

Abstract
Neuroinflammation occurs in acute and chronic CNS injury, including stroke, traumatic brain injury and neurodegenerative diseases. Microglia are specialized resident myeloid cells that mediate CNS innate immune responses. Disease relevant stimuli such as reactive oxygen species (ROS) can influence microglia activation. Previously we observed that p53, a ROS responsive transcription factor, modulates microglia behaviors in vitro and in vivo, promoting pro-inflammatory functions and suppressing down-regulation of the inflammatory response and tissue repair. Here we describe a novel mechanism by which p53 modulates the functional differentiation of microglia both in vitro and in vivo. Adult microglia from p53deficient mice have increased expression of the anti-inflammatory transcription factor c-Maf. To determine how p53 negatively regulates c-Maf, we examined the impact of p53 on known c-Maf regulators. MiR-155 is a microRNA (miRNA) that targets c-Maf. We observed that cytokine induced expression of miR-155 was suppressed in p53 deficient microglia. Furthermore, Twist2, a transcriptional activator of c-Maf, is increased in p53 deficient microglia. We identified recognition sites in the 3′ untranslated region of Twist2 mRNA that are predicted to interact with two p53 dependent miRNAs: miR-34a and miR-145. Here we demonstrate that miR-34a and -145 are regulated by p53 and negatively regulate Twist2 and c-Maf expression in microglia and the RAW macrophage cell line. Taken together, these findings support the hypothesis that p53 activation induced by local ROS or accumulated DNA damage, influences microglia functions and that one specific molecular target of p53 in microglia is c-Maf.
Keywords: c-Maf, microglia, miRNAs, neuroinflammation, p53, Twist2
INTRODUCTION
Acute and chronic injury of the central nervous system (CNS) is associated with neuroinflammation (1). Microglia, the resident immunocompetent cells of the CNS, participate in a neuroinflammatory response to ischemic or traumatic injury. In the uninjured CNS, microglia express little or no mRNA for genes associated with active inflammation such as CD45, MHC class II, co-stimulatory molecules (CD80 and CD86) and integrins (2–4). During the response to injury, microglia become activated, demonstrating increased expression of activation markers (CD45, MHC class II, CD86 etc.). They also change in morphology from cells with a small soma and fine ramifications to larger ameboid cells capable of migrating toward a site of injury or infection.
Microglia, like macrophages, are capable of a variety of inflammatory responses to altered environmental stimuli (5). In macrophages, classical activation develops in response to pro-inflammatory cytokines such as interferon-γ(INF-γ) and toll like receptor (TLR) ligands. Classically activated macrophages perform functions important for pathogen suppression such as generation of ROS and secretion of pro-inflammatory mediators, activities that may cause injury to bystander cells. In contrast, anti-inflammatory signals such as interleukins (IL)-4, -10 or -13, immune complexes, transforming grown factor (TGF)-β and glucocorticoids lead to type II inflammation, in which pro-inflammatory activities are suppressed and inflammatory cells promote tissue remodelling. Microglia expression of activation markers differs quantitatively but not qualitatively from that observed in peripheral macrophages (4). The classical activation state of myeloid cells has been linked with promoting inflammation (6), whereas the alternative phenotype is anti-inflammatory and promotes tissue repair (7).
Although the roles microglia play in the injured CNS are still under investigation, an emerging theme is that glia contribute to the pathogenesis of neurological diseases via a non cell-autonomous mechanism (8). Microglia behavior during inflammation can lead to: i) loss of normal function in other neuroglia that subsequently trigger downstream damage to vulnerable neurons; and ii) direct neuronal toxicity (9). The response to CNS injury may trigger both processes if neural injury leads to promotion of neurotoxic, tissue destructive inflammatory responses while concurrently suppressing a potentially neuroprotective inflammatory response.
The transcription factor p53 is best known as a regulator of cell-cycle control and apoptosis in response to discrete cellular insults (8). Under basal conditions, p53 is ubiquitnated and rapidly degraded. However, p53 activity can be rapidly increased in response to a wide range of stresses, including DNA damage, hypoxia, and oxidative damage (10). Previously, we identified p53 as a novel candidate modulator of microglia behavior and demonstrated ongoing p53 transcriptional activity in microglia cultured under basal conditions (11–13). Genetic knockout or pharmacological inhibition of p53 results in microglia that are less neurotoxic in response to pro-inflammatory stimuli (11, 14). We also observed that p53 deficient microglia express genes associated with the anti-inflammatory/tissue repair phenotype of alternatively activated macrophages (13). Additionally, p53 is activated in a sub-population of microglia in the inflamed human brain (12) but p53 activation is excluded from the population of microglia labeled by CD163 (13), a marker of macrophages that have adopted anti-inflammatory or tissue repair phenotypes (15).
MicroRNAs (miRNAs) are small non-coding RNAs (~22 nucleotides long) involved in many physiological and pathological processes (16–18). A single miRNA can regulate hundreds of genes and up to 90% of human genes are potentially regulated by miRNAs (19, 20). MiRNAs downregulate the expression of mRNA targets by binding to complementary sequences in the 3-prime untranslated region (UTR). Several reports suggest that miRNAs play an important role in regulating the responses of innate immune cells (21) including microglia (18). For example, recent reports suggest that miR-124 is a key regulator of microglia quiescence in the CNS, thereby helping to prevent CNS inflammation (18, 22). Since a single miRNA can modulate expression of many genes during inflammation, miRNAs may serve as epigenetic regulators of microglia behavior (23).
As a transcriptional activator, p53 exerts its function through promoting expression of target genes to initiate cellular responses. Recent work has revealed that miRNAs are important components in the p53 transcriptional network (24). Maturation of miRNAs is promoted by p53-mediated transcription (25) and p53 also induces expression of specific miRNAs. For example, several groups reported that p53 directly regulates the expression of the miR-34 family (26–30). The miR-34 family consists of miR-34a, miR-34b and miR-34c, which are encoded by two different genes. In transformed cells, ectopic expression of miR-34a promotes p53-mediated apoptosis, cell cycle arrest and senescence, whereas inactivation of endogenous miR-34a strongly inhibits p53-dependent apoptosis (24). In addition to the miR-34 family, p53 also directly regulates expression of several additional miRNAs, including miR-145 (24, 25). Over-expression of miR-145 reduces c-Myc expression, whereas suppression of endogenous miR-145 enhances c-Myc expression (24), which partially accounts for miR-145-mediated inhibition of tumor cell growth both in vitro and in vivo (31, 32).
We previously reported that microglia from p53 deficient mice had altered gene expression and inflammatory behaviors that suggested p53 transcriptional activity is a critical activator of pro-inflammatory responses in microglia (13). Here, we identify c-Maf, a transcription factor known to promote functional differentiation and anti-inflammatory responses in both lymphocytes and myeloid cells (33, 34), as a downstream p53 target in microglia. We determined that p53 negatively regulates c-Maf in microglia by enabling the induction of miR-155, a miRNA that suppresses c-Maf. Additionally Twist2, a transcriptional activator of c-Maf, is suppressed by p53-regulated miR-34a and miR-145. Taken together, we propose that p53 activation may influence microglia responses to injury by suppressing c-Maf expression and the subsequent protective inflammatory response.
MATERIALS AND METHODS
Animals
Mice deficient in p53 (p53−/−) (35) and miR-155 (miR-155 KO) (36), and strain-matched p53 expressing mice (p53+/+) as well as miR-155 expressing mice (C57/BL6 from Jackson Laboratories) were maintained in a specific pathogen-free facility under guidance of an IACUC approved protocol.
Middle Cerebral Artery Occlusion (MCAO)
MCAO was performed on anesthetized 12–16 week old male C57/BL6 mice obtained from Jackson Laboratories using the transient intraluminal filament method (37) with laser Doppler cerebral blood flow (CBF) monitoring. The filament was inserted to obtain a 70% or greater occlusion for 15 min, followed by filament removal and documentation of reperfusion, a paradigm developed to result in ischemic preconditioning (38). Mice with CBF profiles outside a predetermined expected range were excluded. Mice were followed by neurobehavioral assessments using a published scoring system (39) for 72 hours following the lesion. Animals with a detectable behavioral deficit were excluded from further study. Using this paradigm, there is no detectable region of infarction in cortex by TTC staining.
Ex Vivo Flow Cytometry
Adult mice (12–16 weeks old, n=4 mice per genotype) were intracardially perfused with cold saline solution. Cortical tissue was separated from whole brain and dissociated using the MACS Neural Dissociation Kit (Miltenyi) and a modified version of the Miltenyi gentleMACS protocol “Preparation of single-cell suspensions from mouse neural tissue” according to the manufacturers instructions. Depletion of myelin and positive selection of myeloid cells was performed in sequential myelin and CD11b magnetic selection steps (Miltenyi). The remaining cells were centrifuged at 300g for 10 min at 4 °C, the pellet was resuspended in FACS medium (FM) [10% FCS (Atlanta Biologicals), 10 mM HEPES pH7.3 (Gibco), in HBSS buffer without calcium or magnesium (Gibco)] and incubated with FC block (BD Pharmingen). Cells were then stained with fluorochrome conjugated antibodies according to manufacturer’s protocol: FITC-CD45 (BD Pharmigen, Transduction Labs), APC-F4/80 (Affymetrix eBioscience) or isotype control and DAPI (Sigma-Aldrich). Cells with antibodies were incubated on ice for 45 min, washed 3 times with FM and resuspended in FM. All cells were sorted on a BD ARIA II cell sorter directly into TRIzol LS reagent (Invitrogen). Microglia were defined as the population of cells that were F4/80positive, and CD45intermediate using a previously published approach (40).
Cell Culture
The RAW cell line was grown in high glucose DMEM supplemented with 10% FBS (Gibco), 25 U/ml penicillin and 25 mg/ml streptomycin and incubated in a 37 °C cell incubator with 5% CO2 supplemented. RAW cells were plated onto 24-well dish at a density of 5×104, transfected with 5 pmol of Mission miRNA mimics (Sigma-Aldrich) using jetPEI (Polyplus Transfection) reagents according to manufacturer’s protocol. For IL-4 treatment, cells were treated with 10 ng/mL IL-4 for 24 hours before harvest. For Nutlin treatment, cells were treated with DMSO, 10 μM, 40 μM, or 80 μM Nutlin for 6 hours before harvest. Cells were lysed for total RNA 24 hours post-transfection and for total protein 72 hours post-transfection. Mixed glia cultures were generated using previously published methods from cortical tissue dissected on postnatal day 3 or 4 (13). Cells were cultured in high glucose DMEM (Hyclone, Thermo Scientific) supplemented with 10% heat inactivated horse serum (Gibco), 10% nutrient mixture F-12 ham (Sigma-Aldrich), 2mM L-glutamine (Sigma-Aldrich), 10mM HEPES (Sigma-Aldrich), and 20% L929 conditioned medium (41). Microglia were isolated from the cultures 7–10 days post-dissection by collecting floating cells. Primary microglia were plated on poly-d-lysine coated plates at a density of 5×105 per 35mm plate or 1×106 per 60mm plate in D10C media. Cells were treated with IFN γ(10 U/ml) for 24 hours, Nutlin-3 (40uM) for 6 hours, or vehicle control.
Real-time Quantitative Reverse Transcriptase Polymerase Chain Reaction
Total RNA was extracted using the High Pure RNA Isolation Kit (Roche) or miRNeasy kit (Qiagen) according to manufacturers’ instructions. cDNA was generated using the High Capacity cDNA Reverse Transcriptase Kit (Applied Biosciences). cDNA from adult mRNA transcripts was amplified using the Ovation qPCR system (NuGEN) at the University of Washington CHDD Microarray and Bioinformatics Core Laboratory. cDNA from adult mouse miRNA was generated using miRNA specific primers. Quantitative PCR was performed using the StepOnePlus Real Time PCR Instrument (Applied Biosciences) with the Taqman Gene Expression Assay, Taqman MicroRNA Assay, or Roche Universal Probe Library. Taqman probe and primers used are as follows: cMaf: Mm02581355_s1, Twist2: Mm00492147_m1, miR-34a: 000426, miR-145: 002278, miR-155: 002571. PCR products were normalized to TBP (Mm01277042_m1) or 18S (Mm03928990_g1) for Taqman gene expression assays, and Sno202 (001232) and Sno234 (001234) for microRNA assays. Roche UPL primer probe sets are listed in table 1.
Table 1.
Roche Primer and Probe sets utilized for qPCR
| Gene Name | Forward Primer (5′-3′) | Reverse Primer (5′-3′) | Roche Probe# |
|---|---|---|---|
| GAPDH | AGCTTGTCATCAACGGGAAG | TTTGATGTTAGTGGGGTCTC | 9 |
| TBP | TGTGCACAGGAGCCAAGA | CCCCACCATGTTCTGGAT | 33 |
| IL1α | TTGGTTAAATGACCTGCAACA | GAGCGCTCACGAACAGTTG | 52 |
| IL1β | AGTTGACGGACCCCAAAAG | AGCTGGATGCTCTCATCAG | 38 |
| MARCO | ATGCTCGGTTACTCCAGAGG | ATTGTCCAGCCAGATGTTC | 10 |
| c-Maf | CCTTCCTCTCCCGAATTTTT | CCACGGAGCATTTAACAAGG | 33 |
| Twist2 | CATGTCCGCCTCCCACTA | GATGTGCAGGTGGGTCCT | 10 |
qPCR primer sequences and corresponding probes were designed using the ProbeFinder Software available at http://www.roche-applied-science.com
Western Blot
RAW cells were lysed with RIPA buffer (Thermo-scientific) containing protease inhibitor cocktail (Sigma). 10 μg of total proteins were run on NuPage 4–12% Bis Tris gel (Invitrogen) and transferred according to manufacturer’s protocol (Invitrogen). Membranes were probed for Twist2 (Abcam, Ab66031, Rabbit polyclonal), c-Maf (Abcam, Ab76817, mouse monoclonal), and β-actin (Santa Cruz, SC-47778, mouse monoclonal) as primary antibodies (1:1000 dilution), and anti-Rabbit (Santa Cruz, SC2301) or anti-mouse (Santa Cruz, SC2302) as secondary antibodies (1:5000 dilution). Twist2, c-Maf protein levels were normalized to β-actin. All films were scanned and analyzed using ImageJ 1.47 software.
RESULTS
c-Maf expression is suppressed by p53 in microglia
We previously reported that p53 supports the a proinflammatory response in microglia (13). We observed that p53 knockout (p53−/−) microglia in neuron/microglia co-cultures are functionally distinct from wild-type primary microglia in response to HIV gp120 coat protein (11). We further found that p53−/− microglia demonstrated a blunted response to interferon-γ (IFNγ), failing to increase expression of genes associated with classical macrophage activation or secrete proinflammatory cytokines, and tend to have gene expression patterns and functions similar to alternatively activated macrophages (13).
The c-Maf transcription factor has been reported to promote expression of anti-inflammatory cytokines and suppress expression of pro-inflammatory cytokines in macrophages (33, 34, 42). We hypothesized that p53 promotes microglia pro-inflammatory behaviors by supressing c-Maf mediated gene transcription. We therefore measured c-Maf mRNA in microglia cultured from day 3–4 postnatal p53+/+ or p53−/− mice, and found that c-Maf was elevated by two fold in p53−/− cells compared to p53+/+ cells (data not shown), indicating a regulatory connection between p53 and c-Maf. However, cultured microglia are exposed to cytokines that promote cell division and c-Maf expression may be regulated by these cytokines (43). To determine if the regulation of c-Maf expression by p53 in cultured microglia was unique to the in vitro environment we asked whether c-Maf expression was specifically regulated by p53 in microglia from the adult brain. We obtained microglia from 12–16 weeks old adult mice by ex-vivo flow cytometry, based on previously published criteria (44–46) and measured c-Maf mRNA by quantitative RT-PCR. Compared to p53+/+ cells, c-Maf mRNA was elevated by 2.5 fold in p53−/− microglia (Fig. 1), suggesting that basal p53 activity in normal microglia is sufficient to suppress c-Maf expression in vivo.
Figure 1. c-Maf is negatively regulated by p53 in adult mice microglia.
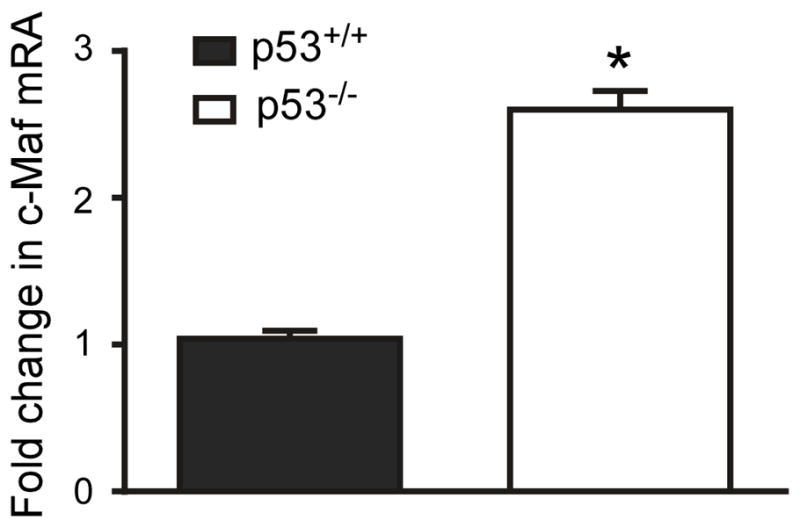
Expression of c-Maf mRNA is elevated in p53−/−mice. Quantitative RT-PCR for c-Maf mRNA was performed on total RNA obtained from p53+/+ and p53−/− adult mouse brain microglia extracted by ex-vivo flow cytometry (n= 3 adult mice brains were used in ex-vivo flow cytometry. Statistics were determined from separate qPCR runs, p<0.005 by unpaired t-test).
MiR-155, a suppressor of c-Maf, is regulated by p53
Since c-Maf is a potential downstream target of p53 in microglia, next we asked how p53 influences the expression of c-Maf. The transcriptional repertoire of p53 has been intensely studied, but direct binding of p53 to the c-Maf gene had not been identified (47). To assess whether p53 directly suppresses expression of c-Maf mRNA, we evaluated the c-Maf genomic sequence and observed several p53 consensus binding sites 2 Kb upstream of the translational start site. One recent study reported that p53 promoted transcriptional activation of c-Maf during the process of lens development (48). Since this report suggested that a direct interaction between p53 and the c-Maf gene led to regulation in the opposite direction of what was observed in microglia, we hypothesized that p53 represses c-Maf expression in microglia through an alternate mechanism. To determine how c-Maf expression levels are negatively regulated by p53, we assessed whether other known regulators of c-Maf expression are influenced by p53 in microglia.
Since p53 generally acts as a transcriptional activator, we hypothesized that suppression of c-Maf was mediated by a p53 induced miRNA. The pro-inflammatory microRNA miR-155 negatively regulates c-Maf expression in T-cells (36). We therefore evaluated miR-155 expression in cultured primary microglia by quantitative RT-PCR. Under resting conditions, very little miR-155 is expressed in microglia (Fig. 2A). Treatment with classical activation cytokine IFNγ leads to a robust induction of miR-155 in p53+/+ cells. In contrast, induction of miR-155 in p53−/− cells was significantly repressed. Comparing IFNγ-induced miR-155 expression between p53+/+ and p53−/−microglia cultures revealed that p53 significantly augments the induction of miR-155 (Fig. 2A). We next measured c-Maf mRNA expression in microglia cultures from miR-155 knocked-out (KO) mice and found that c-Maf mRNA was elevated by 4.5 fold over WT microglia (Fig. 2B). We also observed a 1.5 fold increase of c-Maf mRNA in microglia collected by ex-vivo flow cytometry from miR-155 KO adult mice compared to WT mice (Fig. 2C), demonstrating that even under basal conditions c-Maf expression is suppressed by this pathway in vivo. Taken together, these data suggest that p53 negatively regulates c-Maf expression by transcriptional activation of miR-155.
Figure 2. miR-155, a negative suppressor of c-Maf, requires p53 for induction in microglia.

(A): miR-155 expression in microglia cultured from p53+/+ and p53−/− neonatal mice were determined by miRNA qRT-PCR assay. Primary cultured microglia were treated with 10U/ml IFNγ or vehicle control for 24 hr. before total RNAs were extracted. (n=3 from separate culture experiments and qPCR runs, p<0.001 for miR-155 level in p53+/+ vs. p53−/−cells upon IFNγ treatment by two-way ANOVA post hoc comparison). (B): c-Maf mRNA level in microglia cultured from JAX WT and miR-155 KO neonatal mice were detected by qRT-PCR. (n≥3 from separate culture experiments and qPCR runs, p<0.05 by unpaired t-test with Welch’s correction). (C): c-Maf mRNA level were determined by qRT-PCR in microglia collected from adult JAX WT and miR-155 KO mice by ex-vivo flow cytometry. (n= 3 adult mice brain were used in ex-vivo flow cytometry. Statistics were determined from separate qPCR runs, p<0.05 by unpaired t-test).
miR-155 KO recapitulates a portion of the p53 KO phenotype in microglia
Previously we reported that p53−/−microglia did not promote HIV-gp120 neurotoxicity (11) and have a muted transcriptional response to a classical activation signal (13). If miR-155 is the key mediator of the p53-dependent microglia pro-inflammatory response, we hypothesized that mediators of classical activation influenced by p53 would be similarly regulated by miR-155. To test this hypothesis, primary microglia cultured from WT and miR-155 KO mice were treated with 10 U/ml of IFNγ for 24h, and mRNAs associated with classical activation were evaluated. We previously reported that three genes associated with a type I inflammatory response: IL-1α, IL-1β and MARCO (49), are not effectively induced by IFNγ in p53−/− microglia (13). Similar results were observed in miR-155 KO microglia for IL-1α (Fig. 3A) and MARCO (Fig. 3C), but not for IL-1β (Fig. 3B). These results suggest that regulation of IL1α and MARCO by p53 in microglia is mediated through miR-155; however, an alternative pathway might influence microglia IL1β expression in response to IFNγ.
Figure 3. Classical activation of microglia is suppressed in miR-155 KO cells.
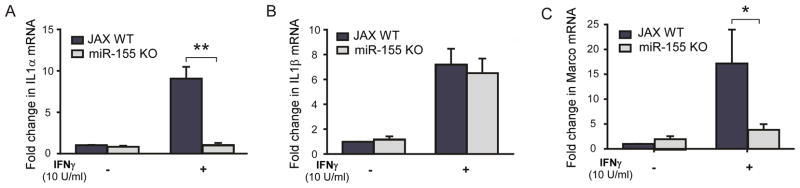
qRT-PCR was performed on WT and miR-155 KO microglia treated with IFN-γ or control for interleukin-1α (IL-1lα) (A), interleukin-1β (IL-1β) (B), and the murine scavenger receptor MARCO (C). (n=3 separate cultures and qPCR runs, *p<0.01, **p<0.0001by two way ANOVA comparison)
Twist2, a positive regulator of c-Maf, is regulated by p53-inducible miRNAs
c-Maf expression is also transcriptionally regulated by Twist2, a basic helix-loop-helix (bHLH) transcription factor. Twist2 promotes myeloid differentiation and interacts with the 5′ regulatory region of the c-Maf gene to increase c-Maf expression (50). We therefore investigated the relationship among p53, Twist2 and c-Maf in microglia. We observed that Twist2 was up-regulated by 2.5 fold in cultured p53−/− microglia compared to p53+/+ cells on the microarray analysis previously reported (13). An even larger increase in Twist2 expression was confirmed by qPCR on mRNA isolated from p53−/− and p53+/+ microglia (Fig. 4). This finding suggests an alternate mechanism by which c-Maf expression is regulated by p53 via p53-dependent suppression of Twist2 expression.
Figure 4. Expression of Twist2, a positive regulator of c-Maf, is influenced by p53 genotype.
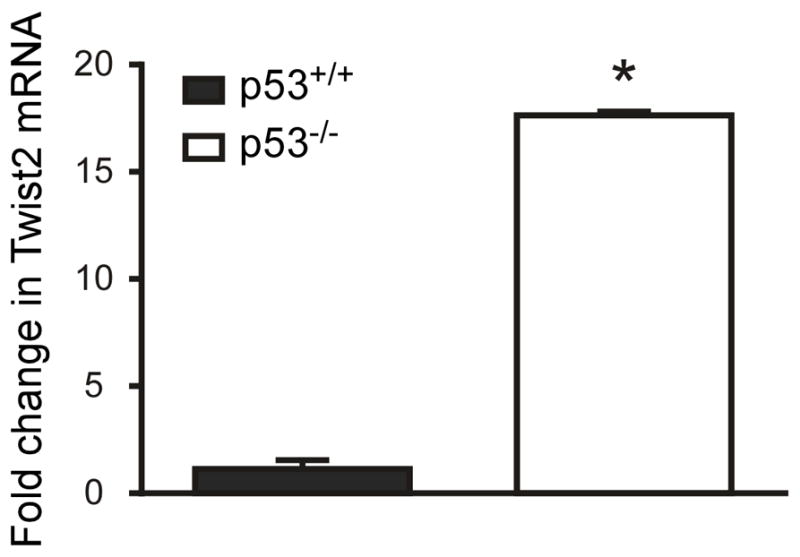
Expression of Twist2 mRNA is elevated in p53−/−microglia. Quantitative RT-PCR for Twist2 mRNA on total RNA extracted from microglia cultured from p53+/+ and p53−/− neonatal pups. (n= 3 separate cultures and qPCR runs, *p<0.0001 by unpaired t-test)
Since p53 is known as a transcriptional activator, we hypothesized that p53 also induces the expression of miRNAs that negatively regulate Twist2. The 3′-UTR of Twist2 was analyzed in silico for the identification of miRNA targets. Using two independent software platforms, we identified binding regions for two specific miRNAs, miR-34a and miR-145, which are known p53 transcriptional targets (26, 27, 31). We performed RT-PCR for these two miRNAs in both cultured microglia (Fig. 5A–B) and adult microglia extracted by ex-vivo flow cytometry (Fig. 5C–D) and observed p53-dependent expression of these two miRNAs in both systems. Interestingly, in vitro these two p53 dependent miRNAs are differentially regulated by p53, with miR-34a being turned on by basal p53 activity and miR-145 being induced by higher level p53 activity following treatment with the MDM2 inhibitor, nutlin. However, both miR-34a and miR-145 were significantly reduced in adult p53−/− microglia isolated by ex vivo sorting. Taken together, these findings support our hypothesis that p53 can suppress c-Maf expression through a pathway independent of miR-155: inducing miRNAs that suppress the c-Maf activator Twist2.
Figure 5. miRNAs that target Twist2 for suppression are targeted by p53 in microglia both in vivo and in vitro.
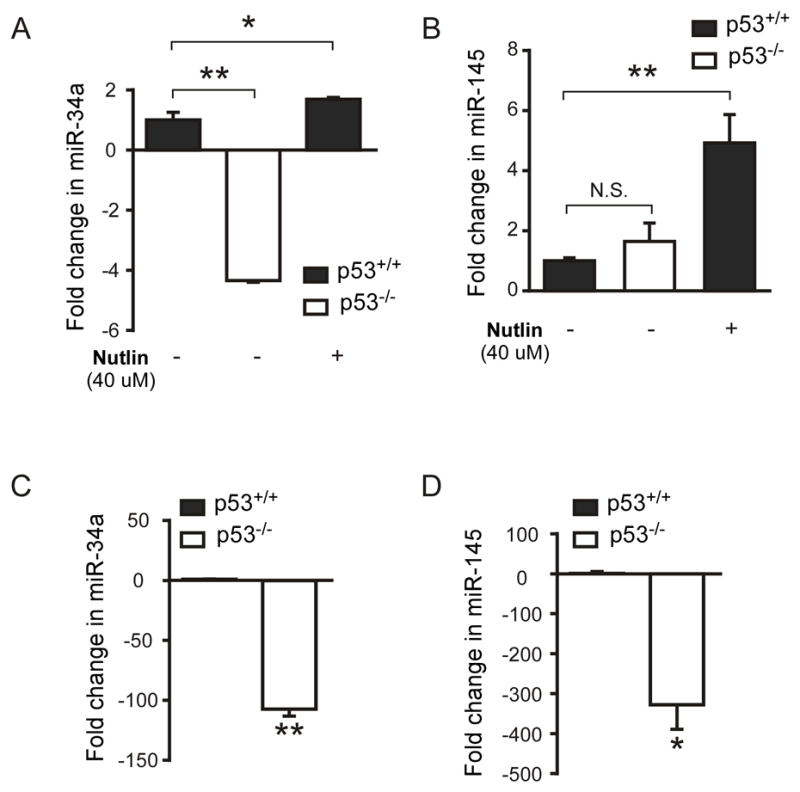
(A) and (B): qRT-PCR was performed to detect miR-34a and miR-145 on total RNA extracted from cultured microglia, that were either p53−/−, p53+/+ or p53+/+ following p53 induction with nutlin for 6 hours. (n=3 separate cultures, statistics were determined from separate qPCR runs, p<0.0001 by one-way ANOVA). (C) and (D): qRT-PCR was performed for miR-34a and miR-145 on adult microglia extracted from p53−/− and p53+/+ mice. (n=3 from separate qPCR runs, *p<0.02, **p<0.002 by unpaired t-test).
To further examine the hypothesis that p53 regulates Twist2 via induction of miR-145 and miR-34a, we employed the mouse leukemic monocyte macrophage cell line (RAW cells). To determine if activation of p53 lessened Twist2 protein in a myeloid cell line, we exposed RAW cells to increasing concentrations of nutlin. We observed greater p53 stabilization (Fig. S1) and decreasing Twist2 protein detected by Western blot with increasing nutlin concentration (Fig. 6A–B). Next, miR-145 and miR-34a mimics (Sigma) were transfected into RAW cells, and Twist2 mRNA levels were determined by qRT-PCR. Twist2 mRNA levels were significantly reduced when either miR-145 or miR-34a was overexpressed in RAW cells. As shown in Figure 6, 24 hours post transfection, Twist2 mRNA was knocked down by 66% and 64%, when miR-145 and miR-34a were expressed, respectively (Fig. 6C). The levels of miR-145 and miR-34a before and after transfection were determined by the manufacturer’s recommended miRNA RT-PCR assay from the same extracted RNA samples (Fig. S2). Additionally, we checked c-Maf mRNA level in RAW cells transfected with either miR-145 or miR-34a mimics, and observed an over 60% reduction in c-Maf mRNA level in both conditions (Fig. 6D). To ensure that the observed level of reduction in mRNA lead to a physiologically relevant change in Twist2 or c-Maf protein, we performed Western blot for Twist2 and c-Maf on lysates from RAW cells treated with miR-34a and miR-145 mimics and compared the observed change in protein relative to a physiological inducer of c-Maf, IL-4 (Fig. 6EG). The reduction in c-Maf protein by miR-34a and miR-145 in RAW cells was significant, though less profound. Taken together, our data strongly suggest that the p53-induced miRNAs, miR-145 and miR-34a, target Twist2 mRNA for suppression resulting in decreased c-Maf expression.
Figure 6. p53 activation suppresses Twist-2 and c-Maf expression and transfection of miR-145 or miR-34a down regulates Twist2 and c-Maf in RAW cells.

(A): p53 activation by nutlin causes a dose-dependent reduction of Twist2 protein in RAW cells. (B): Densitometry of Twist2 labeled bands relative to actin labeled bands on Western blot of RAW cell lysates after treatment with DMSO or 10 μM, 40 μM, or 80 μM of Nutlin. Data were plotted from 3 separate experiments, p<0.001 by one-way ANOVA. (C): Transfection of miRNA mimics miR-145 and miR-34a significantly decreases Twist2 mRNA level in RAW cells. 24 hours post transfection, (n=3 from separate transfections and qPCR runs, p<0.001 by one-way ANOVA). (D): c-Maf mRNA levels were determined by qRT-PCR in samples when cells were transfected with or without miRNA mimics (n=3 from separate transfections and qPCR runs, p<0.01 by one-way ANOVA) (E): Transfection of miR-145 and miR-34a mimics significantly decreased Twist2 and c-Maf protein in RAW cells, while IL-4 treatment increased both Twist2 and c-Maf protein. (F): Densitometry of Twist2 protein level in RAW cells transfected with miR-145 and miR-34a mimics, or treated with IL-4. Data were plotted from 4 separate experiments, *p<0.05 **p<0.001 by one-way ANOVA. (G): c-Maf protein level in RAW cells when cells were transfected with miR-145 and miR-34a mimics, or treated with IL-4. Data were plotted from 4 separate experiments, p<0.05 by one-way ANOVA).
The in vivo response to CNS ischemia involves p53 activation, induction of p53 dependent miRNAs and suppression of c-Maf
The data presented demonstrate that p53 influences c-Maf expression in several paradigms including cultured microglia, RAW cells and microglia extracted from normal adult mouse brain. The question remains whether these miRNAs modulate inflammatory gene expression in vivo during neuroinflammation. To address this, we induced neuroinflammation by transient middle cerebral artery occlusion (MCAO), a model of CNS ischemia in which we have previously demonstrated that loss of p53 leads to increased microglia expressing CD206, a marker of the anti-inflammatory phenotype (13). We performed a Pscan promoter analysis (51) on microarray data obtained from forebrain microglia ex vivo extracted 3 days following 15 min. MCAO (a less severe ischemic pulse that does not induce cortical infarction but does result in ischemic preconditioning (38)) or sham surgery. We observed that 15 min. ischemia lead to a significant (P<0.02) induction of p53 transcriptional targets in ex vivo sorted cortical microglia 3 days after the ischemic insult. These findings demonstrate that the pro-inflammatory response supported by p53 could be detected 3 days following this injury paradigm. To determine if this model of neuroinflammation involves activation of miR-155, miR-145 and miR-34a in microglia, we obtained forebrain microglia by ex-vivo flow cytometry 3 days after 15 minute MCAO. The presence of an inflammatory response to ischemia was confirmed by a 30% increase in microglia and a 10 fold increase in macrophages obtained from cortex ipsilateral to the MCAO compared to the contralateral side. We observed an increase in the level of miR-155 (Fig. 7A). MiR-34a and MiR-145 were not induced. In addition, the expression of c-Maf mRNA was suppressed in microglia from the ischemic side of the brain at both time points (Fig. 7B). Twist2 mRNA was not detected. In summary, these data suggest that neuroinflammation in vivo is associated with induction of at least one p53 dependent miRNA that suppresses c-Maf expression in microglia. The finding also suggests that miR-155 is the dominant mediator of c-Maf suppression during the response to CNS ischemia.
Figure 7. In vivo CNS inflammation induces activation of miR-155 and suppression of c-Maf in microglia.
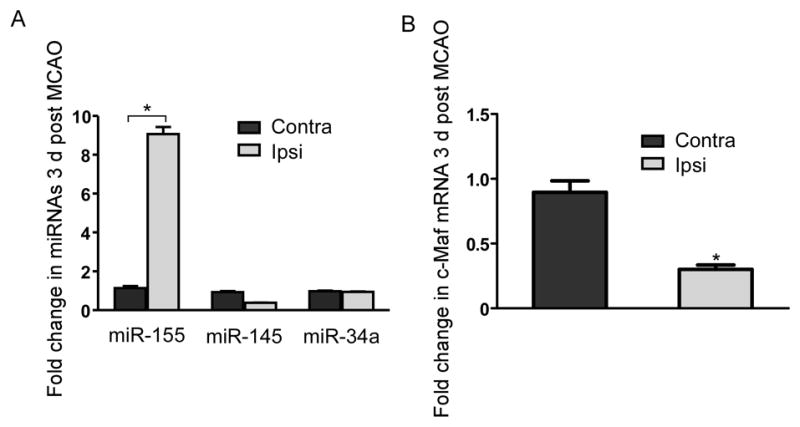
(A) qPCR for miR-155, miR-145, and miR-34a expression in microglia extracted by ex vivo flow cyometery from mice that were 3 days (A) after 15 min. transient MCAO. miRNA levels were normalized to both Sno202 and Sno234 RNAs, p<0.001 by two-way ANOVA. (B) qPCR for c-Maf mRNA relative to housekeeping gene expression in microglia extracted 3 days following 15 min. transient MCAO, p<0.001 by unpaired t-test.
Discussion
Many studies suggest that inflammation influences tissue injury and degeneration in the CNS. It is now believed that neuroinflammation is an important contributor to neurodegeneration. Neurons are susceptible to oxidative damage from ROS induced by inflammation and inflammatory mediators can be directly neurotoxic or attract leucocytes with cytotoxic properties. However, microglia are also capable of promoting tissue repair in the CNS, thus the modulation of microglia behavior may have important influences on long term functional outcome in CNS disease or injury states. Several studies have shown that microglia exhibit patterns of functional differentiation similar to those described for macrophages (52). Microglia express marker proteins associated with specific differentiation states in a number of CNS diseases and disease models (53, 54), demonstrating that microglia are capable of heterogeneous functional differentiation responses to disease-relevant stimuli.
Inflammation is a complex response that evolved to restore homeostasis after infection or injury, and microglia are key regulators of the inflammatory responses in CNS. However, little is known regarding the endogenous molecular signals that determine which components of the microglia functional repertoire will be expressed in response to a changing neural environment. Identifying the molecular signals that control whether microglia exhibit destructive or protective functions is likely to suggest novel therapeutic targets that may improve functional outcome in diseases associated with neuroinflammation such as Alzheimer’s disease (55, 56), Parkinson’s disease (57), ALS (58), multiple sclerosis (1), traumatic brain injury (59), and stroke (60). We previously identified p53 as a transcriptional regulator of microglia behavior (13) and now report that p53 acts through miRNA dependent regulation of c-Maf, a known transcriptional regulator of the inflammatory response as diagramed in Figure 8. p53-mediated transcriptional activity is required for the induction of p53-dependent miRNAs (miR-145 and miR-34a). These miRNAs target a transcriptional regulator, Twist2, for suppression, thereby inhibiting the expression of the anti-inflammatory transcription factor c-Maf. In the absence of p53, microglia also fail to induce expression of miR-155, a miRNA that directly targets c-Maf. Thus, when p53 is activated in microglia by ROS, spontaneous DNA damage, or cellular stress associated with CNS disease and injury, the effectors of such activation work together to suppress the expression of c-Maf, thereby suppressing the anti-inflammation and tissue repair behaviors of microglia and making it more likely for microglia to adopt pro-inflammatory behaviors that can be injurious to surrounding neural cells.
Figure 8. Proposed model for p53 modulation of c-Maf expression.
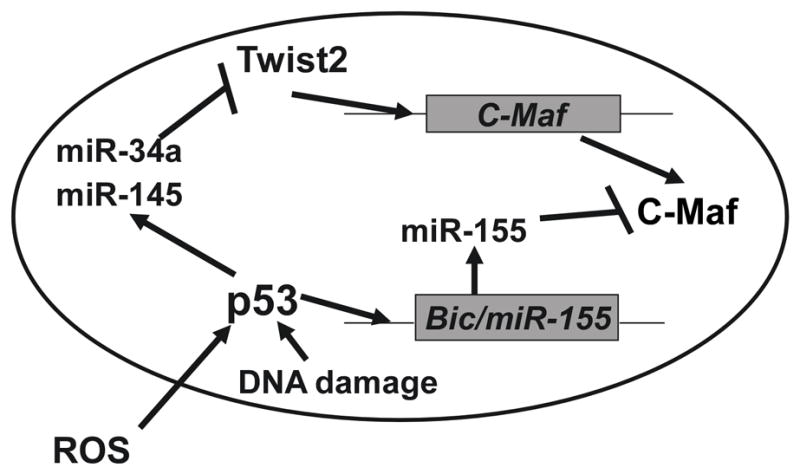
p53-mediated transcriptional activity is required for the induction of p53-dependent miRNAs (miR-145 and miR-34a). These miRNAs target a transcriptional regulator, Twist2, for suppression, thereby inhibiting the expression of the anti-inflammatory transcription factor c-Maf. In the absence of p53, microglia also fail to induce expression of miR-155, a miRNA that directly targets c-Maf. When p53 is activated in microglia by ROS, spontaneous DNA damage, or cellular stress associated with CNS disease and injury, the effectors of such activation work together to suppress the expression of c-Maf, thereby suppressing the anti-inflammation and tissue repair behaviors of microglia and making it more likely for microglia to adopt pro-inflammatory behaviors that can be injurious to surrounding neural cells.
A new role for p53 in promoting a pro-inflammatory response
P53 is a key modulator of stress responses becoming activated by physiological stressors, including but not limited to DNA damage (induced by UV, ionizing radiation (IR), or chemical agents such as hydrogen peroxide), oxidative stress, osmotic shock, ribonucleotide depletion, and deregulated oncogene expression (61, 62). Such activation is marked by two major events: a dramatic increase in the half-life of the p53 protein, and a conformational change in the structure that activates p53 transcriptional activity through phosphorylation of its N-terminal domain (63, 64). In microglia however, it appears that p53 influences gene transcription even in the absence of cellular stress. We previously reported that cultured microglia have demonstrable p53 transcriptional activity under basal conditions (13) and here we show that microglia extracted from uninjured adult brain demonstrate differential expression of c-Maf and its regulatory miRNAs. We and others have reported that p53 also influences microglia responses to inflammatory stimuli including the regulation of cytokine expression (13), inducible nitric oxide synthase expression and tumor necrosis factor alpha secretion (14). Inhibiting p53 mediated transcription in microglia prevented neurotoxicity suggesting that targeting of p53-mediated pathways in microglia may have therapeutic benefit in CNS injury and disease exacerbated by an over-exuberant inflammatory response.
c-Maf transcription factor and its role in anti-inflammatory responses in CNS
The Maf family of transcription factors consists of long and short members as well as a viral oncogene v-Maf (65). This family of Maf transcription factor has a variety of reported roles in regulating cellular differentiation (66). c-Maf is a long Maf family member that promotes the differentiation of Th2 lymphocytes (67) as well as expression of F4/80 (mouse homologue of EMR1: EGF-like module-containing mucin-like hormone receptor-like 1) (68) and an anti-inflammatory cytokine pattern in macrophages (33, 34, 42). It was also demonstrated that c-Maf plays a role in regulation of cell fate decision (69). Thus c-Maf transcriptional activity is a critical molecular determinant of functional differentiation in two different classes of leukocytes. As a critical transcriptional activator of anti-inflammatory cytokine genes, c-Maf must be suppressed during pro-inflammation and activated during anti-inflammation. Oxidative stress has been shown to negatively regulate c-Maf expression, but the precise mechanism by which this occurs has not been determined (69). Our findings suggest that repression of c-Maf is one means by which p53 influences the functional differentiation of microglia.
p53 promotes pro-inflammatory behavior in microglia through miR-155
Initially miR-155 was identified as a proinflammatory miRNA that contributes to macrophage activation by targeting anti-inflammatory genes (4, 22). Recent studies suggest that miR-155 promotes skewing toward pro-inflammatory behaviors by targeting anti-inflammatory genes like the IL-13 receptor (IL13Ra1) for degradation (4). MiR-155 also targets SMAD2, which is involved in the TGF-β anti-inflammatory signaling pathway (70, 71). Lastly, miR-155 inhibits CEBPβ, which was recently shown to be important for the expression of a number of genes associated with anti-inflammatory or “alternative” activation states such as Arg1, IL-10, IL13Rα1, and CD206 (72). In microglia, miR-155 has been shown to downregulate the expression of the suppressor of cytokine stimulus (SOCS)-1 expression (73), which also serves to promote pro-inflammatory behaviors. Here we show that in microglia, miR-155 is induced in a p53 dependent manner by pro-inflammatory cytokines. We also observed that in the absence of miR-155, c-Maf expression is increased, confirming that the previously reported role for miR-155 in lymphocytes is recapitulated in microglia. Thus, in the setting of oxidative stress (as occurs in chronic ischemia and neurodegenerative diseases), chronic p53 activation in microglia may suppress the anti-inflammatory response by promoting expression of miR-155. Here we demonstrate that neuroinflammation caused by brief CNS ischemia leads to activation of p53 mediated gene expression, induction of miR-155 and suppression of c-Maf mRNA in microglia. These findings support the hypothesis that p53 has a functional impact on an in vivo inflammatory response.
How p53 may regulate miR-155
MicroRNAs have an established role in regulating inflammatory responses (23). For example, bic/miR155 plays an important role in the differentiation of B, T, and Dendritic cells (36). Here we observed that induction of miR-155 expression by IFNγ treatment was significantly higher in p53+/+ microglia compared to p53−/− microglia. This suggests that induction of miR-155 is highly dependent on p53. The mechanism by which p53 regulates miR-155 expression has not been determined. It has been reported that p53 can directly regulate the transcription of certain miRNAs, but there is no clear consensus sequence for p53 binding to regulatory elements in proximity to the Bic/miR155 gene. An alternative mechanism by which p53 may modulate miR-155 expression is that p53 promotes post-transcriptional maturation of miRNAs (25). Drosha is a protein that mediates the processing of pri-miRNA transcripts into pre-miRNAs. For the maturation of a subset of miRNAs, Drosha requires RNA-associated proteins such as DEAD box RNA helicases p68 and p72 (also known as DDX17) to carry out its function. p53 promotes the Drosha-mediated processing of certain miRNAs with growth-suppressive functions, such as miR-16-1, miR-143 and miR-145, in cells in response to DNA damage (25). Therefore, the regulation of miR155 by p53 in microglia could be through post-transcriptional modulation.
p53 modulates c-Maf in part through Twist2
The Twist family of basic helix-loop-helix (bHLH) transcription factors, including Twist1 and Twist2, are highly conserved, key regulators of mesodermal differentiation, embryogenesis (74), epithelial-mesenchymal transition (EMT) (75), myeloid lineage development, as well as inflammatory responses of mature myeloid cells (50). Deficiency in Twist-1 or Twist-2 results in different phenotypes. Absence of Twist-1 are embryonic lethal in mice, due to a failure in neural tube closure, whereas Twist-2 KO mice develop a severe inflammatory syndrome and die within 2 weeks after birth (76, 77).
Here we report a novel discovery that p53 regulates c-Maf expression in microglia through inducing miRNAs that target a c-Maf transcriptional activator, Twist2. Several groups have reported that p53 can directly promote the transcription of the miR-34 family members (26, 27, 30) and our data demonstrate a functionally significant regulation of miR-34a and miR-145 by p53 in the regulation of Twist2. While Twist2 has not been specifically studied as a p53 target or in neuroinflammation, it was previously reported that Twist2 promotes the production of the anti-inflammatory cytokine IL-10 (50). Twist-2 KO mice demonstrated enhanced production of proinflammatory cytokines in combination with the reduced production of IL-10 and develop uncontrolled and eventually lethal inflammation (76). Several recent studies showed that the transcription factor c-Maf promotes expression of IL-10 as well as IL-4 (42, 78). Accordingly, it was also found that c-Maf mRNA levels in Twist2 KO dendritic cells were significantly lower than those in wild type cells (50). These studies strongly suggest that Twist2 is involved in suppressing inflammation by promoting the transcription of c-Maf. Our findings demonstrate that Twist2 is negatively regulated by p53 in cultured microglia and RAW cells.
We were not able to detect Twist2 expression in adult microglia, suggesting that this regulator of c-Maf may not be active in the healthy adult CNS. Additionally, though p53 targets were activated and miR-155 induced at 3 days following the ischemic insult, the miRNAs regulating Twist2 were not induced. Thus, the Twist2 arm of this regulatory mechanism may require longer survival times or alternate inflammatory stimuli to become active in the regulation of microglia c-Maf expression in vivo. Additionally, while experiments reported here demonstrate that miR-34a and miR-145 modulate Twist2 expression, further experimentation is needed to definitively determine if miR-34a and/or miR-145 directly interact with the 3′-UTR of Twist2. Nevertheless, the data presented here support an important functional role for p53 dependent miRNAs in the regulation of anti-inflammatory transcriptional regulators Twist2 and c-Maf; identifying a previously unreported epigenetic mechanism for modulating neuroinflammation.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Drs. Sean P. Murphy and Richard S. Morrison for helpful discussions during the performance of these experiments, Dr. Shahani Noor for training and assistance with ex-vivo flow cytometry, Diana Chao for performing the MCAO procedure, Amanda Case for training and assistance with primary microglia culture and Dr. David B. Wang for assistance with perfusions and editing the manuscript.
Footnotes
This work was supported by the National Institutes of Health (R01NS073848, R03NS70141 and R21NS062269 to G.A.G. with facilities support from P30-HD02274 to the UW Center on Human Development and Disability).
References
- 1.Aktas OUOI-DCNRZF. Neuronal damage in brain inflammation. Archives of Neurology. 2007;64:185–189. doi: 10.1001/archneur.64.2.185. [DOI] [PubMed] [Google Scholar]
- 2.Lynch M. The Multifaceted Profile of Activated Microglia. Molecular Neurobiology. 2009;40:139–156. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- 3.Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. Journal of Neuroscience Research. 2005;81:374–389. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- 4.Ponomarev ED, Veremeyko T, Weiner HL. MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia. 2013;61:91–103. doi: 10.1002/glia.22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rickard AJ, Young MJ. Corticosteroid receptors, macrophages and cardiovascular disease. Journal of Molecular Endocrinology. 2009;42:449–459. doi: 10.1677/JME-08-0144. [DOI] [PubMed] [Google Scholar]
- 6.Weber MS, Prod’homme T, Youssef S, Dunn SE, Rundle CD, Lee L, Patarroyo JC, Stuve O, Sobel RA, Steinman L, Zamvil SS. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat Med. 2007;13:935–943. doi: 10.1038/nm1620. [DOI] [PubMed] [Google Scholar]
- 7.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of Two Distinct Macrophage Subsets with Divergent Effects Causing either Neurotoxicity or Regeneration in the Injured Mouse Spinal Cord. The Journal of Neuroscience. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jebelli JD, Hooper C, Garden GA, Pocock JM. Emerging roles of p53 in glial cell function in health and disease. Glia. 2012;60:515–525. doi: 10.1002/glia.22268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lobsiger CS, Cleveland DW. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci. 2007;10:1355–1360. doi: 10.1038/nn1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendrysa SM, Ghassemifar S, Malek R. p53 in the CNS: Perspectives on Development, Stem Cells, and Cancer. Genes & Cancer. 2011;2:431–442. doi: 10.1177/1947601911409736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garden GA, Guo W, Jayadev S, Tun C, Balcaitis S, Choi J, Montine TJ, Möller T, Morrison RS. HIV associated neurodegeneration requires p53 in neurons and microglia. The FASEB Journal. 2004 doi: 10.1096/fj.04-1676fje. [DOI] [PubMed] [Google Scholar]
- 12.Jayadev S, Yun B, Nguyen H, Yokoo H, Morrison R, Garden G. The Glial Response to CNS HIV Infection Includes p53 Activation and Increased Expression of p53 Target Genes. Journal of Neuroimmune Pharmacology. 2007;2:359–370. doi: 10.1007/s11481-007-9095-x. [DOI] [PubMed] [Google Scholar]
- 13.Jayadev S, Nesser NK, Hopkins S, Myers SJ, Case A, Lee RJ, Seaburg LA, Uo T, Murphy SP, Morrison RS, Garden GA. Transcription factor p53 influences microglial activation phenotype. Glia. 2011;59:1402–1413. doi: 10.1002/glia.21178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davenport CM, I, Sevastou G, Hooper C, Pocock JM. Inhibiting p53 pathways in microglia attenuates microglial-evoked neurotoxicity following exposure to Alzheimer peptides. Journal of Neurochemistry. 2010;112:552–563. doi: 10.1111/j.1471-4159.2009.06485.x. [DOI] [PubMed] [Google Scholar]
- 15.Moestrup SK, Møller HJ. CD163: a regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Annals of medicine. 2004;36:347–354. doi: 10.1080/07853890410033171. [DOI] [PubMed] [Google Scholar]
- 16.Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, Sharon E, Spector Y, Bentwich Z. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- 17.Perruisseau-Carrier C, Jurga M, Forraz N, McGuckin C. miRNAs Stem Cell Reprogramming for Neuronal Induction and Differentiation. Molecular Neurobiology. 2011;43:215–227. doi: 10.1007/s12035-011-8179-z. [DOI] [PubMed] [Google Scholar]
- 18.Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-[alpha]-PU.1 pathway. Nat Med. 2011;17:64–70. doi: 10.1038/nm.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis BP, Burge CB, Bartel DP. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 20.Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A Pattern-Based Method for the Identification of MicroRNA Binding Sites and Their Corresponding Heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 21.He M, Xu Z, Ding T, Kuang DM, Zheng L. MicroRNA-155 Regulates Inflammatory Cytokine Production in Tumor-associated Macrophages via Targeting C/EBP[beta] Cell Mol Immunol. 2009;6:343–352. doi: 10.1038/cmi.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bird L. Immune regulation: MicroRNAs keep microglia quiet. Nat Rev Immunol. 2011;11:76–77. doi: 10.1038/nri2924. [DOI] [PubMed] [Google Scholar]
- 23.Dai R, Ahmed SA. MicroRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Translational Research. 2011;157:163–179. doi: 10.1016/j.trsl.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng Z, Zhang C, Wu R, Hu W. Tumor suppressor p53 meets microRNAs. Journal of Molecular Cell Biology. 2011;3:44–50. doi: 10.1093/jmcb/mjq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 26.Chang T-C, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee Kwang H, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, Arking Dan E, Beer MA, Maitra A, Mendell JT. Transactivation of miR-34a by p53 Broadly Influences Gene Expression and Promotes Apoptosis. Molecular Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional Activation of miR-34a Contributes to p53-Mediated Apoptosis. Molecular Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 29.Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G, Hermeking H. Differential Regulation of microRNAs by p53 Revealed by Massively Parallel Sequencing: miR-34a is a p53 Target That Induces Apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–1593. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 30.Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proceedings of the National Academy of Sciences. 2007;104:15472–15477. doi: 10.1073/pnas.0707351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K, Mo YY. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proceedings of the National Academy of Sciences. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sachdeva M, Mo YY. MicroRNA-145 Suppresses Cell Invasion and Metastasis by Directly Targeting Mucin 1. Cancer Research. 2010;70:378–387. doi: 10.1158/0008-5472.CAN-09-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Homma Y, Cao S, Shi X, Ma X. The Th2 transcription factor c-Maf inhibits IL-12p35 gene expression in activated macrophages by targeting NF-kappaB nuclear translocation. J Interferon Cytokine Res. 2007;27:799–808. doi: 10.1089/jir.2007.0006. [DOI] [PubMed] [Google Scholar]
- 34.Cao S, Liu J, Song L, Ma X. The Protooncogene c-Maf Is an Essential Transcription Factor for IL-10 Gene Expression in Macrophages. The Journal of Immunology. 2005;174:3484–3492. doi: 10.4049/jimmunol.174.6.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS, Allan B. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Dougan G, Turner M, Bradley A. Requirement of bic/microRNA-155 for Normal Immune Function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 38.Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, Chu X, Simon RP. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet. 2003;362:1028–1037. doi: 10.1016/S0140-6736(03)14412-1. [DOI] [PubMed] [Google Scholar]
- 39.Clark W, Gunion-Rinker L, Lessov N, Hazel K. Citicoline treatment for experimental intracerebral hemorrhage in mice. Stroke. 1998;29:2136–2140. doi: 10.1161/01.str.29.10.2136. [DOI] [PubMed] [Google Scholar]
- 40.de Haas AH, Boddeke HW, Brouwer N, Biber K. Optimized isolation enables ex vivo analysis of microglia from various central nervous system regions. Glia. 2007;55:1374–1384. doi: 10.1002/glia.20554. [DOI] [PubMed] [Google Scholar]
- 41.Möller T, Hanisch UK, Ransom BR. Thrombin-Induced Activation of Cultured Rodent Microglia. Journal of Neurochemistry. 2000;75:1539–1547. doi: 10.1046/j.1471-4159.2000.0751539.x. [DOI] [PubMed] [Google Scholar]
- 42.Cao S, Liu J, Chesi M, Bergsagel PL, Ho IC, Donnelly RP, Ma X. Differential Regulation of IL-12 and IL-10 Gene Expression in Macrophages by the Basic Leucine Zipper Transcription Factor c-Maf Fibrosarcoma. The Journal of Immunology. 2002;169:5715–5725. doi: 10.4049/jimmunol.169.10.5715. [DOI] [PubMed] [Google Scholar]
- 43.Rani A, Afzali B, Kelly A, Tewolde-Berhan L, Hackett M, Kanhere AS, Pedroza-Pacheco I, Bowen H, Jurcevic S, Jenner RG, Cousins DJ, Ragheb JA, Lavender P, John S. IL-2 Regulates Expression of C-MAF in Human CD4 T Cells. J Immunol. 2011;187:3721–3729. doi: 10.4049/jimmunol.1002354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proceedings of the National Academy of Sciences. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. Journal of Leukocyte Biology. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 46.Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 47.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 48.Liu Y, Tang FXC, Deng M, Chen P, Ji W, Zhang X, Gong L, Woodward Z, Liu J, Zhang L, Sun S, Liu JP, Wu K, Wu MX, Liu XL, Yu MB, Liu Y, Li DW-C. The Tumor Suppressor p53 Regulates c-Maf and Prox-1 to Control Lens Differentiation. Curr Mol Med. 2012;12:917–928. doi: 10.2174/156652412802480835. [DOI] [PubMed] [Google Scholar]
- 49.Mukhopadhyay S, Chen Y, Sankala M, Peiser L, Pikkarainen T, Kraal G, Tryggvason K, Gordon S. MARCO, an innate activation marker of macrophages, is a class3A scavenger receptor for Neisseria meningitidis. European Journal of Immunology. 2006;36:940–949. doi: 10.1002/eji.200535389. [DOI] [PubMed] [Google Scholar]
- 50.Sharabi AB, Aldrich M, Sosic D, Olson EN, Friedman AD, Lee SH, Chen SY. Twist-2 Controls Myeloid Lineage Development and Function. PLoS Biol. 2008;6:e316. doi: 10.1371/journal.pbio.0060316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zambelli F, Pesole G, Pavesi G. Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res. 2009;37:W247–252. doi: 10.1093/nar/gkp464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colton C, Mott R, Sharpe H, Xu Q, Van Nostrand W, Vitek M. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. Journal of Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Colton C. Heterogeneity of Microglial Activation in the Innate Immune Response in the Brain. Journal of Neuroimmune Pharmacology. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Colton C, Wilcock DM. Assessing activation states in microglia. CNS Neurol Disord Drug Targets. 2010;9:174–191. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- 55.Arnaud L, Robakis N, Figueiredo-Pereira M. It May Take Inflammation, Phosphorylation and Ubiquitination to “Tangle” in Alzheimer’s Disease. Neurodegenerative Dis. 2006;3:313–319. doi: 10.1159/000095638. [DOI] [PubMed] [Google Scholar]
- 56.Salmina AB. Neuron-Glia Interactions as Therapeutic Targets in Neurodegeneration. J Alzheimers Dis. 2009;16:485–502. doi: 10.3233/JAD-2009-0988. [DOI] [PubMed] [Google Scholar]
- 57.Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br J Pharmacol. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 59.Schmidt OI, Heyde CE, Ertel W, Stahel PF. Closed head injury—an inflammatory disease? Brain Res Rev. 2005;48:388–399. doi: 10.1016/j.brainresrev.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 60.Muir KW, Tyrrell P, Sattar N, Warburton E. Inflammation and ischaemic stroke. Curr Opin Neurol. 2007;20:334–342. doi: 10.1097/WCO.0b013e32813ba151. doi:310.1097/WCO.1090b1013e32813ba32151. [DOI] [PubMed] [Google Scholar]
- 61.Gottlieb TM, Oren M. p53 in growth control and neoplasia. Biochim Biophys Acta. 1996;1287:77–102. doi: 10.1016/0304-419x(95)00019-c. [DOI] [PubMed] [Google Scholar]
- 62.Eizenberg O, Faber-Elman A, Gottlieb E, Oren M, Rotter V, Schwartz M. p53 plays a regulatory role in differentiation and apoptosis of central nervous system-associated cells. Mol Cell Biol. 1996;16:5178–5185. doi: 10.1128/mcb.16.9.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haapajärvi T, Pitkänen K, Tsubari M, Laiho M. p53 transactivation and protein accumulation are independently regulated by UV light in different phases of the cell cycle. Mol Cell Biol. 1997;17:3074–3080. doi: 10.1128/mcb.17.6.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sakaguchi K, Herrera JE, Saito Si, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. DNA damage activates p53 through a phosphorylation–acetylation cascade. Genes Dev. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Motohashi H, Shavit JA, Igarashi K, Yamamoto M, Engel JD. The world according to Maf. Nucleic Acids Research. 1997;25:2953–2959. doi: 10.1093/nar/25.15.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kataoka K. Multiple Mechanisms and Functions of Maf Transcription Factors in the Regulation of Tissue-Specific Genes. Journal of Biochemistry. 2007;141:775–781. doi: 10.1093/jb/mvm105. [DOI] [PubMed] [Google Scholar]
- 67.Agnello D, Lankford CR, Bream J, Morinobu A, Gadina M, O’Shea J, Frucht D. Cytokines and Transcription Factors That Regulate T Helper Cell Differentiation: New Players and New Insights. Journal of Clinical Immunology. 2003;23:147–161. doi: 10.1023/a:1023381027062. [DOI] [PubMed] [Google Scholar]
- 68.Nakamura M, Hamada M, Hasegawa K, Kusakabe M, Suzuki H, Greaves DR, Moriguchi T, Kudo T, Takahashi S. c-Maf is essential for the F4/80 expression in macrophages in vivo. Gene. 2009;445:66–72. doi: 10.1016/j.gene.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 69.Nishikawa K, Nakashima T, Takeda S, Isogai M, Hamada M, Kimura A, Kodama T, Yamaguchi A, Owen MJ, Takahashi S, Takayanagi H. Maf promotes osteoblast differentiation in mice by mediating the age-related switch in mesenchymal cell differentiation. The Journal of Clinical Investigation. 2010;120:3455–3465. doi: 10.1172/JCI42528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Louafi F, Martinez-Nunez RT, Sanchez-Elsner T. MicroRNA-155 Targets SMAD2 and Modulates the Response of Macrophages to Transforming Growth Factor-β. Journal of Biological Chemistry. 2010;285:41328–41336. doi: 10.1074/jbc.M110.146852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martinez-Nunez RT, Louafi F, Sanchez-Elsner T. The Interleukin 13 (IL-13) Pathway in Human Macrophages Is Modulated by MicroRNA-155 via Direct Targeting of Interleukin 13 Receptor α1 (IL13Rα1) Journal of Biological Chemistry. 2011;286:1786–1794. doi: 10.1074/jbc.M110.169367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, Nerlov C. A CREB-C/EBPβ cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proceedings of the National Academy of Sciences. 2009;106:17475–17480. doi: 10.1073/pnas.0908641106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology. 2012;135:73–88. doi: 10.1111/j.1365-2567.2011.03514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 75.Ansieau S, Bastid J, Doreau A, Morel A-P, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, Maestro R, Voeltzel T, Selmi A, Valsesia-Wittmann S, Caron de Fromentel C, Puisieux A. Induction of EMT by Twist Proteins as a Collateral Effect of Tumor-Promoting Inactivation of Premature Senescence. Cancer Cell. 2008;14:79–89. doi: 10.1016/j.ccr.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 76.Šošić D, Richardson JA, Yu K, Ornitz DM, Olson EN. Twist Regulates Cytokine Gene Expression through a Negative Feedback Loop that Represses NF-κB Activity. Cell. 2003;112:169–180. doi: 10.1016/s0092-8674(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 77.Chen ZF, Behringer RR. twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes & Development. 1995;9:686–699. doi: 10.1101/gad.9.6.686. [DOI] [PubMed] [Google Scholar]
- 78.Ho IC, Lo D, Glimcher LH. c-maf Promotes T Helper Cell Type 2 (Th2) and Attenuates Th1 Differentiation by Both Interleukin 4–dependent and –independent Mechanisms. The Journal of Experimental Medicine. 1998;188:1859–1866. doi: 10.1084/jem.188.10.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.