

Abstract
A combination of experimental and density functional theory (DFT) investigations suggests that the Cu-catalyzed fluorination of unsymmetrical diaryliodonium salts with general structure [Mes(Ar)I]+ in N,N′-dimethylformamide proceeds through a CuI/CuIII catalytic cycle. A low concentration of fluoride relative to combined iodonium reagent plus copper ensures that [Mes(Ar)I]+ is available as the reactive species for oxidative “Ar+” transfer to a CuI center containing one or two fluoride ligands. A series of different possible CuI active catalysts (containing fluoride, triflate, and DMF ligands) have been evaluated computationally, and all show low-energy pathways to fluorinated products. The oxidation of these CuI species by [Mes(Ar)I]+ to form cis-Ar(F)CuIII intermediates is proposed to be rate-limiting in all cases. Ar–F bond-forming reductive elimination from CuIII is computed to be very facile in all of the systems examined. The conclusions of the DFT experiments are supported by several experimental studies, including tests showing that CuI is formed rapidly under the reaction conditions and that the fluoride concentration strongly impacts the reaction yields/selectivities.
Introduction
Diaryliodonium salts1 are widely used as electrophilic arylating reagents in both metal-free2 and transition-metal-catalyzed reactions.3 In particular, there has been significant recent attention to the development of Cu-catalyzed cross-couplings of diaryliodonium salts with diverse coupling partners, including phosphonates,3b,3e CF3SO2Na,3d fluoride,4 and nitrogen3a,3c,3f and carbon5 nucleophiles. Although a number of literature reports have probed the mechanisms of metal-free reactions of diaryliodonium salts with nucleophiles,6 there is still little known about the detailed mechanism of aryl transfer from diaryliodonium salts to transition metals such as Cu.7 For example, the nature of the active Cu catalyst that reacts with the diaryliodonium salt has not been elucidated in most systems. Furthermore, the mechanistic origin of the selectivity of aryl transfer from unsymmetrical IIII reagents to transition metal centers is poorly understood.1,6
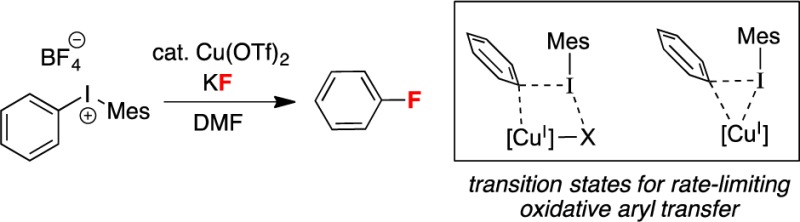
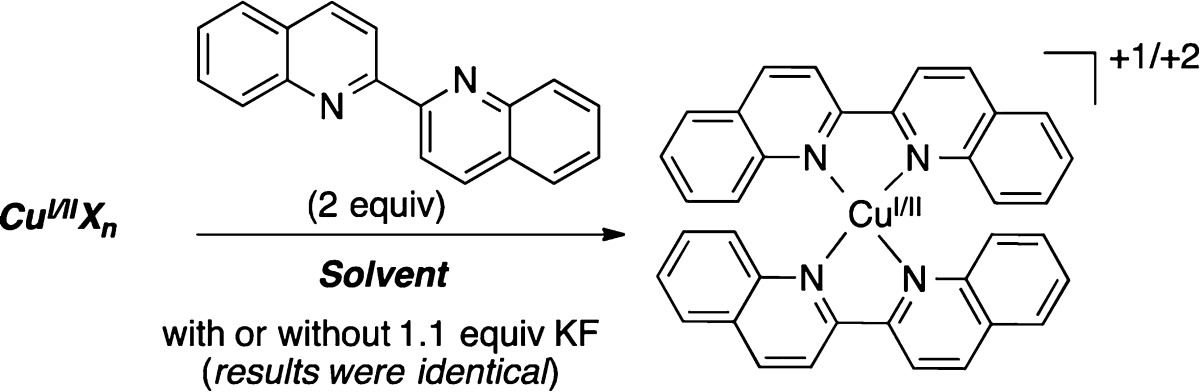
We have recently disclosed the Cu-catalyzed fluorination of diaryliodonium salts with potassium fluoride.4a In the presence of 20 mol % of Cu(OTf)2, the unsymmetrical iodonium reagents [Mes(Ar)I]+ react with high selectivity to generate Ar–F and Mes–I, regardless of the electronic properties of the aryl group. This selectivity is complementary to that observed in the uncatalyzed fluorination of [Mes(Ar)I]+.4a In our original communication, we proposed that this transformation occurs through a CuI/III catalytic cycle (Scheme 1) in which the reaction of a CuI catalyst with [Mes(Ar)I]+ generates a CuIII(Ar)(F) intermediate that undergoes reductive elimination8 to liberate the aryl fluoride product.
Scheme 1. Proposed CuI/III Catalytic Cycle.
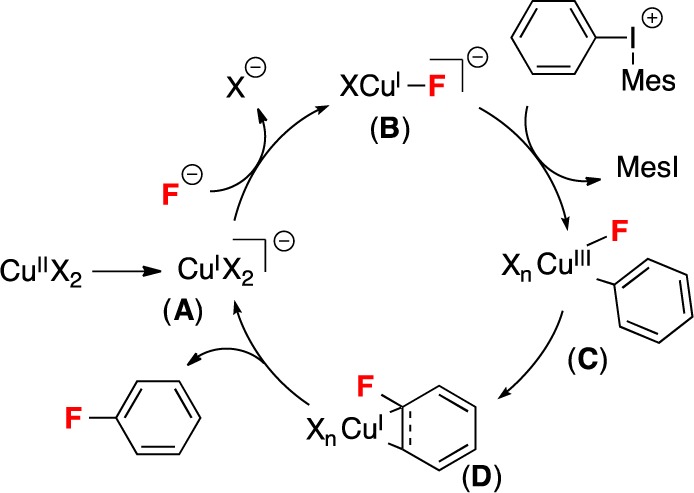
X– = OTf– or F–.
Herein we describe a detailed investigation of the mechanism of this Cu-catalyzed fluorination, using a combination of density functional theory (DFT) calculations and experimental studies. These investigations provide detailed insights into many features of the reaction, including (i) the role of the Cu precatalyst, (ii) possible structures of the active Cu catalyst, (iii) the impact of changing ratios of reagents and Cu precatalyst, (iv) the nature of initial interactions between CuI and the iodonium reagent, and (v) the subsequent sequence of intermediates leading to Ar–F bond-forming reductive elimination from CuIII.
Results and Discussion
Oxidation State of the Copper Catalyst
Before conducting DFT calculations, we sought to gain experimental insights into the oxidation state of the active Cu species during this transformation. As shown in Table 1, the CuII salt Cu(OTf)2 and the CuI salts Cu(OTf)(CH3CN)4, Cu(OTf)(tBuCN)2, and Cu(OTf)·benzene all afford good yield and high selectivity in this transformation under the standard conditions (DMF, 60 °C, 18 h). The lower yield with the CuI precatalysts is due to competing formation of side products (predominantly biphenyl, along with traces of mesitylene, benzene, and diphenyl ether). With both CuII and CuI precatalysts, the reaction is highly solvent dependent, with the best yields and selectivities obtained in DMF.9 As shown in Figure 1, the initial rate of product formation in DMF at 60 °C is slightly faster with Cu(OTf)(CH3CN)4 than with Cu(OTf)2.
Table 1. Cu-Catalyzed Fluorination of [Mes(Ph)I]+ as a Function of Cu Precatalyst and Solventa.
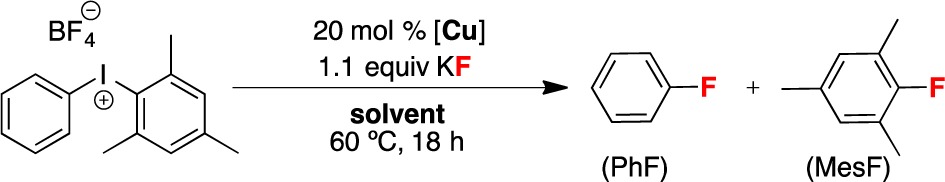
| [Cu] | solvent | yieldb | PhF:MesF |
|---|---|---|---|
| Cu(OTf)2 | DMF | 85% | 98:2 |
| Cu(OTf)(MeCN)4 | DMF | 73% | 99:1 |
| Cu(OTf)(tBuCN)2 | DMF | 71% | 96:4 |
| Cu(OTf)·benzene | DMF | 57% | 96:4 |
| Cu(OTf)2 | NMPc | 38% | 95:5 |
| Cu(OTf)(MeCN)4 | NMPc | 55% | >99:1 |
| Cu(OTf)2 | EtOAc | 39% | 13:87 |
| Cu(OTf)(MeCN)4 | EtOAc | 39% | 13:87 |
| Cu(OTf)2 | toluene | 35% | 14:86 |
| Cu(OTf)(MeCN)4 | toluene | 34% | 13:87 |
Conditions: [Mes-I-Ph]BF4 (0.05 mmol, 1 equiv), [Cu] (0.01 mmol, 0.2 equiv), KF (0.055 mmol, 1.1 equiv), solvent (0.1 M), 60 °C.
Combined yield of PhF and MesF as determined by 19F NMR spectroscopic analysis.
NMP = N-methylpyrrolidinone.
Figure 1.
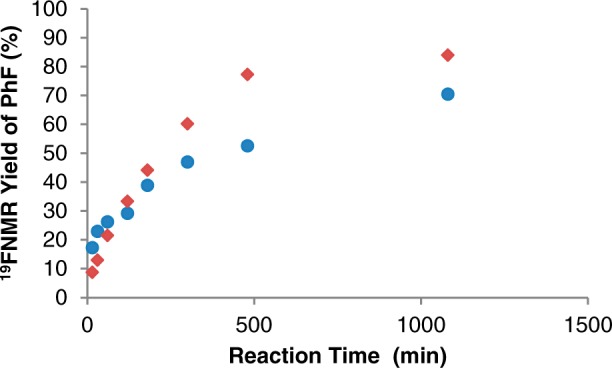
PhF formation as a function of time in the reaction of [Mes(Ph)I]+ with KF catalyzed by Cu(OTf)2 and Cu(OTf)(CH3CN)4 in DMF at 60 °C. Conditions: [Mes-I-Ph]BF4 (0.1 mmol, 1 equiv), [Cu] (0.02 mmol, 0.2 equiv), KF (0.11 mmol, 1.1 equiv), DMF (0.1 M), 60 °C. Yields of PhF at each time point were determined by 19F NMR spectroscopic analysis and represent an average of two separate runs. ◆ = Cu(OTf)2; ● = Cu(OTf)(CH3CN)4.
On the basis of the results in Table 1 and Figure 1, we hypothesized that the two precatalysts might be operating via a similar CuI active species. In the case of Cu(OTf)2, reduction to CuI could be occurring in situ, accounting for the slower initial rate with this precatalyst. Importantly, DMF is well known as a reductant for transition metals.10,11 To test for this possibility, we performed CuI trapping experiments, using 2,2′-biquinoline (biq) as a ligand for colorimetric detection of CuI. Lockhart has shown that biq has a strong binding affinity for CuI, and the resulting complexes exhibit a characteristic intense purple color (λmax = 540 nm).12 Thus, we examined the speciation of Cu(OTf)2 in the presence of biq in a variety of solvents (Table 2). An intense purple color was observed in DMF and NMP within 5 min at room temperature in both the presence and absence of 1.1 equiv of KF, consistent with the formation of CuI in these solvents. UV–vis spectroscopic analysis of these purple solutions showed a λmax between 540 and 550 nm, further consistent with the formation of CuI under these conditions. In contrast, when Cu(OTf)2 and biq were stirred in EtOAc or toluene, an orange precipitate was observed. This is indicative of the CuII complex, [CuII(biq)2]+2.13 However, treatment of this orange precipitate with DMF at room temperature resulted in rapid dissolution and a concomitant color change to intense purple. Overall, these experiments suggest that DMF promotes the reduction of CuII to CuI at 60 °C. Thus, we conclude that CuI species are available with both the Cu(OTf)2 and Cu(OTf)(CH3CN)4 precatalysts and, as discussed below, are likely to be the active catalysts in these systems.14,15
Table 2. CuI/II Trapping Experiment.
| [Cu] | solvent | λmax | color |
|---|---|---|---|
| Cu(OTf)2 | DMF | 540 | dark purple solution |
| Cu(OTf)(MeCN)4 | DMF | 540 | dark purple solution |
| Cu(OTf)2 | NMP | 550 | light purple solution |
| Cu(OTf)2 | EtOAc | na | orange precipitate |
| Cu(OTf)2 | toluene | na | orange precipitate |
Preliminary Considerations for Computational Studies of Copper Triflate Catalyzed Reactions
In our original communication, we conducted DFT calculations on the reaction between the symmetrical iodonium cation [Ph2I]+ and the CuI anion [CuF(OTf)]−.4a In the present study, we have expanded these calculations to examine the unsymmetrical diaryliodonium reagent [Mes(Ph)I]+.16 In addition, other possible CuI catalysts, including CuF(DMF)17,18 and [CuF2]−, have been assessed in detail, following the consideration of computed thermodynamic data for the interaction of [Mes(Ph)I]+ and CuI with triflate, fluoride, and DMF as donor ligands (shown in Table 3). T-shaped and linear geometries are anticipated for IIII and CuI, respectively. For IIII species, only data for ligands trans to Ph are presented, as the alternative isomers with the ligands trans to Mes are marginally less stable [by 0.7 (DMF) and 0.3 kcal/mol (F–)] or exhibit identical ΔG (OTf–).
Table 3. Computation for Reactions of CuI and [Mes(Ph)I]+ with Donor Ligands Present during Cu Catalysis.
| ligand exchange at IIII and CuI centers | ΔG (kcal/mol) | entry |
|---|---|---|
| [Mes(Ph)I]+ + DMF → [Mes(Ph)I(DMF)]+ | 2.7 | 1 |
| [Mes(Ph)I]+ + OTf– → Mes(Ph)I(OTf) | 1.6 | 2 |
| [Mes(Ph)I]+ + F– → Mes(Ph)IF | –15.7 | 3 |
| [Cu(DMF)2]+ + OTf– → Cu(OTf)(DMF) + DMF | 0.7 | 4 |
| Cu(OTf)(DMF) + OTf– → [Cu(OTf)2]− + DMF | 2.0 | 5 |
| [Cu(DMF)2]+ + F– → CuF(DMF) + DMF | –20.7 | 6 |
| CuF(DMF) + F– → [CuF2]− + DMF | –16.4 | 7 |
| [Cu(OTf)2]+ + F– → [CuF(OTf)]− + OTf– | –20.8 | 8 |
| [CuF(OTf)]− + F– → [CuF2]− + OTf– | –19.1 | 9 |
The data in entries 1 and 2 show that DMF and triflate are relatively poor ligands for [Mes(Ph)I]+. In contrast, fluoride binding to [Mes(Ph)I]+ is highly thermodynamically downhill (entry 3). Additionally, the complexation of fluoride with a variety of CuI starting materials is even more thermodynamically favorable (entries 6–9). Assuming that all of the copper is present as CuI, and noting that the reaction of F– with CuI is thermodynamically favored over that with [Mes(Ph)I]+ (compare entries 3 and 6–9), an inspection of the ratio of [Mes(Ph)I]+ to CuI to F– under the catalysis conditions (1 to 0.2 to 1.1, Table 1) shows that there is insufficient F– present to form an adduct with all of the [Mes(Ph)I]+ reagent. This deficiency is retained until close to completion of catalysis.
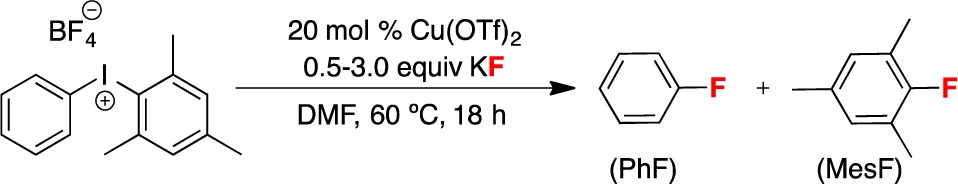
We reasoned that this should be an important factor in catalysis, as [Mes(Ph)I]+ is a significantly more reactive electrophile than Mes(Ph)IF. Under conditions where the resting state of the IIII reagent is Mes(Ph)IF, an extra 15.7 kcal/mol would need to be added to the ΔG⧧ for Cu catalysis compared to the analogous reaction with [Mes(Ph)I]+. As a result, Cu catalysis is expected to be dramatically slower when [KF] ≥ (2[Cu(OTf)2)] + [Mes(Ph)I]+). To test this proposal experimentally, we examined the impact of the ratio of [KF] to ([Cu(OTf)2)] + [Mes(Ph)I]+) on catalysis. These studies were conducted using 20 mol % of Cu(OTf)2 and 1 equiv of [Mes(Ph)I]+, and the amount of KF was varied from 0.5 to 3.0 equiv relative to the iodonium reagent. Under these conditions, the iodonium reagent would be fully complexed with fluoride at 1.4 equiv of KF, assuming that all of the KF is soluble. The extent of Cu catalysis can be estimated based on the ratio of products PhF:MesF. Under Cu-catalyzed conditions, PhF is favored by ≥96:4, while the uncatalyzed reaction affords an approximately 20:80 ratio of PhF:MesF. As shown in Table 4, selectivity consistent with Cu catalysis was observed up to 1.25 equiv of KF. However, as predicted, significant erosion of selectivity was observed at 1.5 equiv of KF. Furthermore, with 2.0 or more equiv of KF, the observed selectivity was identical to that of the uncatalyzed reaction.
Table 4. Impact of KF Stoichiometry on Yield and Selectivity.
| KF (equiv) | yield (PhF) | yield (MesF) | PhF:MesF |
|---|---|---|---|
| 0.5 | 37% | <1% | >99:1 |
| 1.0 | 83% | 1% | 99:1 |
| 1.2 | 74% | 2% | 98:2 |
| 1.5 | 25% | 22% | 53:47 |
| 2.0 | 10% | 40% | 20:80 |
| 3.0 | 9% | 34% | 21:79 |
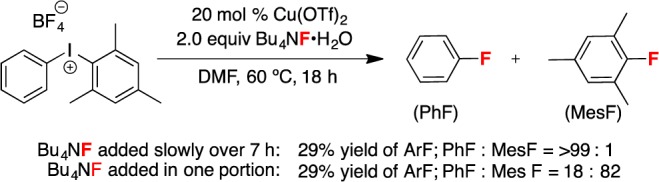
As discussed above, we hypothesize that the dramatic change in selectivity in the presence of ≥1.5 equiv of F– is due to a change in the resting state of the diaryliodonium salt from the cation [Mes(Ph)I]+ to the neutral species Mes(Ph)IF. Under these conditions, the uncatalyzed reaction is proposed to predominate over Cu catalysis. We hypothesized that this situation could be remedied by slow addition of F– over the course of the reaction. Indeed, as shown in Scheme 2, the slow addition of 2 equiv of a soluble fluoride source (TBAF·H2O)19 over 7 h resulted in >99:1 selectivity for PhF over MesF (29% yield).20 For comparison, 18:78 selectivity (and 29% yield)20 was observed when the identical reaction was conducted in a single pot. These experimental results are fully consistent with the calculations presented in Table 3.
Scheme 2. Slow Addition of TBAF·H2O.
The computations in Table 3 also illustrate the competitive nature of DMF and triflate as ligands for CuI (entries 4 and 5). This prompted us to examine the possibility of three different CuI complexes as reactants in the oxidation step: [CuF(OTf)]−, CuF(DMF), and [CuF2]− (Figure 2). Notably, in eq 2 of Figure 2, F– is shown as accessed from Mes(Ph)IF, as there is no free F– present. Importantly, these data only approximate the actual speciation in solution due to difficulties in computation of relative energies when charge separation is involved (Figure 2, eq 2) and the very high concentration of DMF present (12.9 M) relative to OTf– and F– (Figure 2, eq 1). Keeping these considerations in mind, pathways from all of these CuI species are potentially competitive.
Figure 2.

Relationship between potential fluorocopper(I) reactants. Energies ΔG (ΔH) are in kcal/mol.
We have also found that Ar–F bond-forming reductive elimination is facile from all of the arylcopper(III) species explored (vide infra). Thus, attempts to ascertain the identity of the catalytically relevant CuI intermediate(s) rely upon DFT and experimental examination of the oxidation step. The general strategy used for exploring oxidation mechanisms is discussed first, together with computation for the [CuF(OTf)]− system, as it is more complicated than the others and illustrates concepts displayed in the other systems. Gaussview structures for selected intermediates and transition states are shown to illustrate general features occurring in all cases.
General Strategy for Searching for Mechanisms in Reaction of [CuF(OTf)]− with [Mes(Ph)I]+
Computation of the oxidation mechanism was guided by our recent DFT study of aryl transfer from diaryliodonium reagents to palladium(II) centers.7b,21 In particular, we modeled transition structures based on the reaction of binuclear [Pd(C∼N)(μ-O2CMe)]2 (C∼N = 3-methyl-2-phenylpyridinyl) with [Ph2I]+. Here, an initial interaction of [Ph2I]+ with a donor atom of an acetate ligand to give T-shaped IIII22 is followed by a transition structure with a four-centered “Pd···(μ-Ph)···I···O” motif (A). This then leads to “Ph+” transfer to Pd (B) with release of PhI. Thus, our initial searches for transition structures in Cu-catalyzed fluorination were modeled on motifs containing “I···F” (C) or “I···O” interactions (D).
Scheme 3. Transition Structure (A) and Product (B) for the Arylation of PdII7b and Model Transition Structures Explored in the Current Study.

Computation for Ph group transfer from [Mes(Ph)I]+ to [CuF(OTf)]− resulted in two different transition structures. The first conforms to motif C (TS_IIa), containing a semibidentate triflate ligand. The second is based on motif F (TS_IIb) (Figure 3). Vibrational frequency calculations were employed to ascertain the structures of their precursors (IIa, IIb) and to establish the identity of the CuIII products (IIIa, IIIb). The precursor structures IIa and IIb have strong interactions between the reactants, illustrated by O–Cu–F angles of 127.6° and 110.5°, respectively. These results led us to a search for even earlier transition structures and accompanying precursors with weaker interactions. These were identified and supported by intrinsic reaction coordinate (IRC)/vibrational frequency calculations to be linked to IIa and IIb. Energies in kcal/mol [ΔG(ΔH)] are presented in Figure 3, although discussion is confined to considerations of ΔG.
Figure 3.

Energy profile for the reaction of [Mes(Ph)I]+ with [CuF(OTf)]−, illustrating oxidative transfer of “Ph+” to give the CuIII complexes IIIa and IIIb, followed by reductive elimination. The final CuI species is arbitrarily chosen as [Cu(OTf)2]−. Selected interatomic distances and angles are reported (∑(van der Waals radii): I + O = 3.50 Å; I + F = 3.45 Å; I + Cu = 3.38 Å).23 Energies ΔG (ΔH) are reported in units of kcal/mol. Gaussview structures are shown for the low-energy pathway.
The “I···F” interaction in Ia (2.580 Å) is ∼0.4 Å shorter than the “I···O” interaction in Ib. Ia lies below Ib and essentially at the same level as the reference point “[CuF(OTf)]− + [Mes(Ph)I]+”, noting uncertainty associated with ion separation in this step. Both pathways may be considered competitive, as they differ by only 1.2 kcal/mol in reaching transition structures TS_IIa and TS_IIb (with TS_IIb being slightly favored).
The “I···F” interaction is retained through TS_Ia to TS_IIa, but the “I···O” interaction ceases after IIb, being replaced by an “I···Cu” interaction in TS_IIb. The linear geometry at Cu in Ia and Ib becomes markedly nonlinear in subsequent species. The developing interaction between [Mes(Ph)I]+ and [CuF(OTf)]− is reflected in shorter “Cu···C” distances upon going from TS_Ia to TS_IIa and from intermediate IIb to TS_IIb. Consistent with the assignment of “Cu···C” distances as indicative of bonding in TS_Ia, IIa, TS_Ib, and IIb, the hydrogen or iodine atoms attached to these carbon atoms are displaced above the plane of the phenyl ring. The geometries at Cu for Ia, Ib through to IIa, IIb are linear (Ia, Ib) or approximately trigonal planar (IIa, IIb), as expected for CuI species.
Both TS_IIa and TS_IIb can be regarded as late transition states, in view of the very long “I···C(Mes)” distances of 2.769 (TS_IIa) and 2.561 Å (TS_IIb). These can be compared with 2.148 (Ia) and 2.139 Å (Ib) for the initial interaction of the two molecules. In addition, the “Cu···C(μ-Ph)” distances of 1.967 (TS_IIa) and 1.965 Å (TS_IIb) are only ∼0.05 Å longer than those in the CuIII products [1.911 (IIIa) and 1.924 Å (IIIb)]. Similarly, the “Cu···I” distance for TS_IIb (2.583 Å) is only 0.03 Å longer than that for coordinated iodomesitylene in the CuIII product IIIb. Structure TS_IIa is approximately square-planar at Cu, with the Cu center bearing a Ph ligand, a fluoride ligand, and a semibidentate triflate group (Cu–O = 2.061, Cu···O = 2.300 Å).
Reductive elimination is computed to be facile for both pathways, occurring directly from IIIa and IIIb with activation energies of only 4.4 and 4.5 kcal/mol, respectively, to give CuI products with an η2-bound fluorobenzene ligand.
We also explored mesityl transfer to gain insights into the experimentally observed selectivity for Ph group fluorination. We began by searching for transition structures for the reaction of [Mes(Ph)I]+ with [CuF(OTf)]− that contain a mesityl bridge in configurations TS_IIa (“I···F”) and TS_IIb (“I···Cu”). Placement of methyl groups in the 2- and 6-positions of the bridging aryl group, with the leaving group now as PhI, gave structures with unfavorable steric interactions in the copper coordination sphere. Thus, for an analogue of TS_IIa with a bridging Mes group, there is a methyl “C···O” interaction of 2.83 Å and a methyl “C···F” interaction of 2.36 Å, which are both much shorter than the corresponding sum of van der Waals radii (Me + O = 3.52 Å; Me + F = 3.47 Å).23,24 Optimization of this structure led to the transition structure TS_IIa_Mes (Figure 4). This is analogous to TS_IIa, with approximate square-planar coordination at Cu, but with a different orientation of the bridging aryl group. The orientation of the Mes group is altered in a manner that results in the 2- and 6-positions being further removed from coordinated oxygen and fluorine atoms. Also, the “Cu···C···I” angle for the bridging aryl group is increased from 74.8° in TS_IIa to 87.9° in TS_IIa_Mes. The overall sequence leading to a CuIII species is very similar to Ia → IIIa. However, analogues of TS_Ia and IIa display the Mes group interacting with Cu in an η1 manner, presumably due to unfavorable steric interactions with the 2,6-methyl groups. An additional 2.6 kcal/mol is required to access transition structure TS_IIa_Mes compared with the Ph-bridged analogue. Furthermore, an additional 3.8 kcal/mol is required relative to TS_IIb (see below and Supporting Information for full details of this sequence). This is consistent with the high selectivity observed experimentally.
Figure 4.
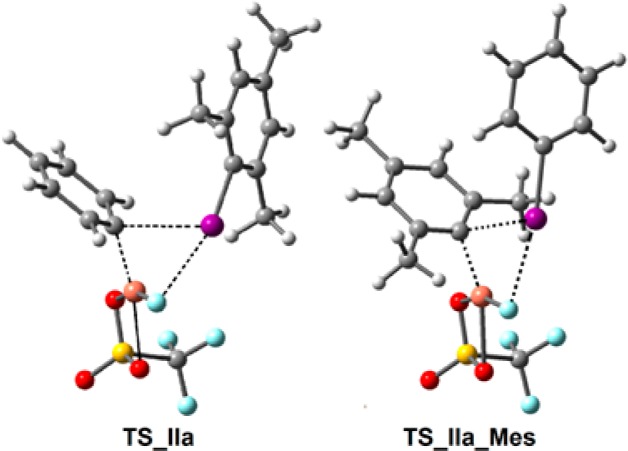
Optimized structures for TS_IIa with a bridging Ph group and TS_IIa_Mes with a bridging Mes group, oriented to illustrate the difference in orientation of bridging aryl groups and in “Cu···C···I” angles.
Modeling of a bridging Mes analogue of TS_IIb containing an “I···Cu” interaction prior to geometry optimization revealed a short methyl “C···O” interaction (2.47 Å). Attempted computation for this transition structure led smoothly away from this motif to TS_IIa_Mes, which contains an “I···F” interaction (vide supra).
Reaction of CuF(DMF) with [Mes(Ph)I]+
Following an analogous approach to that used for [Cu(F)(OTf)]−, a cationic transition structure TS_IIa_DMF directly analogous to the neutral triflate species TS_IIa (with an “I···F” interaction) was identified (Figure 5). Here, DMF is present as a monodentate O-donor ligand in place of the semibidentate triflate in TS_IIa. The PhCuIII product IIIa_DMF then undergoes facile reductive elimination (ΔG⧧ = 3.6 kcal/mol).25 Interestingly, a corresponding transition structure for Mes transfer at CuF(DMF), explored in a similar manner to that for [CuF(OTf)]−, could not be identified.
Figure 5.
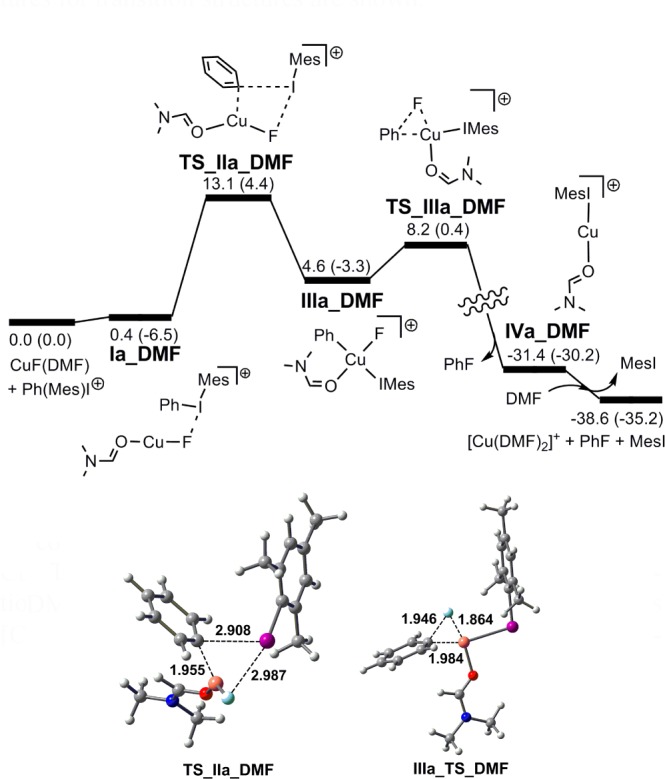
Energy profile for the reaction of [Mes(Ph)I]+ with CuF(DMF), illustrating oxidative transfer of “Ph+” to give the CuIII complex IIIa_DMF and subsequent reductive elimination. The final CuI species is arbitrarily chosen as [Cu(DMF)2]+. Energies ΔG (ΔH) are in kcal/mol. Gaussview pictures for transition structures are shown.
Reaction of [CuF2]− with [Mes(Ph)I]+
An analogous approach was used to elucidate the reaction pathways for [CuF2]−, and the results are shown in Figure 6. Ph and Mes transfer pathways were identified, and both proceed via “I···F” interactions analogous to those found for [CuF(OTf)]− in Figure 3. For mesityl transfer (red pathway), a T-shaped CuIII intermediate (IIIa′-F_Mes) is formed upon loss of PhI. The addition of free triflate to this structure would then enable formation of a transition structure for reductive elimination from IIIa_F_Mes.
Figure 6.

Energy profile for the reaction of [Mes(Ph)I]+ with [CuF2]−, illustrating oxidative transfer to give ArCuIII species IIIa_F and IIIa′_F_Mes, and subsequent reductive elimination. The final CuI species is arbitrarily chosen as [CuF(OTf)]−. Energies ΔG (ΔH) are in kcal/mol. Gaussview structures for “Ph+” transfer are shown.
Computation for the Oxidation Step with Variation of the Aryl Group for the Three CuI Reactants
We next explored a series of different [Mes(Ar)I]+ reagents in order to compare the calculated selectivity for oxidative transfer of Ar versus Mes to CuI to the experimentally observed selectivity as a function of Ar group substitution. Table 5 shows the energy requirement to achieve transition structures from “[CuF(OTf)]− + [Mes(Ar)I]+”, “CuF(DMF) + [Mes(Ar)I]+2”, “[CuF2]− + [Mes(Ar)I]+”, or the relevant precursor adducts if these are at lower energy than the reactant pair. The experimental ratio of products (ArF:MesF) is shown, together with Cu-free ratios (columns 2 and 3). Note that computation for CuF(DMF) is shown, even though we cannot detect a pathway for mesityl transfer in this system.
Table 5. Energy Values (kcal/mol) Required to Access Transition Structures for ArF and MesF Coupling from the Reagent Pairs (“[CuF(OTf)]− + [Mes(Ar)I]+” or “CuF(DMF) + [Mes(Ar)I]+” or “[CuF2]− + [Mes(Ar)I]+”) or Their Respective Precursor Adducts When These Are at Lower Energy.
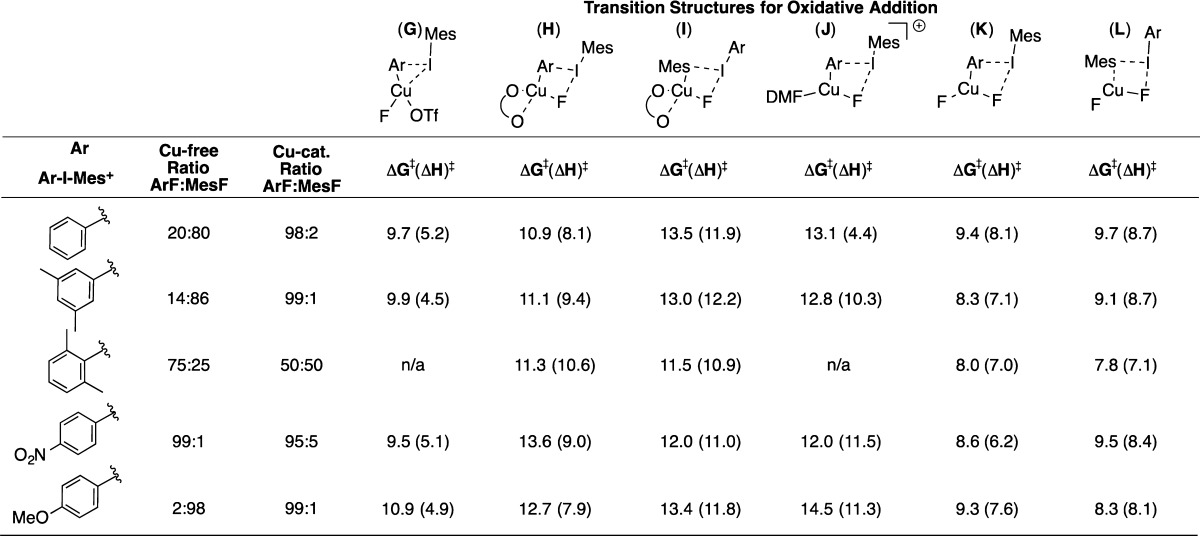
For [CuF(OTf)]− (columns G–I), the activation energies in column G are consistently lower (by 1–3 kcal/mol) than those in columns H and I. Also supportive of transition structure G as a favored pathway, these transition structures exhibit “I···C” distances for the departing aryl group (2.764–2.893 Å) that are shorter than those for H and I (2.936–3.097 Å), suggesting that G is an earlier transition state than H/I. The data are consistent with the observed selectivities for coupling products, where Ar transfer (G) is preferred over Mes transfer (I). For competition between 2,6-Me2C6H3 and Mes transfer, essentially identical energies are obtained, reflecting the similar steric and electronic properties of these groups. Notably, as discussed above, a triangular transition structure analogous to G does not appear to be feasible for 2,6-substituted arenes.
The [CuF2]− system displays the same trends as for [CuF(OTf)]−, showing a competitive process for [Mes(2,6-Me2C6H3)I]+, and favoring aryl transfer for other [Mes(Ar)I]+ reagents. The p-MeOC6H4 derivative is the lone exception. In this case, Mes transfer is computed to be favored by 1 kcal/mol, which is inconsistent with the experimental selectivity. Although the energy difference is small, it is notable that computation for the [CuF2]− pathway shows much smaller contributions for the steric impact of 2,6-Me2 substitution across the board. For instance, ΔΔG⧧ for XC6H4 transfer versus Mes transfer is +0.3 and +0.8 kcal/mol for the first two entries in Table 3 with [CuF2]− (compare columns K and L). In contrast, the corresponding ΔΔG⧧ values in the [CuF(OTf)]− pathways are 3.8 and 3.1 kcal/mol (compare columns G and I).
For CuF(DMF) (column J), activation energies for all transfer reactions are higher than the lowest energy pathways for [CuF(OTf)]− and [CuF2]−, noting the uncertainties in comparing energy barriers between the three systems.
All of the calculated activation energies in Table 5 (ΔG⧧ = 7.8–14.5 kcal/mol) are lower than those calculated for Ar–F and Mes–F coupling under Cu-free conditions (15.3–22.2 kcal/mol, vide infra). Thus, it appears that a Cu-catalyzed route might also account for formation of the minor product (MesF) under catalysis conditions, rather than this being formed as a product of Mes(Ph)IF decomposition. It is particularly notable that low activation energies are computed for both 2,6-Me2C6H4 and mesityl transfer in the [CuF(OTf)]− and [CuF2]− systems.
Comparison of Possible Oxidation Pathways
Similar reaction manifolds are obtained for all three CuI catalysts, and Figure 7 (related to Figure 2) provides an overview in which the reference energy is now assigned to the adduct formed between [CuF2]− and [Mes(Ph)I]+. This is the only pathway for which the adduct is significantly lower than the reactant pair. Thus, this adduct could be considered as a resting state. However, as noted earlier, caution is required in interpretation because of both the presence of DMF at 12.9 M [which may lead to the formation of CuF(DMF)] and also the moderate reliability of DFT when ion separation is involved.
Figure 7.
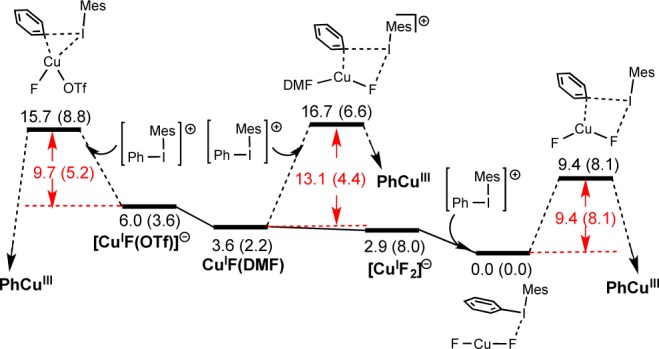
Overview of reaction profile to afford PhCuIII species. The reference energy is taken as the adduct CuF2·Mes(Ph)I. Energies ΔG (ΔH) are in kcal/mol.
Experiments designed to test the viability of CuF226 as a precatalyst show that triflate is not necessary for Cu-promoted coupling. Specifically, the reaction of 1 equiv of CuF2 with [Ph(Mes)I]+ at 60 °C in DMF afforded a >99:1 ratio of PhF to MesF in quantitative yield.27 Although CuF2 has low solubility in DMF, it is sufficiently soluble to give a relatively weak positive color test for CuI. Under these conditions, in the absence of triflate, the reaction likely occurs via either CuF(DMF) or [CuF2]−.
Overall, the similarities between the reaction barriers for the three CuI species examined herein suggest that all three pathways could be competitive. The most favorable one is likely to depend on the reaction conditions and the speciation of Cu in solution.
Copper-Free Decomposition of Mes(Ar)IF
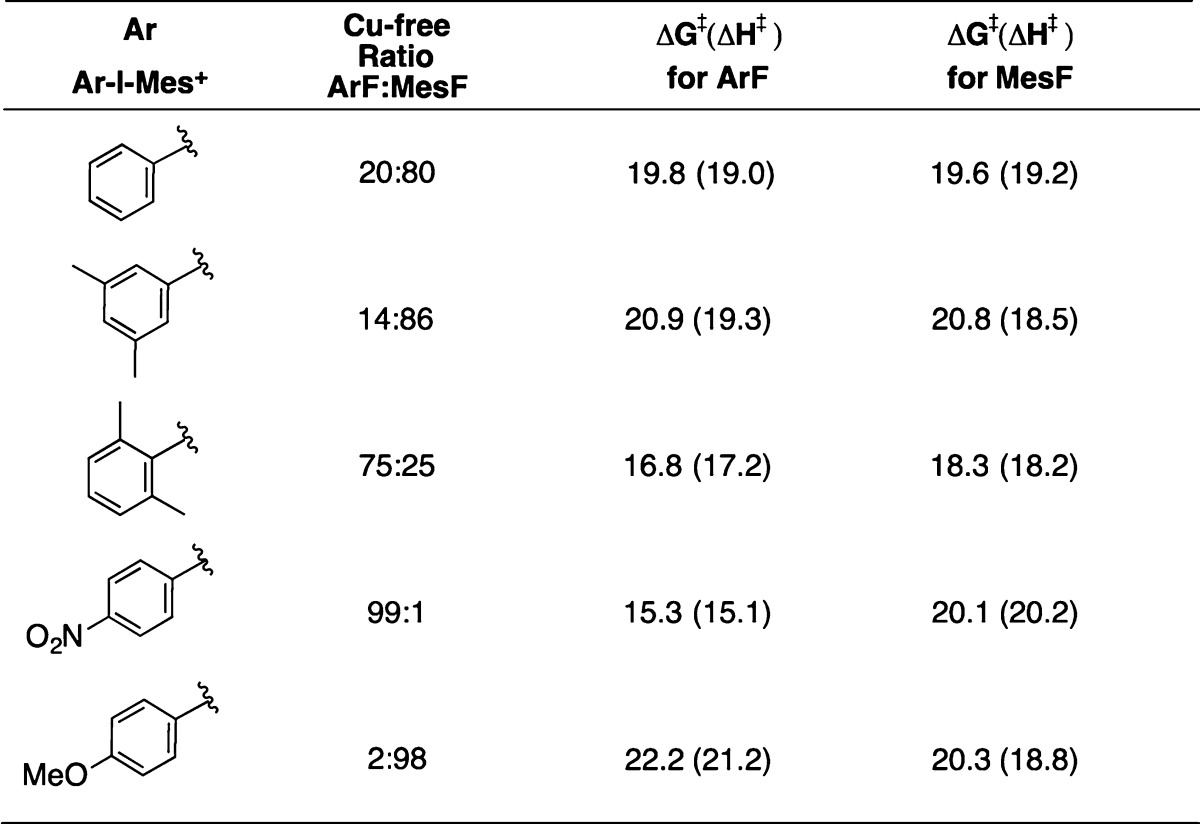
Experimental evidence for the role of copper catalysis rests mainly on the observation of faster rates and very different product ratios of ArF:MesF upon the addition of Cu. To explore the difference in selectivity for ArF and MesF, we also examined C–F bond formation from Mes(Ar)IF under uncatalyzed conditions. The Mes(Ar)IF reagents examined are those discussed above for Cu catalysis. Computation for Mes(Ar)IF followed the approach documented by De Lüthli28 and Olofsson6a for related unsymmetrical diaryliodine(III) species. An example is shown in Figure 8, illustrating the transition structure TS_isom for isomerization of T-shaped isomers of Mes(p-NO2C6H4)IF and the transition structures for competing aryl–fluoride coupling pathways.
Figure 8.
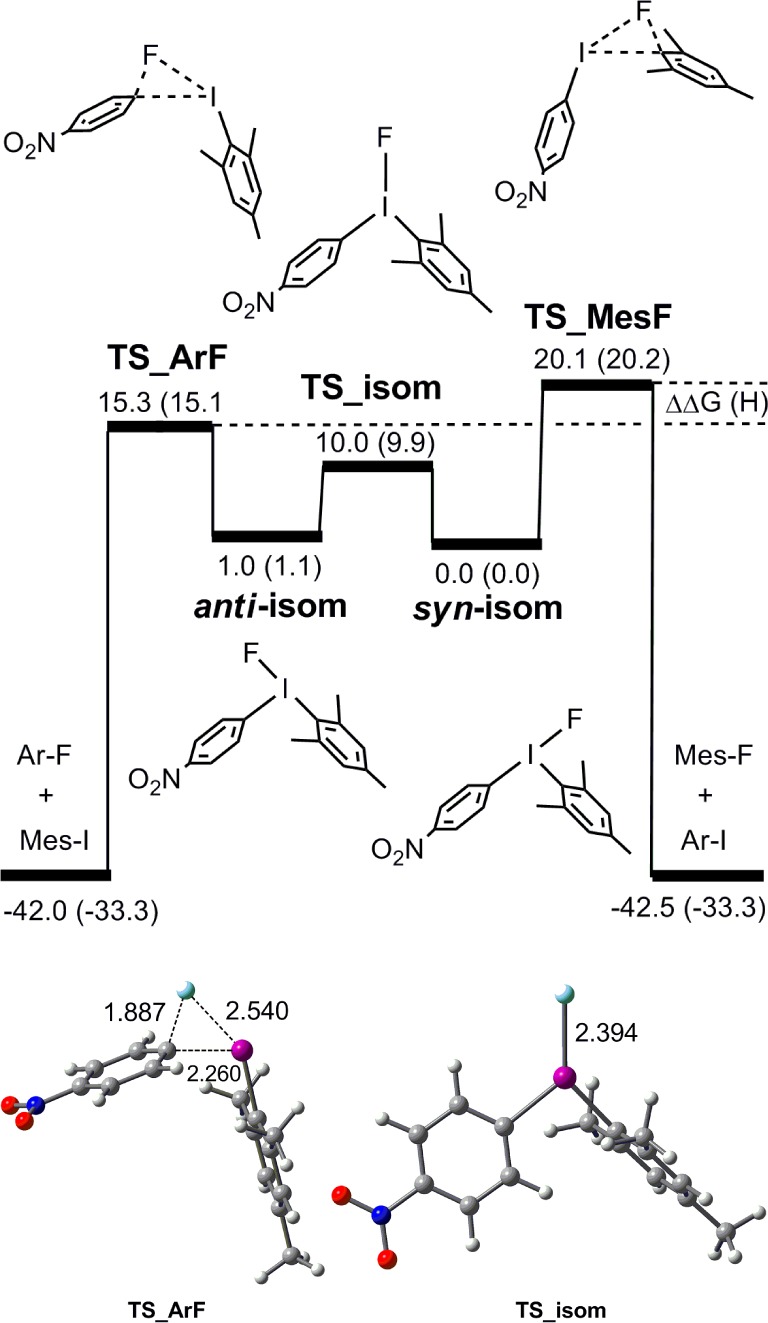
Energy profile and representative Gaussview structures for interconversion of isomers of Mes(p-NO2C6H4)IF. Energies ΔG (ΔH) in kcal/mol are referenced to the lower energy isomer (syn-isom).
Computational results are summarized in Table 6, together with the ratios of products obtained experimentally for Cu-free fluorination. Curtin–Hammett conditions, in which the energies required for isomerization are significantly lower than for reductive elimination, were found in all cases. This allows the reference point for energy to be chosen as that of the more stable isomer. This was calculated to be the syn-isomer in all cases except for Mes(p-MeOC6H4)IF. As noted for related systems,28 the differences in activation energies (ΔΔG⧧ and ΔΔH⧧) for competing transition structures are relatively small, but are broadly consistent with the experimental results. For example, for Mes(3,5-Me2C6H4)IF, the calculated ΔΔH⧧ value (0.8 kcal/mol) is consistent with the experimental product distribution. However, ΔΔG⧧ computes as only 0.1 kcal/mol in this case. Furthermore, the computed ΔΔH⧧ and ΔΔG⧧ values (±0.2 kcal/mol) for Mes(Ph)IF do not discriminate between competing pathways. The lack of sensitivity of computation for the latter two systems does not appear to be a result of the functional, basis set, or solvent applied here. For example, a calculation under our computation conditions for a representative example from the Lüthli study,28 (p-MeC6H4)(Ph)IBr, gives a ΔΔG⧧ of 0.5 kcal/mol, favoring p-MeC6H4Br as product. This compares well with the reported value of 0.62 kcal/mol (B3LYP, aug-cc-pVDZ, gas phase).28
Table 6. Computed Barriers Compared to Observed Selectivities for ArF and MesF Coupling upon Uncatalyzed Decomposition of Mes(Ar)IF.
Reductive Elimination of Aryl Fluorides from ArCuIII Species
Reductive elimination of PhF and MesF from CuIII is computed to involve simple triangular motifs in transition structures with planar four-coordinate geometries at copper (Figures 3, 5, and 6). The activation energies for these processes (3.6–7.7 kcal/mol) are substantially lower than for the oxidation processes in all models examined. As part of a related catalytic process for Ar–F bond formation, Ribas and co-workers have computed the activation parameters for C–F coupling at a five-coordinate CuIII center containing a tetradentate macrocyclic ligand, [CuIIIF(L-C,N,N′,N″)]+.29 They obtained a ΔG⧧ of 16.2 kcal/mol, which is significantly higher than in the present systems. This may be the result of either (i) the extra stability provided by the polydentate ligand in the Ribas system and/or (ii) the differences between reductive elimination from four- versus five-coordinate CuIII centers.
Conclusions
Both experimental and DFT results support the feasibility of a CuI/CuIII catalytic cycle for the reaction of unsymmetrical diaryliodonium cations with potassium fluoride in DMF using CuII(OTf)2 as the precatalyst. Several possible reaction pathways were found, and they exhibit similarities in the manner in which CuI species interact with [Mes(Ar)I]+ cations. In all cases, initial CuI·IIII adduct formation involves an interaction between a donor atom of a ligand on Cu and the iodine(III) center. This is followed by interaction of the Ar group with copper, leading, eventually, to transition structures for rate-limiting Ar transfer (“Cu···(μ-η1-Ar)···I” with an additional “I···Cu” or “I···F” interaction) to form cis-Ar(F)CuIII. The CuIII species then undergo facile ArF bond-forming reductive elimination. The evidence supporting these proposed pathways is as follows:
-
(1)
CuI is detected experimentally when CuII(OTf)2 is added to DMF, but not in solvents that do not support catalysis.
-
(2)
Experimental and DFT studies suggest that the reactive IIII species are cations of general structure [Mes(Ph)I]+.
-
(3)
DFT calculations show four low-energy pathways for oxidative transfer of “Ar+” from [Mes(Ar)I]+ to CuI, forming cis-Ar(F)CuIII species. These pathways proceed from [CuF(OTf)]− (2 pathways), CuF(DMF) (1 pathway), and [CuF2]− (1 pathway). All have activation parameters that are lower than those for decomposition of Mes(Ar)IF in the absence of Cu.
-
(4)
DFT studies of the highest energy pathway [from CuF(DMF)] do not show a viable transition structure for mesityl group transfer. However, pathways commencing with [CuF(OTf)]− and [CuF2]− enabled comparisons of the relative energies of Ar versus Mes transfer from [Mes(Ar)I]+. For the five aryl groups examined, agreement with experiment is found in all cases commencing from [CuF(OTf)]− and in four commencing from [CuF2]−. Notably, with [CuF2]−, smaller ΔΔG⧧ values for Ar versus Mes transfer are observed across the board, suggesting that this single anomaly may not be significant.
Overall, our experimental and DFT studies of this system illustrate flexibility in the ligand environment that will support a CuI/III catalytic cycle for ArF coupling. As such, these studies suggest that there is considerable latitude for the development of CuI/III-catalyzed aryl–X bond-forming reactions beyond the immediate successful protocol for Cu(OTf)2-catalyzed fluorination of diaryliodonium salts.3,8,30
Acknowledgments
We thank the Australian Research Council and the U.S. National Institutes of Health (GM 073836) for financial support, and the Australian National Computational Infrastructure and the University of Tasmania for computing resources.
Supporting Information Available
Complete ref (16), computation for the reaction of [CuF(OTf)]− with [Mes(Ar)I]+ for mesityl transfer, energy parameters, Cartesian coordinates and Gaussview diagrams of all optimized structures, and full details of all experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For reviews on diaryliodonium salts, see:; a Yusubov M. S.; Maskaev A. V.; Zhdankin V. V. Arkivov 2011, 2011, 370. [Google Scholar]; b Merritt E. A.; Olofsson B. Angew. Chem., Int. Ed. 2009, 48, 9052. [DOI] [PubMed] [Google Scholar]; c Canty A. J.; Rodemann T.; Ryan J. H. Adv. Organomet. Chem. 2008, 55, 279. [Google Scholar]; d Zhdankin V. V.; Stang P. J. Chem. Rev. 2008, 108, 5299. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Deprez N. R.; Sanford M. S. Inorg. Chem. 2007, 46, 1924. [DOI] [PubMed] [Google Scholar]; f Zhdankin V. V. Chem. Rev. 2002, 102, 2523. [DOI] [PubMed] [Google Scholar]
- For examples of metal-free nucleophilic reactions with diaryliodonium salts, see ref (1) and:; a Wagner A. M.; Sanford M. S. J. Org. Chem. 2014, 79, 2263. [DOI] [PubMed] [Google Scholar]; b Umierski N.; Manolikakes G. Org. Lett. 2013, 15, 188. [DOI] [PubMed] [Google Scholar]; c Jalalian N.; Ishikawa E. E.; Silva L. F.; Olofsson B. Org. Lett. 2011, 13, 1552. [DOI] [PubMed] [Google Scholar]; d Laudge K. P.; Jang K. S.; Lee S. Y.; Chi D. Y. J. Org. Chem. 2012, 77, 5705. [DOI] [PubMed] [Google Scholar]; e Grushin V. V.; Kantor M. M.; Tolstaya T. P.; Scherbina T. M. Izv. Akad. Nauk SSSR, Ser. Khim. 1984, 2338. [Google Scholar]
- For Cu-catalyzed C-heteroatom coupling reactions with Ar2I+, see:; a Sokolovs I.; Lubriks D.; Suna E. J. Am. Chem. Soc. 2014, 136, 6920. [DOI] [PubMed] [Google Scholar]; b Fañanás-Mastral M.; Feringa B. L. J. Am. Chem. Soc. 2014, 136, 9894. [DOI] [PubMed] [Google Scholar]; c Lv T.; Wang Z.; You J.; Lan J.; Gao G. J. Org. Chem. 2013, 78, 5723. [DOI] [PubMed] [Google Scholar]; d Cullen S. C.; Shekhar S.; Nere N. K. J. Org. Chem. 2013, 78, 12194. [DOI] [PubMed] [Google Scholar]; e Xu J.; Zhang P.; Gao Y.; Chen Y.; Tang G.; Zhao Y. J. Org. Chem. 2013, 78, 8176. [DOI] [PubMed] [Google Scholar]; f Vaddula B.; Leazer J.; Varma R. S. Adv. Syn. Catal. 2012, 354, 986. [Google Scholar]
- a Ichiishi N.; Canty A. J.; Yates B. F.; Sanford M. S. Org. Lett. 2013, 15, 5134. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ichiishi N.; Brooks A. F.; Topczewski J. J.; Rodnick M. E.; Sanford M. S.; Scott P. J. H. Org. Lett. 2014, 16, 3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cu-catalyzed C–C coupling reactions with Ar2I+:; a Baralle A.; Fensterbank L.; Goddard J.-P.; Ollivier C. Chem.—Eur. J. 2013, 19, 10809. [DOI] [PubMed] [Google Scholar]; b Kieffer M. E.; Chuang K. V.; Reisman S. E. J. Am. Chem. Soc. 2013, 135, 5557. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Collins B. S. L.; Suero M. G.; Gaunt M. J. Angew. Chem., Int. Ed. 2013, 52, 5799. [DOI] [PubMed] [Google Scholar]; d Phipps R. J.; McMurray L.; Ritter S.; Duong H. A.; Gaunt M. J. J. Am. Chem. Soc. 2012, 134, 10773. [DOI] [PubMed] [Google Scholar]; e Zhu S.; MacMillan D. W. C. J. Am. Chem. Soc. 2012, 134, 10815. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Duong H. A.; Gilligan R. E.; Cooke M. L.; Phipps R. J.; Gaunt M. J. Angew. Chem., Int. Ed. 2011, 50, 463. [DOI] [PubMed] [Google Scholar]; g Phipps R. J.; Gaunt M. J. Science 2009, 323, 1593. [DOI] [PubMed] [Google Scholar]
- a Malmgren J.; Santoro S.; Jalalian N.; Himo F.; Olofsson B. Chem.—Eur. J. 2013, 19, 10334. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Grushin V. V. Acc. Chem. Res. 1992, 25, 529. [DOI] [PubMed] [Google Scholar]; c Grushin V. V.; Demkina I. I.; Tolstaya T. P. J. Chem. Soc., Perkin Trans. 2 1992, 505. [Google Scholar]; d Chun J. H.; Lu S.; Lee Y. S.; Pike V. W. J. Org. Chem. 2010, 75, 3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DFT calculations on Cu-catalyzed reactions with diaryliodonium salts:; a Chen B.; Hou X.-L.; Li Y.-X.; Wu Y.-D. J. Am. Chem. Soc. 2011, 133, 7668. [DOI] [PubMed] [Google Scholar]; DFT calculations on Pd-catalyzed reactions with diaryliodonium salts:; b Canty A. J.; Ariafard A.; Sanford M. S.; Yates B. F. Organometallics 2013, 32, 544. [Google Scholar]
- a Zhang H.; Yao B.; Zhao L.; Wang D- X.; Xu B- Q.; Wang M.-X. J. Am. Chem. Soc. 2014, 136, 6326. [DOI] [PubMed] [Google Scholar]; b Casitas A.; Ribas X. Chem. Sci. 2013, 4, 2301. [Google Scholar]; c Fier P. S.; Hartwig J. F. J. Am. Chem. Soc. 2012, 134, 10795. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Casitas A.; King A. E.; Parella T.; Costas M.; Stahl S. S.; Ribas X. Chem. Sci. 2010, 1, 326. [Google Scholar]; e Yao B.; Wang D. X.; Huang Z. T.; Wang M. X. Chem. Commun. 2009, 2899. [DOI] [PubMed] [Google Scholar]
- Goodgame D. M. L.; Goodgame M.; Canham G. W. R. Nature 1969, 222, 866. [Google Scholar]
- Muzart J. Tetrahedron 2009, 65, 8313. [Google Scholar]
- For examples of the reduction of CuII with DMF, see:; a Malkhasian A. Y. S.; Finch M. E.; Nikolovski B.; Menon A.; Kucera B. E.; Chavez F. A. Inorg. Chem. 2007, 46, 2950. [DOI] [PubMed] [Google Scholar]; b Teo J. J.; Chang Y.; Zeng H. C. Langmuir 2006, 22, 7369. [DOI] [PubMed] [Google Scholar]
- Lockhart T. P. J. Am. Chem. Soc. 1983, 105, 1940. [Google Scholar]
- Ali B. F.; Al-Sou’od K.; Al-Jaar N.; Nassar A.; Zaghal M. H.; Judeh Z.; Al-Far R.; Al-Refai M.; Ibrahim M.; Mansi K.; Al-Obaidi K. H. J. Coord. Chem. 2006, 59, 229. [Google Scholar]
- We have also tested running the fluorination process in a cosolvent of EtOAc/DMF (0.1 M in PhIMes+, 4:1). This afforded 86% overall yield (98:2 selectivity).
- A possible alternative mechanism would be a SET pathway. We have not conducted an extensive investigation of such processes.
- Gaussian 09 was used at the BP86 level for calculations with N,N-dimethylformamide as solvent and utilizing the quadruple-ξ valence polarized def2-QZVP basis set on Cu and iodine along with the corresponding ECP and the 6-311+G(2d,p) basis set on other atoms. See Supporting Information for full details.
- For literature examples of LnCu(DMF) complexes, see:; a Ding S.; Jiao N. J. Am. Chem. Soc. 2011, 133, 12374. [DOI] [PubMed] [Google Scholar]; b Ishiguro S.-I.; Jeliazkova B. G.; Ohtaki H. J. Solution Chem. 1987, 16, 1. [Google Scholar]
- The crystal structure of CuX(PPh3)(DMF) (X = Cl; Br) was previously reported:Scharfe S.; Fässler T. F. Z. Naturforsch. 2012, 676, 564. [Google Scholar]
- TBAF·H2O was selected because it is fully soluble in DMF, rendering it straightforward to add using a syringe pump.
- Significant quantities of biphenyl and phenol were detected as byproducts, which accounts for the modest yield. This is likely due, at least in part, to the water in the TBAF·H2O.
- For a related study, see:Szabó K. J. J. Mol. Catal. A: Chem. 2010, 324, 56. [Google Scholar]
- Landrum G. A.; Goldberg N.; Hoffmann R.; Minyaev R. M. New J. Chem. 1998, 883. [Google Scholar]
- Bondi A. J. Phys. Chem. 1964, 68, 441. [Google Scholar]
- Pauling L.The Nature of the Chemical Bond; Cornell University Press: Ithaca, NY, 1960; p 262. [Google Scholar]
- For Cu(F)(DMF) as the reactant, an additional pathway via an analogue of TS_IIb (with an “I···Cu” interaction) was found that yields PhCuIII(F)(DMF)(IMes). However, we deem this pathway less likely, as the PhCuIII intermediate has trans-disposed phenyl and fluoro ligands and thus requires isomerization to allow reductive elimination. This complex may undergo reversible oxidation, a feature that has been proposed in other CuI/III-catalyzed reactions (ref (8)).
- CuF2-mediated aromatic fluorination was reported previously. Grushin V. U.S. Patent 7,202,388, 2007.
- The use of substoichiometric CuF2 in combination with 0.5–1.1 equiv of KF afforded selectivity favoring MesF. Ongoing investigations are probing the origins of this result.
- De Magalhães H. P.; Lüthi H. P.; Togni A. Org. Lett. 2012, 14, 3830. [DOI] [PubMed] [Google Scholar]
- Casitas A.; Canta M.; Solà M.; Costas M.; Ribas X. J. Am. Chem. Soc. 2011, 133, 19386. [DOI] [PubMed] [Google Scholar]
- For recent reports of related reactions that likely proceed via similar mechanisms, see:; a Ye Y.; Schimler S.; Hanley P.; Sanford M. S. J. Am. Chem. Soc. 2013, 135, 16292. [DOI] [PubMed] [Google Scholar]; b Ye Y.; Sanford M. S. J. Am. Chem. Soc. 2013, 135, 4648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.