

Abstract
A computational model of cancer progression is used to study how mutations in genes that control tumor initiation and progression accumulate in populations. The model assumes that cancer occurs only after a cell lineage has progressed through a series of stages. The greater the number of stages, the more strongly the individual is protected against cancer. It is shown that an extra stage initially improves the survival of individuals by decreasing mortality from cancer. However, the additional buffering by an extra stage reduces the impact of any single hereditary mutation and therefore allows the accumulation of more nonlethal mutations in the population. Extra stages thereby lead to the evolution of partially decreased cancer mortality and significantly increased genetic predisposition to disease in the population as a whole. In general, the model illustrates how all robust control networks allow the accumulation of deleterious mutations. An increase in the number of buffering components leads to significant mutational decay in the protection provided by each buffering component and increased genetic predisposition to disease. An extra buffering component's net contribution to survival and reproduction is often small.
Cancer develops after somatic mutations overcome the multiple checks and balances on cellular proliferation (1–3). Those normal checks and balances define a robust genetic control system that protects against perturbations. For example, DNA damage enhances expression of p53, a transcription factor that in turn modulates the expression of many other genes (4). If the DNA damage is moderate, p53 causes the cellular system to slow the cell cycle, repair the damage, and then proceed with replication. If the DNA damage is severe, p53 triggers an apoptotic pathway that leads to cell suicide.
p53 functions mainly to protect against damage that arises from the environment during the lifetime of the individual. However, a system that protects against the environment may also buffer against the negative effects of inherited mutations (5). For example, mutations that slow DNA repair or allow greater DNA damage may have less effect because p53 compensates by adjusting the repair process and cell cycle progression. Thus, the buffering effects of p53 can reduce the negative consequences of some inherited mutations, slowing the rate at which natural selection removes those mutations from the population.
It has been noted many times that buffering traits allow the accumulation of mutations (5–8). In this paper, I address two issues. First, I study the process of buffering and mutation accumulation in a computational model of cancer. This study leads to a better understanding of genetic predisposition to cancer and to predictions about the relative levels of genetic predisposition in different cancers.
Second, I study the consequences of different amounts of buffering against environmental perturbation. I use a multistage model of cancer progression (9), in which cancer occurs only after a cellular lineage has passed through a series of stages. The number of stages measures the amount of buffering provided by various checks and balances because cancer arises only after a sufficient number of the checks and balances have been bypassed.
I show that an increase in the number of stages causes a small increase in fitness, a large mutational decay in the performance of each stage, and an increase in the total fraction of cancer risk caused by inherited genetic variation. In Conclusions, I consider how this particular model of cancer progression provides hypotheses about other robust genetic control systems.
The Model
Cancer Progression Within Each Individual. I use the classic Armitage and Doll (9) model of cancer progression. In this model, cancer occurs only after n rate-limiting steps have been passed. Initially, there are x0(0) cell lineages in a tissue. Each cell lineage begins life having passed zero of the n steps. A cell lineage progresses through the first step at rate u0; a cell lineage passes the second step at a rate u1; and so on. These assumptions lead to a simple dynamical system for the progression of cell lineages toward cancer,
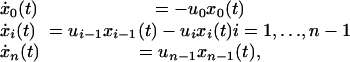 |
where the dots are the derivatives with respect to time, the ui are the constant rates of transition within a particular individual and the xi are the number of cell lineages at age t that have passed i steps. Age is measured in years. I use “cell lineages” rather than “cells” because this model of cancer progression depends on the accumulation of mutations over time in a genome passed down from parent cell to daughter cells; that is, the mutations accumulate in lineages over time rather than to particular cells at a fixed point in time (see ref. 10 for further discussion of this model).
An individual develops cancer if any single cellular lineage passes all n steps. Thus, we can interpret xn(t) as the cumulative probability at age t that an individual has developed cancer. If xn(τ) = 1, then the individual has cancer by age τ with probability one, and ẋn(t) = 0 for t > τ. In other words, once an individual has died of cancer with probability one by age τ, the further rate of change in mortality is zero.
Although a multistage model of progression is generally accepted as the best description of cancer progression (11), the exact meaning of the stages and the transition rates between stages remains poorly understood for most cancers. In colon cancer, there is a tendency for certain morphological stages of tumor formation to follow one after the other (12). Each stage may be associated with particular somatic mutations, or, put another way, the transition rates between stages may be determined in part by the rates of somatic mutations to particular genes.
There are not enough data to argue too finely about the meaning of stages and transitions. For my purposes, I am analyzing the population genetic consequences of a multistage model of progression with regard to the maintenance of inherited deleterious mutations balanced by natural selection.
Age-Specific Fitness Consequences of Cancer. To study how natural selection affects the frequencies of inherited mutations, I need a measure of the fitness consequences of those mutations. I do not need a highly realistic set of assumptions to link genetic variation to cancer mortality because my goal is limited to analyzing how the change in the number of stages or barriers in cancer progression affects genetic variability. At present, there are not enough data to define how all types of mutations influence mortality. In the absence of such data, detailed assumptions are more likely to be wrong than helpful.
Death at different ages has different consequences for fitness. We need an expression, f(t), for the force of selection, the fraction of total fitness lost if an individual dies of cancer at age t. Assume that first reproduction occurs at age F = 15 and maximum age occurs at T = 80. Then, f(t) = 1 for t ≤ F and f(t) = 0 for t ≥ T. Define z = (t – F)/(T – F) as the fraction of reproductive lifespan that has passed between first reproduction, F, and certain death, T. Then, for F ≤ t ≤ T, I set the force of selection at age t to the function f(t) = 1 – 6z2 + 8z3 – 3z4. The curve is shown in Fig. 1. I derived the formula for this shape from a general family of curves based on the beta distribution (see Fig. 1 legend). It is possible to give a family of curves controlled by a shape parameter, but this single curve is sufficient for this particular study.
Fig. 1.

The force of selection at different ages. A general family of curves can be derived by using a form of the beta cumulative distribution function to give different shapes. I used f(t) = 1 – 0.5[(a + 1)(a + 2)za – 2a(a + 2)za + 1 + a(a + 1)za+2], with f(t) = 0 for t < F or t > T, and z = (t – F)/(T – F). The equation for f(t) in the text and the curve illustrated here are obtained with a = 2. The curve shifts to the right as a rises, increasing the force of selection and causing natural selection to push cancer incidence to later ages.
Loss in fitness caused by cancer is the force of selection averaged over the probabilities for death at different ages. This loss is
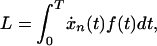 |
and fitness is defined as 1 – L. Here, L can be interpreted as follows. The loss in fitness for death at age t is f(t), and the relative probability of death at age t is ẋn(t), so the integral sums up the loss at each age weighted by the relative chance of death at each age.
Genetic Basis of Transition Rates Between Stages. The rates of transition between stages, ui, determine the dynamics of progression within each individual. To study how genetic variation may cause differences between individuals in progression dynamics, I assume that several genes affect each transition rate.
The logarithm of each rate varies over the range [–b,1], where log10(ui) = –b is the slowest rate, and therefore provides the lowest cancer incidence and the highest fitness. When a transition is at its highest rate, log10(ui) = 1, the transition happens so quickly that it is no longer a rate-limiting step in progression.
Each of the n transitions is affected by a single major diploid locus. This locus suffers recessive loss-of-function mutations, acting as a tumor suppressor gene. If both alleles at the major locus for the ith transition have loss-of-function mutations, then log10(ui) = 1. Typically, if an individual has a single transition at this high rate, that individual would die of cancer at a relatively early age. Alternatively, I could have assumed dominant oncogenic mutations at this single major locus, such that if either allele was mutated to an oncogene, then the transition for the associated step would effectively be passed at birth. Once again, such an individual with the loss of a protective step would tend to die of cancer at a relatively early age. The difference between recessive tumor suppressor loci and dominant oncogenic loci has little effect on this model because dominant and recessive loci would have roughly the same net effect on mortality under the combination of mutation and selection.
Each transition is also affected by k minor diploid loci; thus, there are 2k minor alleles. Each allele has an integer value r in the range [0,255]. Larger values are more deleterious so y = r/255 is the fraction of maximum deleterious effect of an allele. The average value of y over all 2k loci affecting the ith transition, ui, is ȳi, the total deleterious contribution of the minor loci. The actual transition is calculated as log10(ui) =–b + 2bȳi. I used the range (0,255) because that allowed each allele to be stored in one computer byte, which can store integers in the range 0,...,28 – 1, where 28 – 1 = 255.
If log10(ui) > 1, then the value is set to log10(ui) = 1 because this rapid rate of transition is sufficient to make the step very fast and not rate limiting, and larger values make numerical calculations more difficult. This truncation is made only for its computational efficiency and has almost no effect on the quantitative or biological interpretation of the model.
There are a total of n(k + 1) diploid loci. All loci recombine freely. Each allele mutates with probability ν during transmission to a gamete. Functional alleles at major loci mutate to loss-of-function alleles. Loss-of-function alleles back-mutate to functional alleles with probability ν/255. Minor loci alleles mutate to a different integer value in the range [0,255]; each integer not equal to the current allelic value has the same probability of arising by mutation.
At the start of a computer run, the genotype of each individual was initialized as follows. At major loci, each allele is set to the functional state with probability 0.95 and to the loss-of-function state with probability 0.05. At minor loci, each allele is set to the optimum value of zero with probability 0.95; with probability 0.05, each minor allelic value is sampled randomly from the uniform distribution over the integers in the range [0,255].
Note that the transition rates within an individual are not determined by somatic mutation rates or the loss of function of particular tumor suppressors. Instead, the inherited genotype determines the rate at which certain limiting steps occur in progression, without any explicit description or assumptions concerning what those rate-limiting steps are or how they may be passed. It would be easy to make the model in terms of the rates of explicit somatic mutations and genomic changes. But the goal here is to understand how germ-line mutations affect rates of transition through rate-limiting steps, no matter what the details of the rate-limiting steps are and how they are passed. So additional detailed assumptions would detract from the main goal.
Description of the Computer Simulations. I used the following parameters for all runs unless noted otherwise. The population was initialized with genotypes as described above, with 20,000 males and 20,000 females. Fitness was calculated as described above for each individual. Then, an offspring generation was built with 20,000 sons and 20,000 daughters.
For each offspring, a mother was chosen randomly with probability in proportion to fitness relative to the population of females, and a father was chosen randomly with probability in proportion to fitness relative to the population of males. Each mother and father make a haploid gamete by recombining their maternally and paternally inherited alleles. The haploid gametes combine to form the offspring. A simulation continues for 10,000 generations, after which statistics are collected on the final population.
The maximum age of an individual is T = 80 yr, with age of first reproduction at F = 15 yr. An individual starts life with x0(0) = 108 cell lineages, which is approximately the number of stem cells in a human colon. The slowest transition possible is log10(ui) = –b, where b = 3. The minimum transition is not particularly important because mutation will usually decay (raise) the transition rate independently of the minimum set by assumption. The more important consequence of the minimum transition is that it influences the average effect of each mutation (see above).
This study focuses on how the number of steps, n, affects the performance of each component and the level of genetic variation. Component performance in this case is measured by log10(ui), the transition rate for each step on a logarithmic scale. I varied the number of steps over the values n = 6, 7, 8, 9, and 10 in different runs.
To study how the number of minor loci affects genetic variation and component performance, I varied the number of minor loci per step over the values k = 20, 40, and 80.
The five values of n and the three values of k form 15 different combinations. I repeated each of these 15 combinations in 3 replicates, for a total of 45 runs.
Results and Discussion
The performance of a system depends on the performance of its individual components. In this case, fitness measures system performance, and the rates of transition between steps measure component performance. Faster transitions correspond to greater cancer mortality and lower component performance.
Fig. 2 shows that, as the number of components, n, increases, system performance improves and component performance declines. The total improvement in system performance (fitness) is small, on the order of one percent. This small increase in system performance as n rises is associated with a large drop in the performance of individual components.
Fig. 2.

The rise in fitness and decline in component performance as the number of components, n, increases. (A) Average fitness in the population is shown as the deviation from the maximum value of 1.0. The height of the plot shows –log10(s), where average fitness is 1 – s and s measures the deviation from the maximum. As –log10(s) rises, the fitness deviation from the maximum approaches zero at a logarithmic rate. (B) Component performance is shown as –log10(u), where u is the average transition rate between stages and maximum performance occurs when log10(u) is at its minimum value of –3. As –log10(u) declines, component performance declines logarithmically. Different symbols show the varying levels of k (the number of minor loci): k = 20 (#), k = 40 (O), and k = 80 (X).
The transition rates of log10(u) ≈–2.6 for n = 8 illustrate the decline in component performance as n increases. Those transition rates cause negligible fitness loss for n = 8, but those same transition rates with n = 6 would cause widespread cancer mortality early in life and a large loss in fitness. In particular, with n = 6 and log10(u) = –2.6 for all transition rates, everyone dies of cancer by age 57, and fitness is 1 – s = 0.69. Thus, the fitness loss is s = 0.31, and –log10(s) = 0.51, which is nearly two orders of magnitude below the smallest values in Fig. 2 A.
These results show that a rise in component number drives individual components to a poorly adapted state by the accumulation of deleterious mutations. Here, poor adaptation is measured relative to the higher level of component performance attained by systems with fewer components.
Fig. 3 illustrates the increase in genetic variability for cancer risk with a rise in the number of components, n. Fig. 3A plots the percentage of cancer mortality risk concentrated in the 30% of the population most at risk. For example, with n = 8, as much as 85% of the risk concentrates in the top 30% of the population. Fig. 3B shows the standard deviation in log10(ui) values averaged over the n different u values. These results are consistent with a recent study of genetic susceptibility to breast cancer, which found that the half of the population most genetically susceptible to breast cancer accounted for 88% of all cases (13).
Fig. 3.

Rise in the inherited genetic component of cancer predisposition as the number of components, n, increases. (A) The fraction of all cancer mortality among the 30% of the population with the greatest genetic predisposition, labeled as the genetic predisposition index. (B) The standard deviation between individuals in transition rates, log10(ui), averaged over the n different u values. Different symbols show the varying levels of k (the number of minor loci): k = 20 (#), k = 40 (O), and k = 80 (X).
The results in Fig. 3 demonstrate an increase in genetic variability as the system becomes more buffered against perturbations. Increased buffering is a consequence of a rise in the number of components, n. It has been suggested that such increase in genetic variability occurs because buffering against mutational perturbation causes variable alleles to be nearly neutral in their effects (5). The results here do show that system performance (fitness) changes relatively little as buffering and genetic variability increase. However, the variation in performance rises as buffering increases because the enhanced genetic variability is not entirely neutral and causes significant differences between individuals.
Fig. 3 also suggests that a rise in the number of minor loci contributing to quantitative variation causes a decrease in genetic variability. This relation occurs because the sampling variance is higher when a smaller number of loci are sampled.
Fig. 4 shows the frequency of cancer in populations. As n increases, the incidence declines. Major epithelial cancers have mortalities roughly on the order of 10–2, matching the results for n in the range of 6–8. However, not too much should be made of this match because actual progression probably depends on various factors not studied here that modulate transition rates.
Fig. 4.

The frequency of individuals in populations that die from cancer, shown as incidence on a log10 scale. Different symbols show the various levels of k (the number of minor loci): k = 20 (#), k = 40 (O), and k = 80 (X).
One commonly discussed aspect of progression concerns perturbations of DNA repair control systems, leading to faster accumulation of somatic mutations and chromosomal abnormalities as lineages progress toward cancer (14, 15). Similarly, clonal expansion of cellular lineages raises the number of cells that can make the transition into the next stage of progression, raising the effective transition rate (16).
If passing a particular stage in progression did lead to a mutator phenotype or chromosomal instability, then later changes in progression to cancer or disease might happen very rapidly. In that case, the later changes would not be rate-limiting stages in progression; instead, the main rate limiting stages would be the formation of the rapidly mutating phenotype. Thus, the key would be to understand the accumulation of germ-line mutations in DNA repair and cell cycle control systems that determine the rate at which individuals progress to mutator phenotypes or chromosomal instability.
It would be easy to add factors such as mutator phenotypes and chromosomal instability into the computational model used here. But those issues do not change the main conclusions of this article, which focus on how the number of components or rate-limiting stages affect mutational decay and the heritability of disease. Those general issues do not depend on the details of what determines the particular components or rate-limiting stages of a system.
Conclusions
An extra stage in cancer progression initially improves the survival of individuals by decreasing mortality from cancer. However, the additional buffering by an extra stage reduces the impact of any single hereditary mutation and therefore allows the accumulation of more nonlethal mutations in the population. Extra stages thereby lead to the evolution of partially decreased cancer mortality and significantly increased genetic predisposition to disease in the population as a whole.
These conclusions can be put in more abstract terms, to allow comparison with other robust genetic control systems. If a system improves its performance by adding additional buffering components, the evolution of improved system performance leads to an evolutionary decline by mutational decay in the performance of individual components. This decline in component performance maintains significant maladaptation in subsystems of a larger functional system. As systems add additional buffering components and then equilibrate in the face of mutational pressure on components, the net improvement in system performance may be small. In some cases, system performance may ultimately equilibrate to a lower level.
The weakened selective pressure per component with greater buffering also leads to an increase in genetic variability for the performance of each component. Thus, a rise in the number of buffering components may lead to an increase in the genetic variability of system performance.
Turning back to cancer, the model makes some interesting predictions about genetic variability in risk. Some cancers arise after deterioration of a small number of buffering steps whereas progression to other cancers seems to require passing a greater number of buffering stages (1). For example, the age-specific incidence curves for retinoblastoma seem to depend on only two rate-limiting steps whereas the major epithelial cancers seem to depend on roughly six or seven steps. The model here predicts much greater quantitative genetic variability from several minor loci in the multistage epithelial cancers than in cancers with fewer stages, such as retinoblastoma. In addition, there should be greater maladaptation in the components that buffer the multistage cancers than in the components that buffer cancers with fewer stages.
In general, greater robustness of system performance leads to greater maladaptation of component performance.
Acknowledgments
This work was supported by National Science Foundation Grant DEB-0089741 and National Institutes of Health Grant AI24424.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Knudson A. G. (1993) Proc. Natl. Acad. Sci. USA 90, 10914–10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelstein B. & Kinzler K. W., eds. (2002) The Genetic Basis of Human Cancer (McGraw–Hill, New York), 2nd Ed.
- 3.Frank, S. A. & Nowak, M. A. (2004) BioEssays, 26, 291–299. [DOI] [PubMed] [Google Scholar]
- 4.Malkin, D. (2002) in The Genetic Basis of Human Cancer, eds. Vogelstein B. & Kinzler K. W. (McGraw–Hill, New York), 2nd Ed., pp. 387–401.
- 5.de Visser, J. A. G. M., Hermisson, J., Wagner, G. P., Ancel Meyers, L., Bagheri-Chaichian, H., Blanchard, J. L., Chao, L., Cheverud, J. M., Elena, S. F., Fontana, W., et al. (2003) Evolution 57, 1959–1972. [DOI] [PubMed] [Google Scholar]
- 6.Rutherford, S. L. & Lindquist, S. (1998) Nature 396, 336–342. [DOI] [PubMed] [Google Scholar]
- 7.Bergman, A. & Siegal, M. L. (2003) Nature 424, 549–552. [DOI] [PubMed] [Google Scholar]
- 8.Frank, S. A. (2003) J. Evol. Biol. 16, 138–142. [DOI] [PubMed] [Google Scholar]
- 9.Armitage, P. & Doll R. (1954) Brit. J. Cancer 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frank, S. A. (2004) Curr. Biol. 14, 242–246. [DOI] [PubMed] [Google Scholar]
- 11.Weinberg, R. A. (1998) One Renegade Cell (Basic Books, New York).
- 12.Kinzler, K. W. & Vogelstein, B. (2002) in The Genetic Basis of Human Cancer, eds. Vogelstein B. & Kinzler K. W. (McGraw–Hill, New York), 2nd Ed., pp. 583–612.
- 13.Pharoah, P. D. P., Antoniou, A., Bobrow, M., Zimmern, R. L., Easton, D. F. & Ponder, B. A. J. (2002) Nat. Genet. 31, 33–36. [DOI] [PubMed] [Google Scholar]
- 14.Loeb, L. A. (1991) Cancer Res. 51, 3075–3079. [PubMed] [Google Scholar]
- 15.Rajagopalan, H., Nowak, M. A., Vogelstein, B. & Langauer, C. (2003) Nat. Rev. Cancer 3, 695–701. [DOI] [PubMed] [Google Scholar]
- 16.Armitage, P. & Doll, R. (1957) Br. J. Cancer 11, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]