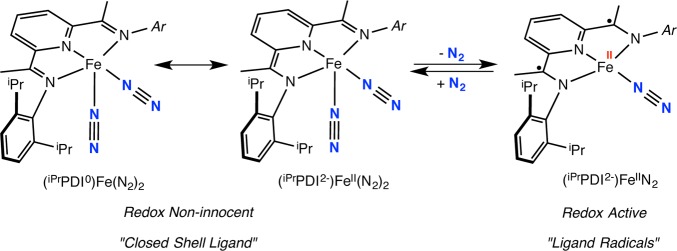
The structural similarity between bis(imino)pyridines and the pyridine N-heterocyclic dicarbenes raised the question if the latter type of iron dinitrogen complexes or catalytic intermediates have redox active chelates with ligand-centered radicals. DFT calculations have been used to explore the electronic structures of (iPrCNC)Fe(N2)2 and (iPrCNC)Fe(CO)2 and variants where methyl groups have been introduced into the meta positions of the central pyridine ring. Computational studies by Danopoulos and Sassmannshausen26 established a classical Fe(0) description for both (iPrCNC)Fe(N2)2 and (iPrCNC)Fe(CO)2 with competitive iron–pyridine π-back-bonding accounting for the relatively high carbonyl stretching frequencies in comparison to iron compounds with dialkylated phosphine-based [PNP] pincers. Subsequent DFT studies by Zhang27 explored the energetics of both singlet and triplet states for (iPrCNC)Fe(N2)2 and found that the singlet was favored for the five-coordinate complex. Dissociation of one of the dinitrogen ligands involved singlet–triplet crossing to produce the high-spin, four-coordinate iron complex (iPrCNC)Fe(N2). Examination of the spin density in the triplet excited states of (iPrCNC)Fe(N2)2 established radical character on the [iPrCNC] ligand, with the principal component in the pyridine ring suggesting that redox activity is possible. Our laboratory recently described investigations into the catalytic alkene hydrogenation activity and electronic structures of (iPrCNC)CoCH328 and (iPrCNC)CoH.29 Both experimental and computational studies supported low-spin Co(II) complexes and the presence of a ligand-centered, essentially pyridine localized radical. Support for this view of the electronic structure was obtained from the observation of Co–H migration into the 4-position of the pyridine ring under a dinitrogen atmosphere to yield the modified cobalt dinitrogen complex. Here we describe an extension of these studies to the iron bis(dinitrogen) derivative (iPrCNC)Fe(N2)2 and related neutral ligand derivatives and report insight into the inability of these compounds to promote catalytic [2π + 2π] cycloaddition chemistry with α,ω-dienes.
Results and Discussion
Spectroscopic Studies on (iPrCNC)Fe(N2)2
Spectroscopic studies were conducted on (iPrCNC)Fe(N2)2 to experimentally establish the electronic structure of the compound and determine the participation of the pyridine N-heterocyclic dicarbene. Because monomeric aryl-substituted bis(imino)pyridine iron complexes undergo dissociation of 1 equiv of N2 upon dissolution in benzene-d6,4,30 similar behavior was probed for (iPrCNC)Fe(N2)2. As reported previously,13 the toluene solution infrared spectrum exhibits two bands for the symmetric and asymmetric dinitrogen stretches centered at 2112 and 2046 cm–1, respectively. No evidence was obtained for the putative four-coordinate compound (iPrCNC)FeN2, even after stirring for extended periods in solution.
Budzelaar31,32 and subsequently our laboratory15 have demonstrated the utility of 1H NMR spectroscopy for identifying redox active chelates in reduced iron and cobalt chemistry. Deviations of the in-plane chelate hydrogens from their diamagnetic reference values arise from either thermal population of a triplet excited state or from temperature-independent paramagnetism and signal participation of the ligand in the electronic structure. To explore this possibility with (iPrCNC)Fe(N2)2, the benzene-d61H NMR spectrum of the iron compound was compared to that of the free pyridine N-heterocyclic dicarbene (see Figure S1, Supporting Information). None of the chemical shifts of (iPrCNC)Fe(N2)2 significantly deviate from those of iPrCNC, establishing that higher spin states of the iron compound are sufficiently energetically separated from the diamagnetic ground state to influence the 1H NMR spectrum of the compound.
The solution behavior of (iPrCNC)Fe(N2)2 was also studied by 15N NMR spectroscopy. The 15N isotopologue was prepared by exposure of the natural-abundance iron dinitrogen compound to 15N2 gas. The benzene-d615N NMR spectrum (Figure S2, Supporting Information) exhibits two broad singlets centered at 323.4 and 348.4 ppm (Δν1/2 = 36 and 33 Hz, respectively). Because the 1H NMR spectrum exhibits the number of resonances consistent with C2v symmetry and hence rapidly interchanging dinitrogen ligands, the two peaks observed are likely due to the α and β nitrogen atoms of a single N2 molecule. The absence of N≡N coupling has been observed previously with iron dinitrogen compounds.9,33
In addition to dinitrogen complexes, four-coordinate bis(imino)pyridine iron compounds with σ-only ligands have also proven useful in establishing the redox activity of this ligand class in reduced iron chemistry.34 One illustrative example is (iPrPDI)Fe(DMAP) (DMAP = 4-N,N-dimethylaminopyridine), an intermediate-spin ferrous compound with a bis(imino)pyridine triplet diradical dianion. Attempts to synthesize the CNC iron analogue by addition of DMAP to (iPrCNC)Fe(N2)2 resulted in loss of only 1 equiv of dinitrogen and furnished a dark crystalline solid identified as (iPrCNC)Fe(DMAP)(N2) in 80% yield as deep red cubes (eq 1).
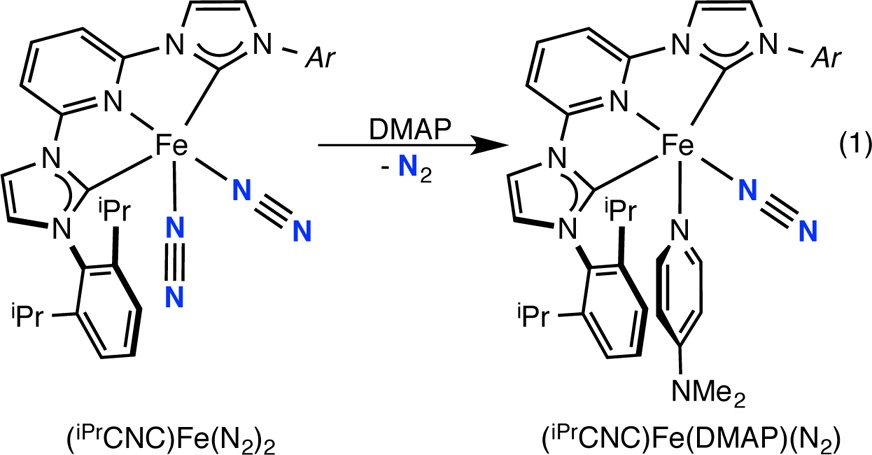 |
1 |
The benzene-d61H NMR spectrum for (iPrCNC)Fe(DMAP)(N2) exhibits the number of resonances expected for a Cs-symmetric iron complex, consistent with coordination of two different neutral ligands. As with (iPrCNC)Fe(N2)2, the chemical shifts of the in-plane hydrogens do not significantly deviate from the reference values of the free chelate. The most notable feature of the spectrum is the upfield shift of the ortho and meta hydrogens on the coordinated DMAP to 6.95 and 5.19 ppm, respectively. (iPrCNC)Fe(DMAP)(N2) was also characterized by a strong N2 absorption at 1998 cm–1 in the pentane solution infrared spectrum. This value is more reduced by ∼10 cm–1 than values for other five-coordinate CNC iron dinitrogen complexes, (iPrCNC)Fe(L)N2 (L = CH2=CH2, PMe3), reported by Danopoulos.13
Single crystals of (iPrCNC)Fe(DMAP)(N2) suitable for X-ray diffraction were obtained from a concentrated diethyl ether solution at −35 °C. A representation of the molecular structure is presented in Figure 1 and confirms the identity of the molecule as a five-coordinate, pseudo-square-pyramidal iron complex with the DMAP ligand occupying the apical position. Selected bond distances and angles are reported in Table 1. No significant distortions in the chelate are observed in either the iPrCNC ligand or the coordinated DMAP.
Figure 1.
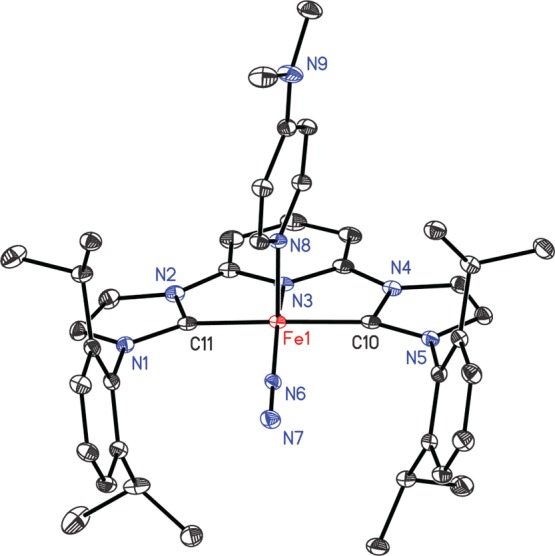
Solid-state structure of (iPrCNC)Fe(DMAP)(N2) with 30% probability ellipsoids. Hydrogen atoms are omitted for clarity.
Table 1. Bond Distances (Å) and Angles (deg) for (iPrCNC)Fe(DMAP)(N2).
| Fe(1)–N(3) | 1.8903(14) | N(1)–C(11) | 1.387(2) |
| Fe(1)–C(10) | 1.9156(17) | C(11)–N(2) | 1.406(2) |
| Fe(1)–C(11) | 1.9179(17) | N(4)–C(10) | 1.405(2) |
| Fe(1)–N(6) | 1.7797(14) | C(10)–N(5) | 1.387(2) |
| Fe(1)–N(8) | 2.0467(16) | N(6)–N(7) | 1.1323(19) |
| N(3)–Fe(1)–N(6) | 160.62(6) | N(1)–C(11)–N(2) | 101.73(13) |
| N(3)–Fe(1)–N(8) | 102.36(6) | N(4)–C(10)–N(5) | 101.55(13) |
Mössbauer, X-ray Absorption, and DFT Studies
The electronic structures of a family of pyridine N-heterocyclic dicarbene iron complexes were also investigated by zero-field 57Fe Mössbauer spectroscopy at 80 K. Representative spectra are presented in Figure 2. To our knowledge, Mössbauer data have not been reported for any iron compound in the series. The experimentally determined isomer shifts (δ) and quadrupole splittings (ΔEQ) along with those for the corresponding bis(imino)pyridine iron compound are reported in Table 2.
Figure 2.
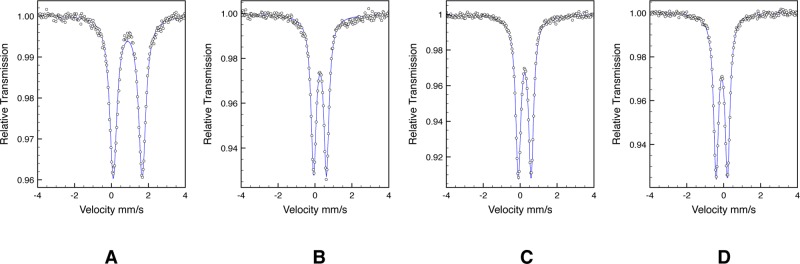
Zero-field 57Fe Mössbauer spectra of (iPrCNC)FeBr2 (A), (iPrCNC)Fe(N2)2 (B), (iPrCNC)Fe(DMAP)(N2) (C), and (iPrCNC)Fe(CO)2 (D) recorded at 80 K.
Table 2. Zero-Field 57Fe Mössbauer Parameters for Pyridine N-Heterocyclic Dicarbene and Pyridine Diimine Iron Complexesa.
| compd | δ (mm/s) | |ΔEQ| (mm/s) |
|---|---|---|
| (iPrCNC)FeBr2 | 0.88 | 1.57 |
| (iPrPDI)FeCl2b | 0.89 | 2.40 |
| (iPrCNC)Fe(N2)2 | 0.27 | 0.69 |
| (iPrPDI)Fe(N2)2b | 0.39 | 0.53 |
| (iPrCNC)Fe(DMAP)(N2) | 0.24 | 0.70 |
| (iPrPDI)Fe(DMAP)b | 0.31 | 1.94 |
| (iPrCNC)Fe(CO)2 | –0.10 | 0.62 |
| (iPrPDI)Fe(CO)2b | 0.03 | 1.17 |
All data were recorded at 80 K, and values are reported relative to α-iron.
Data taken from ref (15).
The parameters observed for (iPrCNC)FeBr2 are typical for a high-spin ferrous compound and are similar to those observed for the bis(imino)pyridine analogue (iPrPDI)FeCl2. For the reduced compounds, pyridine N-heterocyclic dicarbene complexes have isomer shifts consistently lower than those for the corresponding bis(imino)pyridine compounds, a result of increased covalency and relatively short iron–ligand bonds. For (iPrCNC)Fe(N2)2, (iPrCNC)Fe(DMAP)(N2), and (iPrCNC)Fe(CO)2, the data are consistent with iron(0) compounds with the N-heterocyclic carbenes serving as π-acids.35−38
The electronic structures of the pyridine N-heterocyclic dicarbene iron complexes were also investigated by X-ray absorption spectroscopy (XAS).39−41 XAS has proven particularly useful in elucidation of the electronic structure of bis(imino)pyridine iron complexes and has been able to distinguish between redox non-innocent (iPrPDI)Fe(N2)2 and redox active (iPrPDI)Fe(N2)21 as well as detect spin crossover behavior.42 The normalized XAS spectra for (iPrCNC)FeBr2, (iPrCNC)Fe(N2)2, (iPrCNC)Fe(DMAP)(N2), and (iPrCNC)Fe(CO)2 are presented in Figure 3. (iPrCNC)FeBr2 serves as a reference of an established high-spin iron(II) compound, with a pre-edge energy of 7112.0 eV (Table 3), which is in the range for typical ferrous compounds.39 The first pre-edge transitions for the other iPrCNC compounds are shifted by as much as 0.8 eV higher in energy, with the pre-edge for (iPrCNC)Fe(CO)2 at 7112.8 eV. Both (iPrCNC)Fe(DMAP)(N2) and (iPrCNC)Fe(N2)2 have similar pre-edge energies, intensities, and shapes, suggesting that they have similar oxidation states and hence electronic structures. In XAS, shifts of both the pre-edge and rising edge (∼7115–7125 eV) to higher energies are often attributed to a higher metal oxidation state but can also be influenced by metal back-bonding to ligands, resulting in the appearance of a more oxidized metal center.43 In the reduced iPrCNC series of compounds, the rising edge shifts to higher energy as the π-accepting character of the ligands increases, suggesting that ligand back-bonding is the primary contribution to the edge shift and is also consistent with the relatively low isomer shifts observed by Mössbauer spectroscopy.
Figure 3.
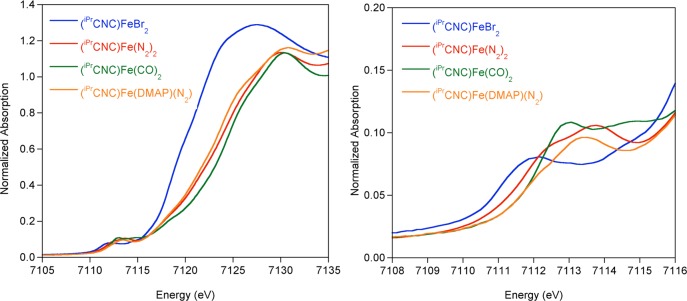
Normalized Fe K-edge XAS spectra of [(iPrCNC)Fe] complexes. Data were collected at 10 K.
Table 3. Experimentally Fit XAS Pre-Edge Positions and Areasa.
| compd | peak 1 (eV); area | peak 2 (eV); area |
|---|---|---|
| (iPrCNC)FeBr2 | 7112.0; 11.9(9) | NA |
| (iPrPDI)FeCl2b | 7111.8; 8(3) | 7113.5(3); 8(4) |
| (iPrCNC)Fe(N2)2 | 7112.4; 13(1) | 7113.8; 8.1(7) |
| (iPrPDI)Fe(N2)2b | 7111.9; 6.4(1) | 7114.0; 19.8(4) |
| (iPrCNC)Fe(DMAP)(N2) | 7112.0; 2.4(5) | 7113.3; 14(1) |
| (iPrPDI)Fe(DMAP)b | 7111.1; 13.3(3) | 7115.0; 20.2(1) |
| (iPrCNC)Fe(CO)2 | 7112.8; 13.5(1) | 7114.5; 8.0(1) |
| (iPrPDI)Fe(CO)2b | 7112.4; 10.2(1) | 7114.5; 16.6(2) |
The XAS data for (iPrCNC)Fe(N2)2 and (iPrCNC)Fe(CO)2 were compared to those for the structurally similar bis(imino)pyridine compounds in an attempt to delineate the relative contributions from π-back-bonding of the CNC versus PDI chelate from the CO or N2 supporting ligands (Figure 4, Table 3).21 The overall spectral shapes for the [(iPrPDI)Fe] and [(iPrCNC)Fe] compounds are similar but the edges for the iPrCNC examples are generally shifted to higher energies. As with the [iPrPDI] compounds, the pre-edges of (iPrCNC)Fe(N2)2 and (iPrCNC)Fe(CO)2 exhibit two features. On the basis of previous assignments for [iPrPDI] compounds,21 it is likely the lower energy pre-edge feature corresponds to a metal 1s to 3d transition with strong contributions from the chelate π* orbital. The first pre-edge feature is shifted 0.5 eV higher in energy for (iPrCNC)Fe(N2)2 in comparison to (iPrPDI)Fe(N2)2 and 0.4 eV to higher energy for (iPrCNC)Fe(CO)2 in comparison to (iPrPDI)Fe(CO)2, suggesting that the iPrCNC ligand affects a 0.4–0.5 eV shift to higher energy in the first pre-edge feature in comparison to iPrPDI. Therefore, the high pre-edge energy of 7112.8 eV for (iPrCNC)Fe(CO)2 is best attributed to increased chelate π-back-bonding and not a higher metal oxidation state. The second pre-edge feature in the iPrPDI compounds results from transitions to N2 or CO π* orbitals.21 For (iPrCNC)Fe(CO)2, the second pre-edge feature at 7114.5 eV is the same as that of the iPrPDI analogue, supporting the assignment as a predominantly CO π* transition. The 0.9 eV pre-edge shift to higher energy of (iPrCNC)Fe(DMAP)(N2) versus (iPrPDI)Fe(DMAP) may be rationalized by combined contributions from the iPrCNC ligand and the coordinated N2 ligand. The similarities of the iPrCNC iron XAS spectra to those of the [(iPrPDI)Fe] compounds support that the main contributions to spectral energy and shape result from back-bonding to the N2 and CO ligands. While the spectra for the iPrCNC ligand are shifted 0.4–0.5 eV to higher energies than those for iPrPDI, the overall electronic structural descriptions for (iPrCNC)Fe(N2)2 and (iPrCNC)Fe(CO)2 appear similar to those for the [(iPrPDI)Fe] compounds.
Figure 4.

Normalized Fe K-edge XAS spectra of [(iPrCNC)Fe] complexes and comparison to the bis(imino)pyridine analogues. Data were collected at 10 K.
Although (iPrCNC)Fe(N2)2 has been the subject of previous computational studies, to our knowledge broken-symmetry solutions corresponding to redox active ligand radical electronic structures have not been investigated. The acquisition of 57Fe Mössbauer and XAS spectral data provides experimental parameters from which to calibrate the computational results. Broken-symmetry possibilities44 were explored for (iPrCNC)Fe(N2)2 at the B3LYP level of DFT. In the broken symmetry notation BS(m,n) describes a state in which there are m unpaired spin-up electrons and n unpaired spin-down electrons on separate fragments.44−46
Full-molecule calculations were performed on three models for (iPrCNC)Fe(N2)2: a spin-restricted RKS and spin unrestricted BS(2,2) and BS(1,1). The two broken-symmetry calculations converged to the same BS(1,1) description with an overlap of S = 0.75, which was 2.2 kcal/mol lower in energy than the RKS solution. Molecular orbital diagrams from both the RKS and BS(1,1) outputs are presented in Figure 5. Also presented in Figure 5 is the spin density plot generated from the BS(1,1) solution. The calculated geometric parameters from both the BS(1,1) and RKS solutions (Table S1, Supporting Information) are in excellent agreement with the experimental data and are within experimental error from each other. The 57Fe Mössbauer parameters were also computed from the two solutions but the values (RKS, δ = 0.26 mm/s, ΔEQ = +0.74 mm/s; BS(1,1), δ = 0.34 mm/s, ΔEQ = +0.61 mm/s) are indistinguishable on the basis of the error in the calculations (δ = ±0.1 mm/s; ΔEQ = ±0.5 mm/s)47 and the experimental values (δ = 0.27 mm/s; |ΔEQ| = 0.69 mm/s). On the basis of energetic, structural, and spectroscopic parameters, DFT calculations are unable to distinguish the classical π-back-bonding description versus the redox active alternative. Analogous calculations were performed on (iPrCNC)Fe(DMAP)(N2), and the results are presented in the Supporting Information. As with (iPrCNC)Fe(N2)2, the energetics of the RKS and BS(1,1) solutions were indistinguishable, with the broken-symmetry solution being 3 kcal/mol lower in energy. However, the computed 57Fe Mössbauer parameters (RKS, δ = 0.28 mm/s, ΔEQ = +0.73 mm/s; BS(1,1), δ = 0.36 mm/s; ΔEQ = +0.50 mm/s) favor the RKS description.
Figure 5.
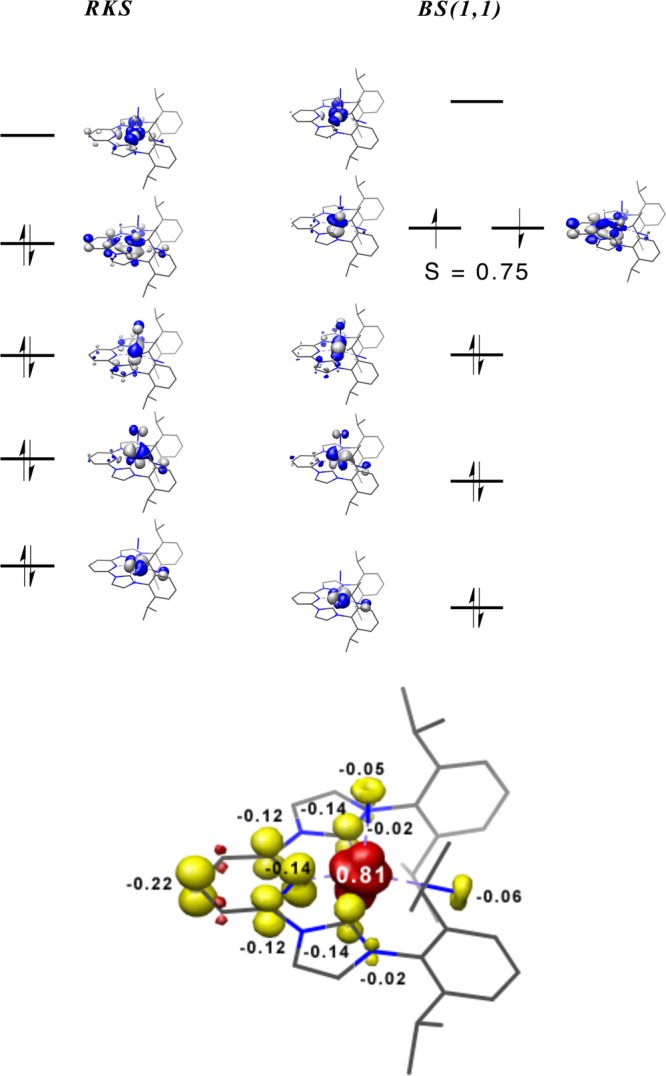
(top) Qualitative MO diagrams for (iPrCNC)Fe(N2)2 obtained from RKS (left) and BS(1,1) (right) DFT calculations at the B3LYP level . (bottom) Spin density plot for (iPrCNC)Fe(N2)2 obtained from the BS(1,1) solution.
As an additional comparison for the calculated electronic structure descriptions, the XAS spectra were calculated using TD-DFT methods. In both coordination compounds and enzymes, calculated TD-DFT XAS spectra have shown good agreement with experiment.21,48 However, the calculated XAS spectra for this series were plagued by charge transfer transitions. This is a limitation of DFT that has been previously described due to the strong dependence of calculated charge transfer bands on the degree of Hartree–Fock mixing.21,49 A full examination of Hartree–Fock mixing in these compounds is beyond the scope of this study. Most notably, the combined computational and experimental results for both (iPrCNC)Fe(N2)2 and (iPrCNC)Fe(DMAP)(N2) provide no definitive evidence for a redox active pyridine N-heterocyclic dicarbene with ligand-centered radicals. On the basis of the spectroscopic studies, the computational data, and previous studies on (iPrPDI)Fe(N2)2,21 we favor a more classical, redox non-innocent description for both reduced compounds where the iPrCNC ligand acts as a π-acceptor. Thus, both (iPrCNC)Fe(N2)2 and (iPrCNC)Fe(DMAP)(N2) are best considered iron(0) compounds with contribution from an iron(II) resonance structure where the chelate has been reduced by two electrons and is in its closed-shell form.
Insights into Iron-Catalyzed [2π + 2π] α,ω-Diene Cycloaddition
Aryl-substituted bis(imino)pyridine iron dinitrogen complexes are known to catalyze the [2π + 2π] cycloaddition of α,ω-dienes to yield the corresponding bicycloheptane derivatives.6,7,50 Because (iPrPDI)Fe(N2)2 and (iPrCNC)Fe(N2)2 have similar ground states, the catalytic cycloaddition chemistry of the latter was studied. Monitoring a benzene-d6 solution of N,N-diallyl-tert-butylamine in the presence of 10 mol % of (iPrCNC)Fe(N2)2 by 1H NMR spectroscopy revealed minimal olefin isomerization along with cyclization to the 3-exo-substituted pyrrolidine rather than cycloaddition (Scheme 3).
Scheme 3. Reaction of N,N-Diallyl-tert-butylamine in the Presence of 10 mol % of (iPrCNC)Fe(N2)2.

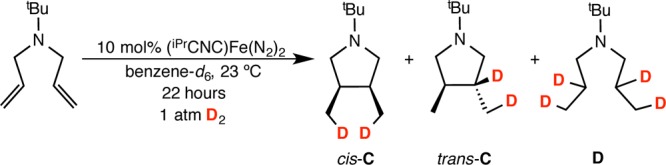
Partial (∼30%) consumption of the starting material was observed over the course of 22 h at 23 °C, and the two products, designated A and B, were formed in an approximate 1:1 ratio. For A, the NMR data are consistent with exclusive formation of the trans isomer of the internal alkene. By comparison, [2π + 2π] cycloaddition promoted by (iPrPDI)Fe(N2)2 is complete in less than 15 min under identical conditions. Repeating the catalytic experiment under 1 atm of H2 at 23 °C resulted in formation of three new products, cis and trans isomers of C as well as D, in a 3:3:4 ratio on the basis of integration of the tert-butyl resonances. Additional analysis by GC-MS established that C was a 1:1 mixture of the diastereomers cis- and trans-3,4-dimethylpyrrolidine. Product D was identified as the open-chain hydrogenation product N-(tert-butyl)-N-propylpropan-1-amine (Scheme 3).
Repeating the hydrogenation experiment with 1 atm of D2 gas placed deuterium exclusively in the methyl positions of the cis isomer of C, suggesting hydrogenation of a putative iron metallacycle (Scheme 4).50 For trans-C, deuterium was located in a 1:1 ratio in both the methyl and methine positions, likely arising from hydrogenation (deuteration) of the 3-exo-substituted pyrrolidine rather than the iron metallacycle. The open-chain product D also contained a 1:1 ratio of deuterium in the methyl and methylene positions, consistent with hydrogenation of the diene substrate and the known performance of (iPrCNC)Fe(N2)2 as an olefin hydrogenation catalyst.12
Scheme 4. Catalytic Deuteration of N,N-diallyl-tert-butylamine in the Presence of (iPrCNC)Fe(N2)2.
Additional insight into the catalytic results was provided by the stoichiometric reaction of N,N-diallyl-tert-butylamine with (iPrCNC)Fe(N2)2. Recrystallization from pentane following addition of the diene to the iron complex yielded brown shards identified as the iron diene complex (iPrCNC)Fe(η2,η2-(CH2=CHCH2)2NtBu). Danopoulos and co-workers have previously reported that addition of ethylene to (iPrCNC)Fe(N2)2 resulted in loss of 1 equiv of dinitrogen and yielded the characterized iron ethylene dinitrogen complex (iPrCNC)Fe(CH2=CH2)(N2).13 Crystallographic characterization established a near-square-pyramidal geometry, with the ethylene ligand occupying the apical position.
The benzene-d61H NMR spectrum of diamagnetic (iPrCNC)Fe(η2,η2-(CH2=CHCH2)2NtBu) exhibited the number of resonances consistent with a compound with C2 symmetry. Diagnostic upfield-shifted resonances for coordination of the diene were observed, with the most extreme case being located at 0.69 ppm. Similar NMR behavior was reported by Danopoulos for (iPrCNC)Fe(CH2=CH2)(N2).13 The solid-state structure of (iPrCNC)Fe(η2,η2-(CH2=CHCH2)2NtBu) was determined by X-ray diffraction, and a representation of the molecular structure is presented in Figure 6. Selected metrical parameters are reported in Table 4. The allyl and tert-butyl groups of the diene were disordered over two positions.
Figure 6.
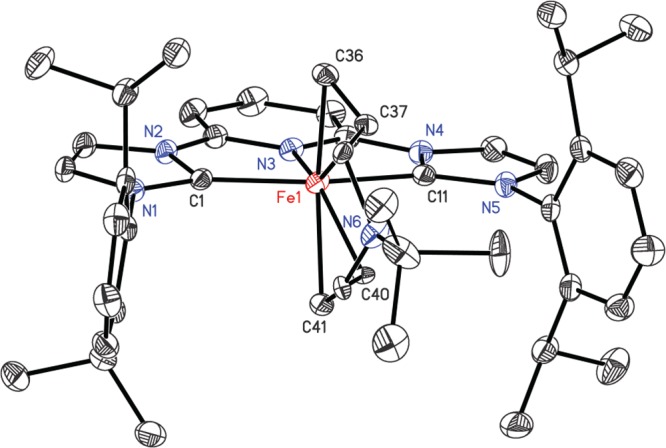
Representation of the molecular structure of (iPrCNC)Fe(η2,η2-(CH2=CHCH2)2NtBu) with 30% probability ellipsoids. Hydrogen atoms are omitted for clarity. The allyl and tert-butyl groups of the diene were disordered over two positions and successfully modeled.
Table 4. Bond Distances (Å) and Angles (deg) for (iPrCNC)Fe(η2,η2-(CH2=CHCH2)2NtBu).
| Fe(1)–N(3) | 1.9008(16) | N(1)–C(1) | 1.398(3) |
| Fe(1)–C(1) | 1.972(2) | C(1)–N(2) | 1.406(2) |
| Fe(1)–C(11) | 1.990(2) | N(4)–C(11) | 1.404(3) |
| Fe(1)–C(36) | 2.079(2) | C(11)–N(5) | 1.400(3) |
| Fe(1)–C(37) | 2.135(4) | C(36)–C(37) | 1.315(5) |
| Fe(1)–C(40) | 2.087(4) | C(40)–C(41) | 1.413(5) |
| Fe(1)–C(41) | 2.076(2) |
| N(1)–C(1)–N(2) | 100.43(16) | N(4)–C(11)–N(5) | 99.81(18) |
The electronic structure of (iPrCNC)Fe(η2,η2-(CH2=CHCH2)2NtBu was investigated by zero-field 57Fe Mössbauer spectroscopy and broken-symmetry DFT calculations.51 The Mössbauer spectrum (Figure 7) exhibits a doublet with an isomer shift of 0.31 mm/s and a quadrupole splitting of 0.60 mm/s. These parameters are indistinguishable from the values for (iPrCNC)Fe(N2)2 and (iPrCNC)Fe(DMAP)(N2) (Table 1) and suggest a high degree of covalency and similar electronic structures among the three reduced compounds. DFT calculations performed at the B3LYP level successfully reproduced the experimental Mössbauer parameters (RKS, δ(calcd) = 0.35 mm/s, ΔEQ(calcd) = 0.18 mm/s) and support a classical closed-shell iron compound with no evidence for ligand-centered radicals. Notably, the electronic structure observed for (iPrCNC)Fe(η2,η2-(CH2=CHCH2)2NtBu) differs from that reported for (iPr(TB)PDI)Fe(η2,η2-(CH2=CHCH2)2NTs) (iPr(TB)PDI = 2,6-(2,6-iPr2-C6H3-N=C-(CH2)3)2(C5HN)), a molecule with an S = 1 ground state (δ = 0.63 mm/s, ΔEQ = 2.47 mm/s).50
Figure 7.
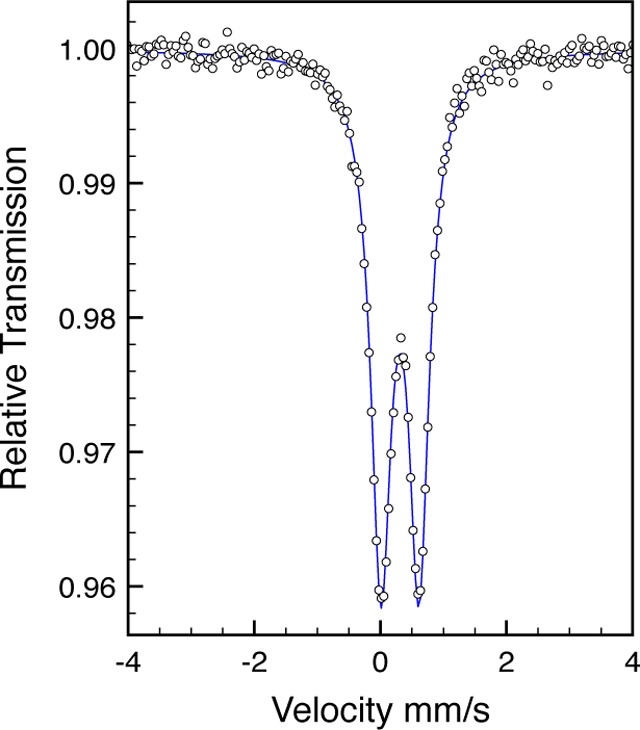
Zero-field 57Fe Mössbauer spectrum of (iPrCNC)Fe(η2,η2-(CH2=CHCH2)2NtBu) at 80 K.
The reactivity of (iPrCNC)Fe(η2,η2-(CH2=CHCH2)2NtBu) with dihydrogen was explored to correlate the intermediacy of the diene complex with the observed hydrogenative cyclization chemistry. Exposure of a benzene-d6 solution of (iPrCNC)Fe(η2,η2-(CH2=CHCH2)2NtBu) to 1 atm of H2 resulted in rapid and selective formation of cis-C. Repeating the experiment with D2 gas and analysis of the products by 1H and 2H NMR spectroscopy revealed formation of the d2 isotopologue of cis-C, where the isotopic label was located 1:1 in the 3,4-methyl groups. Re-exposure of the reaction mixture to 1 atm of dinitrogen resulted in re-formation of (iPrCNC)Fe(N2)2. These results are consistent with previous observations in bis(imino)pyridine iron chemistry where, even if the catalyst resting state is the metal diene complex, the corresponding metallacycle is kinetically accessible and gives rise to cyclization products under catalytic conditions.
Concluding Remarks
The electronic structures of reduced pyridine N-heterocyclic dicarbene iron complexes have been investigated by a combination of structural, spectroscopic, and computational studies. In all cases examined, the [(iPrCNC)Fe] motif prefers five-coordinate bis(neutral) ligand complexes with no experimental evidence for four-coordinate alternatives arising from ligand dissociation. Among these compounds, the tridentate pyridine N-heterocyclic dicarbene chelate acts as a classical π-acceptor with no evidence for ligand-centered radicals, suggesting that an iron(0) oxidation state is the most appropriate description for this family of molecules. Attempts to promote the catalytic [2π + 2π] cyclization of N,N-diallyl-tert-butylamine with (iPrCNC)Fe(N2)2 were unsuccessful, instead producing exo-methylene pyrrolidine and olefin isomerization products in low yield. These results, along with hydrogenation and isotopic labeling experiments, demonstrate that the requisite iron metallacycles for [2π + 2π] cycloaddition are kinetically accessible but, in the absence of ligand radicals and higher oxidation state iron, C–C bond formation via reductive elimination is not observed, highlighting the importance of redox active bis(imino)pyridine ligands for this type of catalysis.
Experimental Section
General Considerations
All air- and moisture-sensitive manipulations were carried out using standard vacuum line, Schlenk, and cannula techniques or in an MBraun inert-atmosphere drybox containing an atmosphere of purified nitrogen. Solvents for air- and moisture-sensitive manipulations were initially dried and deoxygenated using literature procedures.52 Benzene-d6 was purchased from Cambridge Isotope Laboratories and dried over 4 Å molecular sieves. The following compounds were prepared as described previously: (iPrCNC)FeBr2,53 (iPrCNC)Fe(N2)2,13 and (iPrCNC)Fe(CO)2.13
1H NMR spectra were recorded on Varian Mercury 300 and Inova 400, 500, and 600 spectrometers operating at 299.76, 399.78, 500.62, and 599.78 MHz, respectively. 13C NMR spectra were recorded either on an Inova or on a Bruker 500 spectrometer operating at 125.893 and 125.853 MHz, respectively. All 1H and 13C NMR chemical shifts are reported relative to SiMe4 using the 1H (residual) and 13C chemical shifts of the solvent as a secondary standard. For diamagnetic complexes, many assignments were made on the basis of COSY, HSQC, and HMBC NMR experiments. Infrared spectra were collected on a Thermo Nicolet spectrometer. Elemental analyses were performed at Robertson Microlit Laboratories, Inc., in Ledgewood, NJ.
Single crystals suitable for X-ray diffraction were coated with polyisobutylene oil in a drybox, transferred to a nylon loop, and then quickly transferred to the goniometer head. A Bruker APEX2 Duo diffractometer equipped with molybdenum and copper X-ray tubes (λ = 0.71073 and 1.54178 Å, respectively) was used to collect data for (iPrCNC)Fe(DMAP)(N2) (Cu source) and (iPrCNC)Fe(η2,η2-(CH2CHCH2)2NtBu) (Mo source). The space group was identified, and the data were processed using the Bruker SAINT+ program and corrected for absorption using SADABS. The structures were solved using direct methods (SIR92) completed by subsequent Fourier synthesis and refined by full-matrix least-squares procedures.
57Fe Mössbauer spectra were recorded on a SEE Co. Mössbauer spectrometer (MS4) at 80 K in constant acceleration mode. 57Co/Rh was used as the radiation source. WMOSS software was used for the quantitative evaluation of the spectral parameters (least-squares fitting to Lorentzian peaks).54 The temperature of the samples was controlled by a Janis Research Co. CCS-850 He/N2 cryostat within an accuracy of ±1 K. Isomer shifts were determined relative to α-iron at 298 K.
For X-ray absorption spectroscopy, samples were prepared in an inert-atmosphere nitrogen glovebox as finely ground dilutions in boron nitride pressed into 1 mm Al spacers and shipped to the experimental site in a liquid nitrogen Dewar. X-ray absorption spectra were recorded at the Stanford Synchrotron Radiation Lightsource (SSRL) on beamline BL7-3 under standard ring conditions. The beamline optics were optimized, and the monochromator was fully tuned at 7500 eV. The incident energy was calibrated by setting the first inflection of an iron foil to 7111.2 eV. Data were measured in transmission mode. Data were processed using EXAFSPAK55 and pictured using Kaleidagraph.56
All DFT calculations were performed with the ORCA program package.51 The geometry optimizations of the complexes and single-point calculations on the optimized geometries were carried out at the B3LYP level of DFT.57 The all-electron Gaussian basis sets were those developed by Ahlrichs’ group.58 Triple-ζ-quality basis sets def2-TZVP with one set of polarization functions on the metals and on the atoms directly coordinated to the metal center were used. For the carbon and hydrogen atoms, slightly smaller polarized split-valence def2-SV(P) basis sets were used that were of double-ζ quality in the valence region and contained a polarizing set of d functions on the non-hydrogen atoms. Auxiliary basis sets were chosen to match the orbital basis.59 The RIJCOSX60 approximation was used to accelerate the calculations.
Throughout this paper, computational results are described using the BS approach by Ginsberg61 and Noodleman et al.62 Because several BS solutions to the spin-unrestricted Kohn–Sham equations may be obtained, the general notation BS(m,n)63 has been adopted, where m (n) denotes the number of spin-up (spin-down) electrons at the two interacting fragments. Canonical and corresponding64 orbitals, as well as spin density plots, were generated with the program Chimera.65 Nonrelativistic single-point calculations employed the CP(PPP) basis set for iron.66 The Mössbauer isomer shifts were calculated from the computed electron densities at the iron centers as previously described.67 TD-DFT calculations were calculated as previously described.21,48
Preparation of (iPrCNC)Fe(DMAP)(N2)
A 20 mL scintillation vial was charged with 0.100 g (0.155 mmol) of (iPrCNC)Fe(N2)2 in approximately 10 mL of diethyl ether. With stirring, 0.019 g (0.16 mmol) of 4-N,N-dimethylaminopyridine was added and a color change to red-orange was observed. After it was stirred for 30 min, the solution was concentrated and layered with approximately 5 mL of pentane. Storing the solution at −35 °C resulted in crystallization of 0.092 g (80%) of an analytically pure, dark solid identified as (iPrCNC)Fe(DMAP)(N2). Anal. Calcd for C42H51N9Fe: C, 68.38; H, 6.97; N, 17.09. Found: C, 67.98; H, 6.76; N, 16.81. 1H NMR (benzene-d6, 23 °C): δ 1.00 (d, 6 Hz, 6H, CH(CH3)2), 1.05 (d, 6 Hz, 6H, CH(CH3)2), 1.11 (d, 6 Hz, 6H, CH(CH3)2), 1.19 (d, 6 Hz, 6H, CH(CH3)2), 1.93 (s, 6H, N(CH3)2), 3.00 (spt, 6 Hz, 2H, CH(CH3)2), 3.12 (spt, 6 Hz, 2H, CH(CH3)2), 5.19 (d, 6 Hz, 2H, 3-DMAP), 6.83 (s, 2H, imidazolylidene backbone), 6.95 (d, 6 Hz, 2H, 2-DMAP), 7.20–7.33 (m, 6H, m-aryl and 3-pyr), 7.38 (t, 7 Hz, 2H, p-aryl), 7.59–7.70 (m, 3H, imidazolylidene backbone and 4-pyr). 13C NMR {1H} (benzene-d6, 23 °C): δ 23.1 (CH(CH3)2), 23.3 (CH(CH3)2), 25.6 (CH(CH3)2), 26.7 (CH(CH3)2), 28.4 (CH(CH3)2), 28.5 (CH(CH3)2), 38.2 (N(CH3)2), 98.6 (pyr), 106.9 (DMAP), 109.8 (imidazolylidene backbone), 122.9 (aryl), 123.9 (pyr), 124.4 (aryl), 126.2 (imidazolylidene backbone), 129.1 (aryl), 139.0 (aryl), 140.9 (pyr), 146.7 (aryl), 148.8 (aryl), 151.8 (DMAP), 152.0 (DMAP), 203.6 (carbene). IR (pentane, 22 °C): ν(N2) 1998 cm–1. IR (KBr): ν(N2) 1987 cm–1.
Preparation of (iPrCNC)Fe(η2,η2-(CH2=CHCH2)2NtBu)
A 20 mL scintillation vial was charged with a solution of 0.075 g (0.12 mmol) of (iPrCNC)Fe(N2)2 in approximately 5 mL of diethyl ether. With stirring, approximately 0.020 g (0.13 mmol) of N,N-diallyl-tert-butylamine was added, causing an immediate color change to brown. After it was stirred for 30 min, the solution was concentrated and layered with approximately 5 mL of pentane. Storing the solution at −35 °C resulted in crystallization of a brown solid (0.030 g, 35%) identified as (iPrCNC)Fe(η2,η2-(CH2CHCH2)2NtBu). Anal. Calcd for C45H60FeN6: C, 72.95; H, 8.16; N, 11.34. Found, C, 72.81; H, 7.91; N, 11.47. 1H NMR (benzene-d6, 23 °C): δ 0.69 (t, 11 Hz, 2H, N(CH2CHCH2)2), 0.78 (d, 7 Hz, 6H, CH(CH3)2), 0.89 (d, 7 Hz, 6H, CH(CH3)2), 0.94 (s, 9H, NC(CH3)3), 1.20 (d, 7 Hz, 6H, CH(CH3)2), 1.53 (d, 7 Hz, 6H, CH(CH3)2), 2.03 (d, 12 Hz, 2H, N(CH2CHCH2)2), 2.13 (d, 8 Hz, 2H, N(CH2CHCH2)2), 2.42 (spt, 7 Hz, 2H, CH(CH3)2), 2.47 (spt, 7 Hz, 2H, CH(CH3)2), 3.93 (dd, 12 Hz, 3 Hz, 2H, N(CH2CHCH2)2), 4.70–4.76 (m, 2H, N(CH2CHCH2)2), 6.20 (d, 2 Hz, 2H, imidazolylidene backbone), 6.93 (d, 8 Hz, 2H, m-aryl), 6.98 (d, 8 Hz, 1H, 3-pyr), 7.04 (t, 8 Hz, 1H, p-aryl), 7.12 (d, 2 Hz, 1H, imidazolylidene backbone), 7.23 (d, 8 Hz, m-aryl), 7.30 (d, 8 Hz, 1H, 3-pyr), 7.37 (t, 8 Hz, 1H, 4-pyr), 7.40 (t, 8 Hz, 1H, p-aryl), 7.46 (d, 2 Hz, 1H, imidazolylidene backbone). 13C{1H} NMR (benzene-d6, 23 °C): δ 21.6 (CH(CH3)2), 22.2 (CH(CH3)2), 26.8 (CH(CH3)2), 27.0 (CH(CH3)2), 28.4 (CH(CH3)2 or C(CH3)3), 28.5 (CH(CH3)2 or C(CH3)3), 28.6 (N(CH2CHCH2)2), 28.8 (CH(CH3)2 or C(CH3)3), 52.4 (C(CH3)3), 54.0 (N(CH2CHCH2)2), 61.4 (N(CH2CHCH2)2), 99.4 (3-pyr), 100.4 (3-pyr), 110.2 (imidazolylidene backbone), 113.0 (imidazolylidene backbone), 114.8 (4-pyr), 123.2 (m-aryl), 124.1 (m-aryl), 127.0 (imidazolylidene backbone), 127.0 (imidazolylidene backbone), 129.5 (p-aryl), 130.3 (p-aryl), 138.4 (aryl), 139.1 (aryl), 146.2 (pyr), 146.5 (aryl), 147.6 (aryl), 152.4 (pyr), 195.8 (carbene), 208.8 (carbene).
General Procedure for Catalytic Experiments
A 20 mL scintillation vial was charged with 0.010 g (16 μmol) of (iPrCNC)Fe(N2)2 and 0.65 g of benzene-d6. N,N-Diallyl-tert-butylamine (0.024 g, 0.16 mmol) was added, which caused an immediate color change from green to brown. The solution was transferred to a J. Young tube, which was stored in a 23 °C water bath. The reaction was monitored by 1H NMR spectroscopy. For experiments where the solution was exposed to 1 atm of H2 or D2, after transfer to a J. Young tube, the solution was immediately submerged in liquid nitrogen. The headspace was evacuated on a high-vacuum line, and 1 atm of H2 or D2 was introduced. The sample was thawed in a 23 °C water bath.
Acknowledgments
P.J.C. thanks the U.S. National Science Foundation and the Deutsche Forschungsgemeinschaft for a Cooperative Activities in Chemistry between U.S. and German Investigators grant. J.M.D. also acknowledges support from the National Institute of General Medical Sciences (Award Number T32GM008500). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. S.C.E.S. thanks the NSF for a graduate research fellowship (DGE-0646086). Portions of the research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the DOE, BES. The SSRL SMB Program is supported by DOE, BER, and NIH, NCRR, BMTP. We also thank Dr. Carsten Milsmann for computational assistance and helpful discussions.
Supporting Information Available
Figures, tables, and xyz and CIF files giving additional NMR spectroscopic and computational data, crystallographic data for (iPrCNC)Fe(DMAP)(N2) and (iPrCNC)Fe(η2,η2-(CH2=CHCH2)2NtBu), and computational output. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Chirik P. J.; Wieghardt K. Science 2010, 327, 794. [DOI] [PubMed] [Google Scholar]; b Enthaler S.; Junge K.; Beller M. Angew. Chem., Int. Ed. 2008, 47, 3317. [DOI] [PubMed] [Google Scholar]; c Nakazawa H.; Itazaki M. Top. Organomet. Chem. 2011, 33, 27. [Google Scholar]; d Junge K.; Schröder K.; Beller M. Chem. Commun. 2011, 47, 4849. [DOI] [PubMed] [Google Scholar]
- a Bolm C.; Legros J.; Paith J. L.; Zani L. Chem. Rev. 2004, 104, 6217. [DOI] [PubMed] [Google Scholar]; b Bauer E. B. Curr. Org. Chem. 2008, 47, 1341. [Google Scholar]; c Gaillard S.; Renaud J.-L. ChemSusChem 2008, 1, 505. [DOI] [PubMed] [Google Scholar]; d Chirik P. J. In Catalysis Without Precious Metals; Bullock R. M., Ed.; Wiley-VCH: Weinheim, Germany, 2010; Chapter 4. [Google Scholar]
- Blanchard S.; Derat E.; Desage-El Murr M.; Fensterbank L.; Malacria M.; Mouriès-Mansuy V. Eur. J. Inorg. Chem. 2011, 2012, 376. [Google Scholar]
- Bart S. C.; Lobkovsky E.; Chirik P. J. J. Am. Chem. Soc. 2004, 126, 13794. [DOI] [PubMed] [Google Scholar]
- Atienza C. C. H.; Tondreau A. M.; Weller K. J.; Lewis K. M.; Cruse R. W.; Nye S. A.; Boyer J. L.; Delis J. G. P.; Chirik P. J. ACS Catal. 2012, 2, 2169. [Google Scholar]
- Bouwkamp M. W.; Bowman A. C.; Lobkovsky E.; Chirik P. J. J. Am. Chem. Soc. 2006, 128, 13340. [DOI] [PubMed] [Google Scholar]
- Russell S. K.; Lobkovsky E.; Chirik P. J. J. Am. Chem. Soc. 2011, 133, 8858. [DOI] [PubMed] [Google Scholar]
- Sylvester K. T.; Chirik P. J. J. Am. Chem. Soc. 2009, 131, 8772. [DOI] [PubMed] [Google Scholar]
- Russell S. K.; Darmon J. M.; Lobkovsky E.; Chirik P. J. Inorg. Chem. 2010, 49, 2782. [DOI] [PubMed] [Google Scholar]
- Tondreau A. M.; Atienza C. C. H.; Weller K. J.; Nye S. A.; Lewis K. M.; Delis J. G. P.; Chirik P. J. Science 2012, 335, 567. [DOI] [PubMed] [Google Scholar]
- Darmon J. M.; Turner Z. R.; Lobkovsky E.; Chirik P. J. Organometallics 2012, 31, 2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R. P.; Darmon J. M.; Hoyt J. M.; Margulieux G. W.; Turner Z. R.; Chirik P. J. ACS Catal. 2012, 2, 1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danopoulos A. A.; Wright J. A.; Motherwell W. B. Chem. Commun. 2005, 784. [DOI] [PubMed] [Google Scholar]
- Roseblade S. J.; Pfaltz A. Acc. Chem. Res. 2007, 40, 1402. [DOI] [PubMed] [Google Scholar]
- Bart S. C.; Chlopek K.; Bill E.; Bouwkamp M. W.; Lobkovsky E.; Neese F.; Wieghardt K.; Chirik P. J. J. Am. Chem. Soc. 2006, 128, 13901. [DOI] [PubMed] [Google Scholar]
- Chirik P. J. Inorg. Chem. 2011, 50, 9737. [DOI] [PubMed] [Google Scholar]
- de Bruin B.; Bill E.; Bothe E.; Weyhermüller T.; Wieghardt K. Inorg. Chem. 2000, 39, 2936. [DOI] [PubMed] [Google Scholar]
- Budzelaar P. H. M.; de Bruin B.; Gal A. W.; Wieghardt K. W.; van Lenthe J. H. Inorg. Chem. 2001, 40, 4649. [DOI] [PubMed] [Google Scholar]
- Knijnenburg Q.; Gambarotta S.; Budzelaar P. H. M. Dalton Trans. 2006, 5442. [DOI] [PubMed] [Google Scholar]
- Chirik P. J. Inorg. Chem. 2011, 50, 9737. [DOI] [PubMed] [Google Scholar]
- Stieber S. C. E.; Milsmann C.; Hoyt J. M.; Turner Z. R.; Finkelstein K. D.; Wieghardt K.; DeBeer S.; Chirik P. J. Inorg. Chem. 2012, 51, 3770. [DOI] [PubMed] [Google Scholar]
- Russell S. K.; Bowman A. C.; Lobkovsky E.; Wieghardt K.; Chirik P. J. Eur. J. Inorg. Chem. 2012, 3, 535. [Google Scholar]
- Bowman A. C.; Milsmann C.; Atienza C. C. H.; Lobkovsky E.; Weighardt K.; Chirik P. J. J. Am. Chem. Soc. 2010, 132, 1676. [DOI] [PubMed] [Google Scholar]
- Bowman A. C.; Milsmann C.; Bill E.; Lobkovsky E.; Weyhermüller T.; Wieghardt K.; Chirik P. J. Inorg. Chem. 2010, 49, 6110. [DOI] [PubMed] [Google Scholar]
- Jørgensen C. K. Coord. Chem. Rev. 1966, 1, 164. [Google Scholar]
- Danopoulos A. A.; Pugh D.; Smith H.; Sassmannshausen J. Chem. Eur. J. 2009, 15, 5491. [DOI] [PubMed] [Google Scholar]
- Zhang X. Int. J. Quantum Chem. 2010, 110, 1880. [Google Scholar]
- Danopoulos A. A.; Wright J. A.; Motherwell W. B.; Ellwood S. Organometallics 2004, 23, 4807. [Google Scholar]
- Yu R. P.; Darmon J. M.; Milsmann C.; Marguliuex G. W.; Stieber S. C. E.; DeBeer S.; Chirik P. J. J. Am. Chem. Soc. 2013, 135, 13168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer A. M.; Bouwkamp M. W.; Cortez M.-P.; Lobkovsky E.; Chirik P. J. Organometallics 2006, 25, 4269. [Google Scholar]
- Knijnenburg Q.; Hetterscheid D.; Kooistra T. M.; Budzelaar P. H. M. Eur. J. Inorg. Chem. 2004, 1204. [Google Scholar]
- Zhu D.; Thapa I.; Korobkov I.; Gambarotta S.; Budzelaar P. H. M. Inorg. Chem. 2011, 50, 9879. [DOI] [PubMed] [Google Scholar]
- Field L. D.; Hazari N.; Li H. L.; Luck I. J. Magn. Reson. Chem. 2003, 41, 709. [Google Scholar]
- Bart S. C.; Lobkovsky E.; Bill E.; Wieghardt K.; Chirik P. J. Inorg. Chem. 2007, 46, 7055. [DOI] [PubMed] [Google Scholar]
- Nemcsok D.; Wichmann K.; Frenking G. Organometallics 2004, 23, 3640. [Google Scholar]
- Hu X. L.; Castro-Rodriguez I.; Olsen K.; Meyer K. Organometallics 2004, 23, 755. [Google Scholar]
- Tulloch A. A. D.; Danopoulos A. A.; Kleinhenz M. E.; Light M. E.; Hursthouse M. B.; Eastham G. Organometallics 2001, 20, 2027. [Google Scholar]
- Khramov D. M.; Lynch V. M.; Bielawski C. W. Organometallics 2007, 26, 6042. [Google Scholar]
- Westre T. E.; Kennepohl P.; DeWitt J. G.; Hedman B.; Hodgson K. O.; Solomon E. I. J. Am. Chem. Soc. 1997, 119, 6297. [Google Scholar]
- Levina A.; Armstrong R. S.; Lay P. A. Coord. Chem. Rev. 2005, 249, 141. [Google Scholar]
- Shulman G. R.; Yafet Y.; Eisenberger P.; Blumberg W. E. Proc. Natl. Acad. Sci. U.S.A. 1976, 73, 1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmon J. M.; Stieber S. C. E.; Sylvester K. T.; Fernández I.; Lobkovsky E.; Semproni S. P.; Bill E.; Wieghardt K.; DeBeer S.; Chirik P. J. J. Am. Chem. Soc. 2012, 134, 17125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ramallo-López J. M.; Lede E. J.; Requejo F. G.; Rodriguez J. A.; Kim J.-Y.; Rosas-Salas R.; Domínguez J. M. J. Phys. Chem. B 2004, 108, 20005. [Google Scholar]; b Honji A.; Gron L. U.; Chang J.-R.; Gates B. C. Langmuir 1992, 8, 2715. [Google Scholar]
- Noodleman L.; Peng C. Y.; Case D. A.; Mouesca J. M. Coord. Chem. Rev. 1995, 144, 199. [Google Scholar]
- Ginsberg A. P. J. Am. Chem. Soc. 1980, 102, 111. [Google Scholar]
- Kirchner B.; Wennmohs F.; Ye S.; Neese F. Curr. Opin. Chem. Biol. 2007, 11, 134. [DOI] [PubMed] [Google Scholar]
- Römelt M.; Ye S.; Neese F. Inorg. Chem. 2009, 48, 784. [DOI] [PubMed] [Google Scholar]
- a Chandrasekaran P.; Stieber S. C. E.; Collins T. J.; Que L.; Neese F.; DeBeer S. Dalton Trans. 2011, 40, 11070. [DOI] [PMC free article] [PubMed] [Google Scholar]; b DeBeer George S.; Petrenko T.; Neese F. J. Phys. Chem. A 2008, 112, 12936. [DOI] [PubMed] [Google Scholar]
- Roemelt M.; Beckwith M. A.; Duboc C.; Collomb M.-N.; Neese F.; DeBeer S. Inorg. Chem. 2012, 51, 680. [DOI] [PubMed] [Google Scholar]
- Hoyt J. M.; Sylvester K. T.; Semproni S. P.; Chirik P. J. J. Am. Chem. Soc. 2013, 135, 4862. [DOI] [PubMed] [Google Scholar]
- Neese F. WIREs Comput. Mol. Sci. 2011, 2, 73. [Google Scholar]
- Pangborn A. B.; Giardello M. A.; Grubbs R. H.; Rosen R. K.; Timmers F. J. Organometallics 1996, 15, 1518. [Google Scholar]
- Danopoulos A. A.; Tsoureas N.; Wright J. A.; Light M. E. Organometallics 2004, 23, 166. [Google Scholar]
- WMOSS4 Mössbauer Spectral Analysis Software; www.wmoss.org, 2009. –2013.
- George G. N.EXAFSPAK; SSRL, SLAC, Stanford University, Stanford, CA, 2000. [Google Scholar]
- Kaleidagraph: Tools for Discovery, Version 4.03; Synergy Software, Reading, PA, 1986.
- a Becke A. D. J. Chem. Phys. 1986, 84, 4524–4529. [Google Scholar]; b Becke A. D. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]; c Lee C. T.; Yang W. T.; Parr R. G. Phys. Rev. B 1998, 37, 785. [DOI] [PubMed] [Google Scholar]
- a Schäfer A.; Horn H.; Ahlrichs R. J. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar]; b Schäfer A.; Huber C.; Ahlrichs R. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar]; c Weigend F.; Ahlrichs R. Phys. Chem. Chem. Phys. 2005, 7, 3297. [DOI] [PubMed] [Google Scholar]
- a Eichkorn K.; Weigend F.; Treutler O.; Ahlrichs R. Theor. Chem. Acc. 1997, 97, 119. [Google Scholar]; b Eichkorn K.; Treutler O.; Öhm H.; Häser M.; Ahlrichs R. Chem. Phys. Lett. 1995, 240, 283. [Google Scholar]; c Eichkorn K.; Treutler O.; Öhm H.; Häser M.; Ahlrichs R. Chem. Phys. Lett. 1995, 242, 652. [Google Scholar]
- a Neese F.; Wennmohs F.; Hansen A.; Becker U. Chem. Phys. 2009, 356, 98. [Google Scholar]; b Kossmann S.; Neese F. Chem. Phys. Lett. 2009, 481, 240. [Google Scholar]; c Neese F. J. Comput. Chem. 2003, 24, 1740. [DOI] [PubMed] [Google Scholar]
- Ginsberg A. P. J. Am. Chem. Soc. 1980, 102, 111. [Google Scholar]
- Noodleman L.; Peng C. Y.; Case D. A.; Mouesca J. M. Coord. Chem. Rev. 1995, 144, 199. [Google Scholar]
- Kirchner B.; Wennmohs F.; Ye S.; Neese F. Curr. Opin. Chem. Biol. 2007, 11, 134. [DOI] [PubMed] [Google Scholar]
- Neese F. J. Phys. Chem. Solids 2004, 65, 781. [Google Scholar]
- Molekel, Advanced Interactive 3D-Graphics for Molecular Sciences, available at http://www.cscs.ch/molekel/.
- Neese F. Inorg. Chim. Acta 2002, 337, 181. [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. J. Comput. Chem. 2004, 25, 1605. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.