

Abstract
Spinocerebellar ataxia type 7 (SCA7) is unique among CAG / polyglutamine (polyQ) repeat diseases due to dramatic intergenerational instability in repeat length and an associated cone-rod dystrophy retinal degeneration phenotype. SCA7 is caused by a polyQ expansion in the protein ataxin-7. Like other neurodegenerative diseases caused by polyQ expansion mutations, the spectrum of clinical severity and disease progression worsens with increasing polyQ length. Several potential mechanisms for the molecular pathogenesis of polyQ-expanded ataxin-7 have been suggested. These include, but are not limited to, alteration of endogenous ataxin-7 function, abnormal processing and stability of polyQ ataxin-7, and alteration of transcriptional regulation via interaction of polyQ-expanded ataxin-7 with other transcriptional regulators. Ataxin-7's normal function as a transcription factor may contribute to the selective vulnerability of specific cellular populations in SCA7, and the resolution of the mechanistic basis of this pathogenic cascade is a major focus of SCA7 disease research. PolyQ-expanded ataxin-7 can cause non-cell autonomous neurodegeneration in cerebellar Purkinje cells. Advances in understanding SCA7's molecular basis have led to important insights into cell-type specific neurodegeneration. We expect that further study of ataxin-7 normal function, insights into the molecular basis of SCA7 neurodegeneration, and the development of therapeutic interventions for SCA7 will greatly influence related endeavors directed at other CAG / polyQ repeat diseases.
Introduction
SCA7 is an autosomal dominant inherited neurodegenerative syndrome of progressive cerebellar ataxia and retinal degeneration with a wide geographic distribution (1-6). The pronounced anticipation (i.e. worsening disease severity in each successive generation that transmits the mutation) in affected pedigrees made SCA7 a very likely candidate for a causal triplet repeat mutation. Indeed, evidence for a polyglutamine (polyQ) expansion as the cause of SCA7 was established two years before the SCA7 disease gene was cloned (7). Clinical severity in SCA7 ranges from infantile-onset with early death to elderly presentations of slowly progressive isolated ataxia. The breadth of clinical presentation and natural history stems from the marked CAG repeat instability at the SCA7 locus. The unstable CAG repeat region responsible for SCA7 lies within the coding region for a protein known as ataxin-7 (8). Expansion of the polyQ tract in ataxin-7 leads to accumulation of the mutant protein in nuclear inclusions (NIs), and to selective neuronal and photoreceptor degeneration. This review will focus on describing the clinical, radiographic, and pathological features of SCA7, and will discuss current theories regarding how mutant ataxin-7 causes cellular toxicity and how mutant ataxin-7 may promote degeneration of specific hind brain structures and neuronal populations, including cerebellar Purkinje cells (PCs). Due to the focus of this Journal, a description of the cone-rod dystrophy retinal degeneration phenotype and discussion of specific theories of its pathogenesis are not included, but have been reviewed elsewhere (9).
Genetic Features of SCA7
Localization of the gene responsible for the SCA7 phenotype revealed linkage to chromosome 3p12-21.1 (10, 11). Further mapping of this region permitted investigators to test the hypothesis that a CAG repeat expansion was involved (7). Screening of CAG repeat sequences from the region of linkage yielded a novel gene that was named ‘ataxin-7’ (8). Initial studies of the ataxin-7 CAG repeat indicated that expansions beyond 37 CAG's produced the SCA7 disease phenotype, and normal individuals possess ataxin-7 repeats ranging in size from 7 to 34 CAG's (8). The ataxin-7 gene encoded a novel protein of (then) unknown function and typically contains a 10 CAG repeat that is translated into a polyglutamine tract in the ataxin-7 protein. This CAG repeat expands to between 37 and >300 triplets in SCA7 patients, and there is a strong inverse correlation between the age of disease onset and the length of the repeat mutation (Figure 1a), such that longer repeats tend to produce earlier ages of onset (5, 12, 13). There is also a correlation between repeat length and the type of clinical presentation in SCA7, with repeat mutations of < 59 CAGs often yielding initial cerebellar findings and those > 59 CAGs typically producing visual impairment as the first symptom (5).
Figure 1. Genetics and neuropathology of SCA7.


a) Anticipation in SCA7. A plot of age at onset as a function of CAG repeat length for 45 patients reveals a strong inverse correlation between allele size and age at onset (r = 0.74, P < 0.0001). Reprinted from Giunti et al., 1999, Am J Hum Genet 64, 1594. Used with permission.
b) Olivopontocerebellar atrophy in SCA7. MRI scans of two patients with SCA7, one with advanced disease (left) and one with mild disease (right), demonstrate marked and moderate cerebellar atrophy respectively, but show similar degrees of atrophy of the pons (circled). Reprinted from Bang et al., 2004, J Neurol Neurosurg Psych 75, 1452. Used with permission.
The Clinical and Pathological Features of Cerebellar Degeneration in SCA7
Prior to gene discovery, SCA7 was initially differentiated from the larger category of autosomal dominant cerebellar ataxia (ADCA) as ADCA type II, based on the presence of retinopathy (14). Currently, the only identified gene defect associated with ADCA type II is the unstable CAG repeat expansion in the coding region of the ataxin-7 gene (8). Almost all patients with ADCA type II have more than 36 CAG repeats on one ataxin-7 allele, and thus carry the diagnosis of ‘SCA7’. Cerebellar ataxia, which is manifested as difficulty with walking, manual dexterity and speech, is the most common clinical feature of SCA7 and is often the first reported symptom (15). SCA7 patients may eventually develop more extensive neurological deficits, including dysarthria, dysphagia, hypoacusis and eye movement abnormalities (slow saccades; staring) that can progress to frank ophthalmoplegia. Involvement of the cortico-spinal tracts resulting in exaggerated deep tendon reflexes, spasticity and extensor plantar reflexes may be present (13, 15). Because the causal gene in SCA7 is an unstable CAG repeat, SCA7 families demonstrate a pattern of genetic anticipation where subsequent generations develop more severe forms of the disease after inheriting longer repeat expansions than were present in their parents. When very long repeats are inherited, SCA7 may present with an early onset rapidly progressive juvenile form or even a very severe infantile form (12). Infantile-onset SCA7 is remarkable for its widespread disease pathology that includes organ systems outside the central nervous system (CNS).
Magnetic Resonance Imaging (MRI) of SCA7 patients typically reveals marked atrophy of the cerebellum and pons (Figure 1b), while a subset of patients may also demonstrate high T2 signal intensity in transverse pontine fibers (16). MRI-based volumetric analysis demonstrated that SCA7 patients have significantly more pontine atrophy than patients with other SCA's. Interestingly, it appears that volume loss (as measured by these methods) may develop in the pons prior to the development of cerebellar atrophy, suggesting that the primary site of disease onset may be in brainstem structures rather than in the cerebellar folia (17). Patients with SCA7 develop atrophy of the cerebellar cortex, dentate nucleus, inferior olive, subthalamic nucleus and olivocerebellar, spinocerebellar and pyramidal tracts (18, 19). Cerebellar tissue from SCA7 patients has evidence of extensive loss of cerebellar PCs, while only mild changes in the cerebellar granule cell layer are observed. Prominent neuronal loss with gliosis has been noted in the inferior olive (19). Neuronal loss has been reported in brainstem cranial nerve motor nuclei (III, IV, and XII), spinal motor neurons and regions of the basal ganglia, including the substantia nigra (20). Demyelination of the pyramidal tracts and of the posterior columns of the spinal cord may also occur (20).
One pathological feature of SCA7 that is commonly observed in neurodegenerative diseases associated with polyQ-expanded proteins is the presence of nuclear inclusions (NIs) (Figure 2). Mutant ataxin-7 has been localized to NIs, together with a host of other proteins including ubiquitin-proteasome components (21, 22). Several studies of SCA7 neuropathology in patients and in SCA7 mouse models have delineated the pattern of NI formation (21-26). While extensive NI formation in SCA7 is present in exquisitely vulnerable neuronal populations, such as the inferior olivary complex and basis pontis, the presence of NIs extends to other areas of the neuraxis – e.g. cerebral cortex – that are usually not subject to prominent degeneration (21, 27). By documenting a disconnection between NI formation and neuronal pathology, studies on SCA7 patient material contributed to an emerging view that inclusion body formation is not required for neuronal loss in polyQ diseases.
Figure 2. Nuclear inclusions in SCA7.

Here we see ataxin-7 antibody labeling of CNS sections revealing prominent nuclear inclusions (NIs) in a highly representative transgenic mouse model for SCA7 created by inserting an ataxin-7 CAG-92 cDNA into the murine prion protein expression vector (PrP-SCA7-c92Q). NIs form in cerebellar neurons in PrP-SCA7-c92Q mice (a), but not in cerebellar neurons from control transgenic PrP-SCA7-c24Q mice expressing normal length glutamine repeat tracts (b). Regions of the cortex, including the hippocampus, display widespread NI formation in SCA7 transgenic mice expressing ataxin-7 with 92 glutamines (c), but not in PrP-SCA7-c24Q controls (d). The basis pontis (e) and the inferior olivary nucleus (f), structures of pathophysiological relevance to SCA7, contain frequent NIs. At high power, intense nuclear immunoreactivity is noted (arrows) in neurons from PrP-SCA7-c92Q mice (g), but not from PrP-SCA7-c24Q control transgenic mice (h). Adapted from Garden et al., 2002, J Neurosci 22, 4897. Used with permission.
Mechanisms of SCA7 Neurodegeneration
Currently, our understanding of polyQ-associated neurodegeneration is based upon studies at the biochemical, molecular, genetic, and organismal level. Although the CAG / polyQ repeat disorders share many features, the different disease-causing proteins have no identifiable molecular similarities, except for an expanded polyglutamine tract. The threshold for disease-associated polyQ tract length in most of the CAG repeat diseases is nearly identical (between 35 and 40 repeats); and in all cases, the age of onset and severity of disease are inversely related to the repeat length. These observations suggest that long polyQ stretches possess inherent toxicity. It has been proposed that once the number of glutamines crosses a threshold of 35, the polyglutamine tract acquires an altered conformation (i.e. a “polar zipper”) composed of beta-pleated sheets folding back on one another (28). Validation of an unique conformation for long polyQ tracts came with production of the 1C2 antibody that could specifically detect expanded polyQ (7). The hypothesis that abnormally folded polyQ-containing proteins are an important step in disease pathogenesis gained prominence when analysis of human brain material from patients with SCA3 and a mouse model of Huntington's Disease (HD) detected NIs (29, 30). Based upon this work, the theory emerged that polyglutamine expansion tracts misfold, become resistant to degradation and accumulate as proteins aggregate. The ability of Congo Red to prevent polyQ neurotoxicity by inhibiting oligomerization and the toxicity of ‘diffuse’ mutant polyQ-expanded protein (and not ‘inclusions’) to produce cell death in cultured neurons support a model in which adoption of an altered conformation is the crucial event in polyQ disease pathogenesis (31, 32). Visible aggregates (or ‘inclusions’) are not toxic per se but instead may be correlated with the presence of misfolded oligomeric intermediates. Thus, the toxicity of the polyQ expansion tract relates to its propensity to misfold and to the formation of these toxic intermediates (33).
Proteolytic Cleavage and Turnover in SCA7 Pathogenesis
How misfolded and potentially toxic intermediates of mutant ataxin-7 result in specific toxicity in selectively vulnerable populations of neurons is not understood. One feature of polyQ disease pathogenesis, that may account for cell-type specificity in the various diseases, is proteolytic cleavage. Evidence exists for the preferential nuclear accumulation of truncated polyQ-containing fragments of mutant proteins in several polyQ diseases, including HD and SCA7 (24, 30, 34-36). It has been suggested that the production of truncated polyQ disease protein is a key step in disease pathogenesis. The importance of proteolytic cleavage has been most convincingly shown in HD, where protease cleavage processes have been carefully studied (37-41).
Studies of ataxin-7 in mouse models and cell culture support the existence of an amino-terminal truncated fragment in SCA7. An ∼ 55 kDa ataxin-7 fragment was detected with both an amino-terminal directed ataxin-7 antibody and the 1C2 antibody in one SCA7 transgenic mouse model and in SCA7 patients (24). Similar to huntingtin, ataxin-7 can be cleaved by caspase-7 (42), and caspase cleavage can modulate ataxin-7 cellular toxicity in vitro(43). Caspase-7 mediated cleavage would generate a short fragment containing the amino-terminus with the polyQ tract but without the nuclear export sequence (NES), potentially resulting in accumulation of a mutant ataxin-7 fragment in the nucleus. Compared to full-length mutant ataxin-7 protein, expression of a mutant ataxin-7 amino-terminal fragment similar in size to the truncation product found in vivo, had enhanced nuclear localization and cellular toxicity (44). It is not currently understood how the polyQ-expanded form of ataxin-7 causes degeneration in specific neuronal populations. Accumulation of misfolded or fragmented polyQ-expanded ataxin-7 might be envisioned to inhibit proteasome function and thereby produce a toxic cascade, perhaps specifically in certain neuronal cells with high metabolic demand and protein turnover. However, in vivo studies of the SCA7 knock-in mouse model demonstrate that neuronal dysfunction develops in the absence of ubiquitin-proteasome system impairment (45).
To examine how polyQ-expanded ataxin-7 might cause toxicity in cerebellar cells, the mutant protein was over-expressed in cultured cerebellar cells. This resulted in activation of the intrinsic apoptotic pathway with activation of caspase-9 and caspase-3, and ultimately DNA degradation consistent with apoptotic cell death (46). However, a significant component of the neurological dysfunction in SCA7 mice is likely due to reversible physiologic alterations that do not involve apoptotic neuronal loss. In vivo models of SCA7 develop ataxia and Purkinje dendrite degeneration in the absence of Purkinje cell apoptosis (24, 47). Furthermore, electrophysiological examination of hippocampal slice preparations from SCA7 knock-in mice demonstrated normal synaptic connectivity within the hippocampus but mild impairment of the immediate phase of long term potentiation (LTP), due in part to impairment in post-tetanic potentiation (PTP) (48). How the molecular mediators of cell death pathways participate in the sub-apoptotic neurodegeneration observed in mouse models of SCA7, or in the pathogenesis of human disease, remains an active area of research.
Ataxin-7 Normal Function May Relate to SCA7 Pathogenesis
As soon as a disease gene is identified, the next goal of molecular genetic research is to understand the normal biological function of the corresponding protein product. The normal function of a mutated protein often provides insight into the molecular basis of disease pathogenesis and pathways of interest for drug development. When the SCA7 disease gene was discovered, the function of ataxin-7 was unknown. Analysis of the amino acid sequence of ataxin-7 has revealed at least three nuclear localization signals (NLS's) (49, 50). Both nuclear and cytosolic localization of ataxin-7 has been observed (27, 51), and recent data demonstrate that ataxin-7 can shuttle into and out of the nucleus via a classic leucine-type nuclear export signal (NES) (44). The coexistence of functional NLS's and a functional NES in ataxin-7 suggests that it may shuttle another protein or other cargo, or may regulate subcellular localization of a key factor. Although its nuclear localization has been emphasized, ataxin-7 is prominent in the cytoplasm upon immunofluorescence or immunohistochemistry analysis (27, 51), suggesting that cytoplasmic or membrane-associated functions are also possible.
Recently, several studies have definitively shown that at least one function of ataxin-7 is as a core component of a transcription co-activator complex called STAGA. Transcription co-activator complexes are large protein complexes that mediate interactions between upstream transcription activators and the RNA polymerase II transcription complex (52, 53). One well-characterized complex that functions as a mediator between DNA-bound transcription activators and RNA polymerase II is the SAGA complex, originally defined in the yeast Saccharomyces cerevisiae(54). SAGA contains histone acetyltransferase (HAT) activity, mediated by the Gcn5 enzyme. Sgf73, a component of the SAGA complex with essential function in yeast is a homologue of human ataxin-7 (55). While the homology between ataxin-7 and Sgf73 is limited, human ataxin-7 can complement Sgf73 null yeast strains, indicating that the two proteins are functionally interchangeable (56). The identification of Sgf73 as a homologue of ataxin-7 prompted one group to determine if ataxin-7 is a subunit of the mammalian equivalent of the SAGA complex known as ‘STAGA’ (57), while another group's unbiased mass spectrometry sequencing analysis of STAGA-associated factors independently identified a 110 kDa protein as ataxin-7 (58). Biochemical studies of the STAGA complex subsequently confirmed that ataxin-7 is indeed a core component of STAGA and the closely related TATA-binding protein-free TAF containing complex (TFTC) (57, 58). Both STAGA and TFTC mediate transcription activation by promoting histone acetylation; however, STAGA has been shown to interact with certain transcription factors linked to particular nuclear enzymatic activities (59). The assignment of ataxin-7 to STAGA / TFTC raises questions about the effect of mutant ataxin-7 upon STAGA / TFTC complex function. Although the exact function of ataxin-7 in STAGA is unknown, polyQ expansion in ataxin-7 can alter the HAT activity of SAGA / STAGA / TFTC in yeast cells, retinal cells, and neuron-like cells (58, 60, 61). Furthermore, polyQ-expanded ataxin-7 expressed in rod photoreceptors produces severe chromatin decondensation (60). Thus, the polyQ tract expansion in ataxin-7 interferes with normal STAGA / TFTC function, perhaps by diminishing Gcn5 HAT enzymatic activity (58). Of course, one conundrum remains: Why would disruption of a member of a general transcription co-activator complex that is ubiquitously expressed produce a specific pattern of pathology? The answer will require that we identify STAGA-dependent transcription factors whose functional impairment may underlie the production of the SCA7 phenotype.
Transcriptional Regulation and SCA7 Pathogenesis
The realization that ataxin-7 is a transcription co-activator subunit further adds to the increasing body of evidence that many polyQ disease proteins are transcription factors or co-regulators. Indeed, of the nine polyQ disease proteins, seven are associated with prominent NIs, and for seven of nine, evidence for a role in transcription regulation has been uncovered. The huntingtin protein has been shown to regulate transcription through an interaction with repressor element-1 transcription factor (REST) in neurons and to interfere with transcription regulators such as CBP, Sp1, and TAFII-130 (62-64). A close homologue of atrophin-1 (the gene responsible for dentatorubral-pallidoluysian atrophy) in the fruitfly can function as a transcription co-repressor (65), and like the huntingtin protein, human atrophin-1 interferes with CBP-mediated transcription (63). Ataxin-1, the gene with a polyQ expansion in SCA1, may modulate transcription through an interaction with PQBP-1 (66), or function as a transcription co-repressor through its interaction with SMRT (67). Ataxin-3, the gene associated with SCA3, by binding to histone proteins, has been reported to interfere with the chromatin remodeling activities of transcription co-activators with HAT activity, in particular CBP and p300, and may therefore function as a transcription co-repressor (68). Indeed, ataxin-3 appears to directly interact with HDAC3 in the NCoR complex to repress transcription, and polyQ expansion of ataxin-3 may prevent proper recruitment and function of this deacetylase complex, yielding inappropriate transcription activation (69). Given all of these findings, the tendency of polyQ disease proteins to interact with polyQ-containing transcription factors, and the documentation of gene transcription alterations as an early event in disease pathogenesis in both mouse models and human patient material (70, 71), many polyglutamine diseases may be considered “transcriptionopathies” (72). That polyglutamine expansions expressed in yeast yield transcriptional changes resembling those observed in SAGA subunit mutants further supports this view (73).
Selective Neuronal Vulnerability in SCA7
Ataxin-7 is expressed throughout the CNS and in many other non-neuronal organ systems. Yet disease pathology in adult-onset SCA7 is specifically restricted to only certain populations of neurons. Thus, although an appreciation of the general mechanism of polyQ neurotoxicity is important for our understanding of SCA7 disease pathogenesis, accurate animal models of SCA7 have enabled us to address the basis of selective neuronal vulnerability. Since the regulatory elements controlling ataxin-7 gene expression are not well characterized, a number of groups have used heterologous promoter-expression systems to model SCA7 in mice. These models have provided useful insights into the nature of SCA7 neurodegeneration (24-26, 74). The murine prion protein promoter (MoPrP) SCA7 mouse model, in particular, has highlighted the importance of cell-cell interactions in polyQ disease neurodegeneration. Comparison of MoPrP-SCA7 transgenic mice expressing similar levels of ataxin-7-24Q and ataxin-7-92Q protein revealed onset of gait ataxia at 12 weeks of age in the PrP-SCA7-c92Q mice, while PrP-SCA7-c24Q mice are visibly normal beyond two years of age (24). Expression of transgenes under the control of the MoPrP vector is typically not observed in PCs of the cerebellum (75). Thus, in the MoPrP SCA7 mouse model, it was possible to determine if LACK of expression of polyQ-expanded ataxin-7 in PCs would protect them. Immunohistochemical analysis of cerebellar sections from the PrP-SCA7-c92Q line 6076 mice indicated that, under these circumstances, PCs are quite severely affected, with loss of calbindin staining becoming dramatically apparent by 8 weeks of age - even before the onset of a visible phenotype (24). Indeed, by 13 weeks of age, when most of these mice are just becoming visibly ataxic, calbindin staining reveals marked degeneration of PC dendritic arbors (Figure 3a). The dramatic PC degeneration is accompanied by extensive ataxin-7 antibody labeling of NIs in all neurons of the cerebellum, except for PCs. Furthermore, ultrastructural analysis of PCs from the SCA7 mouse model reveals marked histopathology of the PC soma with accompanying degenerative changes (47). These findings led us to conclude that PC degeneration in MoPrP-SCA7 mice is non cell-autonomous, and that non cell-autonomous processes could be operating in this and other polyQ repeat diseases.
Figure 3. Purkinje cell degeneration in SCA7 involves Bergmann glia dysfunction.
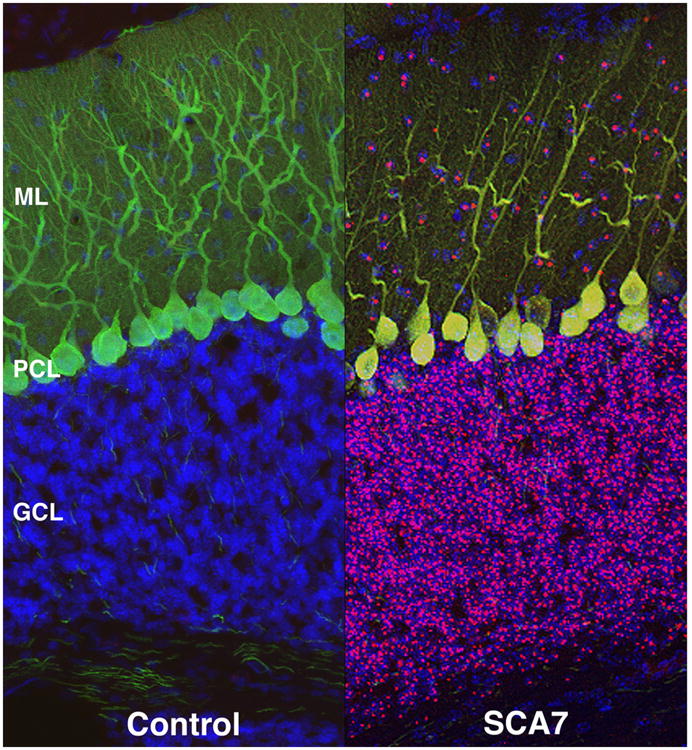

a) Non cell-autonomous SCA7 Purkinje cell degeneration. Confocal microscopy analysis of cerebellar sections from a PrP-SCA7-c92Q mouse (SCA7) and from a non-transgenic littermate (Control). Immunostaining with anti-ataxin-7 antibody (magenta), calbindin antibody (green), and DAPI (blue) yields normally oriented Purkinje cells with extensive dendritic arborization in the ‘Control’ mice. However, SCA7 transgenic mice display decreased dendritic arborization and displacement of Purkinje cell bodies, indicative of degeneration. While neurons in the granule cell layer (GCL) and the molecular layer (ML) contain aggregates of ataxin-7 protein, mutant ataxin-7 protein is not expressed in degenerating Purkinje cells. Degeneration of Purkinje cells without expression of mutant ataxin-7 protein is a so-called “non cell-autonomous” process. Adapted from Garden et al., 2002, J Neurosci 22, 4897. Used with permission.
b) Bergmann glia expression of mutant ataxin-7 protein is sufficient to produce Purkinje cell degeneration. Immunohistochemistry analysis of cerebellar sections from non-transgenic (NT), Gfa2-SCA7-10Q (10Q), and Gfa2-SCA7-92Q (92Q) mice was performed with anti-ataxin-7 (green) and anti-calbindin (red) antibodies. NT mice have normal cerebellar cytoarchitecture, as do 10Q mice. Reduced molecular layer (ML) thickness, misaligned Purkinje cells in the Purkinje cell layer (PCL), and tortuous Purkinje cell dendrites characterize the 92Q cerebellum. Note intense, punctate ataxin-7 immunostaining in the 92Q PCL (arrows), unlike the diffuse ataxin-7 staining observed in the 10Q PCL (arrows). GCL = granule cell layer. Adapted from Custer et al., 2006, Nat Neurosci 9, 1302. Used with permission.
What is the molecular basis of the non cell-autonomous PC degeneration observed in the MoPrP SCA7 mouse model? PCs are the only neurons in the cerebellum not expressing the polyQ-expanded ataxin-7, and yet, they are one of the most severely affected populations. In SCA7 patients, the PCs are also severely affected, and yet, NIs are not prominent in these cells (21). Indeed, examination of cerebellar sections from patients with SCA1, SCA2, SCA3, and DRPLA indicated that NIs are uncommon in PCs (76). Interestingly, in the MoPrP SCA7 mouse model, the surrounding Bergmann glia cells display extensive ultrastructural pathology (47). Since Bergmann glia are the cells that maintain glutamate and amino acid homeostasis in the molecular layer of the cerebellum, we hypothesized that the non-cell autonomous PC injury observed in the PrP-SCA7-c92Q mice might be secondary to Bergmann glia pathology. To test this hypothesis, transgenic mice with polyQ-expanded ataxin-7 under the control of a promoter (Gfa2) that drives expression in only Bergmann glia in the cerebellum were generated (47). Such Gfa2-SCA7-92Q mice do develop ataxia and PC degeneration, suggesting that Bergmann glial dysfunction may contribute to the extra-cellular alterations that produce PC degeneration in SCA7 (Figure 3b). Analysis of the Gfa2-SCA7-92Q mice indicated that mutant ataxin-7 is interfering with Bergmann glia glutamate uptake by impairing expression of the Bergmann glia-specific glutamate transporter GLAST. In this way, glutamate accumulation in PC synaptic clefts could produce excitotoxicity, resulting in PC dysfunction and degeneration, as evidenced by the pattern of dark cell degeneration in both MoPrP and Gfa2-92Q-ataxin-7 mice (Figure 4). However, Gfa2-92Q-ataxin-7 mice develop a later onset and more moderate ataxic phenotype compared to MoPrP-92Q-ataxin-7 mice, suggesting that Bergmann glial dysfunction can not be the only contributor to PC degeneration or the broader neurodegenerative phenotype in SCA7.
Figure 4. Dark cell degeneration in SCA7 transgenic mice.

Ultrastructural analysis of cerebellar sections reveals normal nuclear (Nu) and cytosolic organelles in non-transgenic control mice (a), but detects increased electron density in Purkinje cells from PrP-SCA7-92Q mice (b). Note swollen endoplasmic reticulum denuded of ribosomes (arrow) in inset from diseased 92Q Purkinje cells. Ultrastructural analysis of molecular layer yields Purkinje cell dendrites (PD) that are tightly opposed to Bergmann glia radial fibers in non-transgenic control mice (c), but PD display increased electron density and are adjacent to swollen Bergmann glia fibers (arrows), appearing as electron-transparent gaps, in 92Q cerebellar molecular layer (d). Note that interneuron (IN) dendrites, unlike PD, are not surrounded by electron-transparent gaps in 92Q mice. Adapted from Custer et al., 2006, Nat Neurosci 9, 1302. Used with permission.
Toward rationale therapies
Since SCA7 can cause non-cell autonomous PC degeneration, factors in the PC environment that contribute to normal function and promote survival may be altered by polyQ ataxin-7 expression in surrounding or connecting cells (Figure 5). In addition to the survival promoting effects of Bergmann glia, intact neural circuits are necessary to maintain normal PC structure and function (77, 78). Excitatory afferent input to PC's is provided by climbing fibers from the inferior olive and parallel fibers from granule neurons. In addition to these excitatory inputs, there are also inhibitory inputs from basket and stellate cells. Since glutamate uptake is impaired in ataxin-7-92Q expressing Bergmann glia, an alteration in the balance between excitatory and inhibitory inputs may underlie the more rapid onset of ataxia and neurodegeneration observed in the MoPrP-SCA7-c92Q mice. Future studies, aimed at altering the balance of excitatory to inhibitory neurotransmission by pharmacological means or attempting to increase GLAST expression via ceftriaxone treatment or viral gene transfer (79, 80), are needed.
Figure 5. Model for non-cell autonomous Purkinje cell degeneration in SCA7.

In mice expressing ataxin-7-92Q under the control of the MoPrP promoter, Bergmann glia (purple), granule neurons (blue), and inferior olivary neurons (magenta) all express mutant ataxin-7 and develop nuclear accumulations of mutant protein detectable as aggregates (brown). Purkinje cell neurons (green), however, do not express mutant ataxin-7 protein, but nonetheless degenerate with atrophy, loss of dendrites, and ultrastructural changes, consistent with dark cell degeneration. This is due to a reduction in glutamate uptake by Bergmann glia, together with probable decreases in the trophic support that is normally provided by Bergmann glia (i.e. GDNF) and climbing fibers (i.e. IGF-1) from the inferior olive. In mice expressing ataxin-7-92Q under the control of the Gfa2 promoter, only Bergmann glia express polyQ-expanded ataxin-7 protein. In Gfa2-92Q mice, Purkinje cell neurons still undergo the same pattern of degeneration, though the onset is delayed and progression is retarded.
Another survival promoting action of Bergmann glia that may be altered in SCA7 is the secretion of neurotrophic factors that promote PC survival. Two neurotrophic factors, glial derived neurotrophic factor (GDNF) and insulin like growth factor-1 (IGF-1), play important roles in PC survival. GDNF, synthesized by Bergmann glia, promotes the survival and differentiation of PC's (81, 82). GDNF treatment is protective in several models of excitotoxic neuronal injury (83-86). Chronic intra-ventricular infusion of GDNF also protects shaker mutant rats from PC degeneration (87, 88). Taken together, these studies support the hypothesis that GDNF may not only play an important role in supporting adult PC survival, but may also protect PCs from excitotoxic stimuli. IGF-1 is synthesized both by PCs and inferior olive neurons, which deliver IGF-1 to the molecular layer by anterograde transport (89, 90). Several studies have demonstrated that IGF-1 can be neuroprotective against a variety of neurotoxic insults (91-94). IGF-1 may also modulate the excitatory environment of PCs through the regulation of glial glutamate transporters on Bergmann glia (95). Exogenously delivered IGF-1 promotes renervation and functional recovery after inferior olive lesion (96), and acts synergistically with GDNF to prevent PC degeneration in shaker mutant rats (87). Endogenous IGF-1 may promote PC survival and provide protection from excitotoxic insults, and exogenously administered IGF-1 may generate a neuroprotective feed forward loop by promoting renervation of climbing fiber afferents, thereby bringing more IGF-1 to the molecular layer to promote PC survival and Bergmann glia glutamate uptake. Delivery of exogenous GDNF and IGF-1 to SCA7 mouse models could be employed to determine if the SCA7 phenotype may be delayed (or prevented) by ameliorating environmental changes resulting from loss of normal Bergmann glia function and inferior olive input. If this hypothesis is correct, then it will be possible to prevent PC degeneration and ataxia in SCA7 patients with specific neurotrophic factors.
Acknowledgments
Our research on SCA7 is supported by a grant from the National Institutes of Health (EY14061 to ARL) and the National Organization for Rare Disorders (GAG).
References
- 1.Abe T, Tsuda T, Yoshida M, Wada Y, Kano T, Itoyama Y, et al. Macular degeneration associated with aberrant expansion of trinucleotide repeat of the SCA7 gene in 2 Japanese families. Arch Ophthalmol. 2000;118:1415–21. doi: 10.1001/archopht.118.10.1415. [DOI] [PubMed] [Google Scholar]
- 2.Bryer A, Krause A, Bill P, Davids V, Bryant D, Butler J, et al. The hereditary adult-onset ataxias in South Africa. J Neurol Sci. 2003;216:47–54. doi: 10.1016/s0022-510x(03)00209-0. [DOI] [PubMed] [Google Scholar]
- 3.Gu W, Wang Y, Liu X, Zhou B, Zhou Y, Wang G. Molecular and clinical study of spinocerebellar ataxia type 7 in Chinese kindreds. Arch Neurol. 2000;57:1513–8. doi: 10.1001/archneur.57.10.1513. [DOI] [PubMed] [Google Scholar]
- 4.Jardim LB, Silveira I, Pereira ML, Ferro A, Alonso I, do Ceu Moreira M, et al. A survey of spinocerebellar ataxia in South Brazil - 66 new cases with Machado-Joseph disease, SCA7, SCA8, or unidentified disease-causing mutations. J Neurol. 2001;248:870–6. doi: 10.1007/s004150170072. [DOI] [PubMed] [Google Scholar]
- 5.Johansson J, Forsgren L, Sandgren O, Brice A, Holmgren G, Holmberg M. Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation. Hum Mol Genet. 1998;7:171–6. doi: 10.1093/hmg/7.2.171. [DOI] [PubMed] [Google Scholar]
- 6.Storey E, du Sart D, Shaw JH, Lorentzos P, Kelly L, McKinley Gardner RJ, et al. Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients with spinocerebellar ataxia. Am J Med Genet. 2000;95:351–7. doi: 10.1002/1096-8628(20001211)95:4<351::aid-ajmg10>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 7.Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, et al. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature. 1995;378:403–6. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]
- 8.David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17:65–70. doi: 10.1038/ng0997-65. [DOI] [PubMed] [Google Scholar]
- 9.Garden GA, Truant R, Ellerby LM, La Spada AR. Genetic Instabilities and Hereditary Neurological Diseases. Academic Press; San Diego, CA: 2006. Spinocerebellar Ataxia Type 7: Clinical Features to Cellular Pathogenesis. [Google Scholar]
- 10.Benomar A, Krols L, Stevanin G, Cancel G, LeGuern E, David G, et al. The gene for autosomal dominant cerebellar ataxia with pigmentary macular dystrophy maps to chromosome 3p12-p21.1. Nat Genet. 1995;10:84–8. doi: 10.1038/ng0595-84. [DOI] [PubMed] [Google Scholar]
- 11.Holmberg M, Johansson J, Forsgren L, Heijbel J, Sandgren O, Holmgren G. Localization of autosomal dominant cerebellar ataxia associated with retinal degeneration and anticipation to chromosome 3p12-p21.1. Hum Mol Genet. 1995;4:1441–5. doi: 10.1093/hmg/4.8.1441. [DOI] [PubMed] [Google Scholar]
- 12.Benton CS, de Silva R, Rutledge SL, Bohlega S, Ashizawa T, Zoghbi HY. Molecular and clinical studies in SCA-7 define a broad clinical spectrum and the infantile phenotype. Neurology. 1998;51:1081–6. doi: 10.1212/wnl.51.4.1081. [DOI] [PubMed] [Google Scholar]
- 13.David G, Durr A, Stevanin G, Cancel G, Abbas N, Benomar A, et al. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7) Hum Mol Genet. 1998;7:165–70. doi: 10.1093/hmg/7.2.165. [DOI] [PubMed] [Google Scholar]
- 14.Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–5. doi: 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- 15.Giunti P, Stevanin G, Worth PF, David G, Brice A, Wood NW. Molecular and clinical study of 18 families with ADCA type II: evidence for genetic heterogeneity and de novo mutation. Am J Hum Genet. 1999;64:1594–603. doi: 10.1086/302406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bang OY, Huh K, Lee PH, Kim HJ. Clinical and neuroradiological features of patients with spinocerebellar ataxias from Korean kindreds. Arch Neurol. 2003;60:1566–74. doi: 10.1001/archneur.60.11.1566. [DOI] [PubMed] [Google Scholar]
- 17.Bang OY, Lee PH, Kim SY, Kim HJ, Huh K. Pontine atrophy precedes cerebellar degeneration in spinocerebellar ataxia 7: MRI-based volumetric analysis. J Neurol Neurosurg Psychiatry. 2004;75:1452–6. doi: 10.1136/jnnp.2003.029819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin JJ, Van Regemorter N, Krols L, Brucher JM, de Barsy T, Szliwowski H, et al. On an autosomal dominant form of retinal-cerebellar degeneration: an autopsy study of five patients in one family. Acta Neuropathol (Berl) 1994;88:277–86. doi: 10.1007/BF00310370. [DOI] [PubMed] [Google Scholar]
- 19.Michalik A, Martin JJ, Van Broeckhoven C. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet. 2004;12:2–15. doi: 10.1038/sj.ejhg.5201108. [DOI] [PubMed] [Google Scholar]
- 20.Martin J, Van Regemorter N, Del-Favero J, Lofgren A, Van Broeckhoven C. Spinocerebellar ataxia type 7 (SCA7) - correlations between phenotype and genotype in one large Belgian family. J Neurol Sci. 1999;168:37–46. doi: 10.1016/s0022-510x(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 21.Holmberg M, Duyckaerts C, Durr A, Cancel G, Gourfinkel-An I, Damier P, et al. Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions. Hum Mol Genet. 1998;7:913–8. doi: 10.1093/hmg/7.5.913. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi J, Fujigasaki H, Zander C, El Hachimi KH, Stevanin G, Durr A, et al. Two populations of neuronal intranuclear inclusions in SCA7 differ in size and promyelocytic leukaemia protein content. Brain. 2002;125:1534–43. doi: 10.1093/brain/awf154. [DOI] [PubMed] [Google Scholar]
- 23.Einum DD, Townsend JJ, Ptacek LJ, Fu YH. Ataxin-7 expression analysis in controls and spinocerebellar ataxia type 7 patients. Neurogenetics. 2001;3:83–90. doi: 10.1007/s100480000100. [DOI] [PubMed] [Google Scholar]
- 24.Garden GA, Libby RT, Fu YH, Kinoshita Y, Huang J, Possin DE, et al. Polyglutamine-expanded ataxin-7 promotes non-cell-autonomous purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci. 2002;22:4897–905. doi: 10.1523/JNEUROSCI.22-12-04897.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.La Spada AR, Fu Y, Sopher BL, Libby RT, Wang X, Li LY, et al. Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron. 2001;31:913–27. doi: 10.1016/s0896-6273(01)00422-6. [DOI] [PubMed] [Google Scholar]
- 26.Yvert G, Lindenberg KS, Devys D, Helmlinger D, Landwehrmeyer GB, Mandel JL. SCA7 mouse models show selective stabilization of mutant ataxin-7 and similar cellular responses in different neuronal cell types. Hum Mol Genet. 2001;10:1679–92. doi: 10.1093/hmg/10.16.1679. [DOI] [PubMed] [Google Scholar]
- 27.Lindenberg KS, Yvert G, Muller K, Landwehrmeyer GB. Expression analysis of ataxin-7 mRNA and protein in human brain: evidence for a widespread distribution and focal protein accumulation. Brain Pathol. 2000;10:385–94. doi: 10.1111/j.1750-3639.2000.tb00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994;91:5355–8. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–48. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 30.Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, et al. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–44. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 31.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 32.Sanchez I, Mahlke C, Yuan J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 2003;421:373–9. doi: 10.1038/nature01301. [DOI] [PubMed] [Google Scholar]
- 33.Poirier MA, Li H, Macosko J, Cai S, Amzel M, Ross CA. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J Biol Chem. 2002;277:41032–7. doi: 10.1074/jbc.M205809200. [DOI] [PubMed] [Google Scholar]
- 34.Li M, Miwa S, Kobayashi Y, Merry DE, Yamamoto M, Tanaka F, et al. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol. 1998;44:249–54. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- 35.Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 36.Schilling G, Wood JD, Duan K, Slunt HH, Gonzales V, Yamada M, et al. Nuclear accumulation of truncated atrophin-1 fragments in a transgenic mouse model of DRPLA. Neuron. 1999;24:275–86. doi: 10.1016/s0896-6273(00)80839-9. [DOI] [PubMed] [Google Scholar]
- 37.Gafni J, Ellerby LM. Calpain activation in Huntington's disease. J Neurosci. 2002;22:4842–9. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, et al. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet. 1996;13:442–9. doi: 10.1038/ng0896-442. [DOI] [PubMed] [Google Scholar]
- 39.Kim YJ, Yi Y, Sapp E, Wang Y, Cuiffo B, Kegel KB, et al. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci U S A. 2001;98:12784–9. doi: 10.1073/pnas.221451398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, et al. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem. 1998;273:9158–67. doi: 10.1074/jbc.273.15.9158. [DOI] [PubMed] [Google Scholar]
- 41.Wellington CL, Singaraja R, Ellerby L, Savill J, Roy S, Leavitt B, et al. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem. 2000;275:19831–8. doi: 10.1074/jbc.M001475200. [DOI] [PubMed] [Google Scholar]
- 42.Ellerby LM, Hackam AS, Propp SS, Ellerby HM, Rabizadeh S, Cashman NR, et al. Kennedy's disease: caspase cleavage of the androgen receptor is a crucial event in cytotoxicity. J Neurochem. 1999;72:185–95. doi: 10.1046/j.1471-4159.1999.0720185.x. [DOI] [PubMed] [Google Scholar]
- 43.Young JE, Gouw LG, Propp SS, Lin A, Hermel E, Logvinova A, et al. Proteolytic cleavage of ataxin-7 by caspase-7 modulates cellular toxicity and transcriptional dysregulation. 2005 doi: 10.1074/jbc.M705265200. Submitted for Publication. [DOI] [PubMed] [Google Scholar]
- 44.Taylor J, Grote SK, Xia J, Vandelft M, Graczyk J, Ellerby LM, et al. Ataxin-7 can export from the nucleus via a conserved exportin-dependent signal. J Biol Chem. 2006;281:2730–9. doi: 10.1074/jbc.M506751200. [DOI] [PubMed] [Google Scholar]
- 45.Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet. 2005;14:679–91. doi: 10.1093/hmg/ddi064. [DOI] [PubMed] [Google Scholar]
- 46.Wang HL, Yeh TH, Chou AH, Kuo YL, Luo LJ, He CY, et al. Polyglutamine-expanded ataxin-7 activates mitochondrial apoptotic pathway of cerebellar neurons by upregulating Bax and downregulating Bcl-x(L) Cell Signal. 2005 doi: 10.1016/j.cellsig.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 47.Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, et al. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9:1302–11. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- 48.Yoo SY, Pennesi ME, Weeber EJ, Xu B, Atkinson R, Chen S, et al. SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron. 2003;37:383–401. doi: 10.1016/s0896-6273(02)01190-x. [DOI] [PubMed] [Google Scholar]
- 49.Chen S, Peng GH, Wang X, Smith AC, Grote SK, Sopher BL, et al. Interference of Crx-dependent transcription by ataxin-7 involves interaction between the glutamine regions and requires the ataxin-7 carboxy-terminal region for nuclear localization. Hum Mol Genet. 2004;13:53–67. doi: 10.1093/hmg/ddh005. [DOI] [PubMed] [Google Scholar]
- 50.Kaytor MD, Duvick LA, Skinner PJ, Koob MD, Ranum LP, Orr HT. Nuclear localization of the spinocerebellar ataxia type 7 protein, ataxin-7. Hum Mol Genet. 1999;8:1657–64. doi: 10.1093/hmg/8.9.1657. [DOI] [PubMed] [Google Scholar]
- 51.Cancel G, Duyckaerts C, Holmberg M, Zander C, Yvert G, Lebre AS, et al. Distribution of ataxin-7 in normal human brain and retina. Brain. 2000;123 Pt 12:2519–30. doi: 10.1093/brain/123.12.2519. [DOI] [PubMed] [Google Scholar]
- 52.Blazek E, Mittler G, Meisterernst M. The mediator of RNA polymerase II. Chromosoma. 2005;113:399–408. doi: 10.1007/s00412-005-0329-5. [DOI] [PubMed] [Google Scholar]
- 53.Conaway JW, Florens L, Sato S, Tomomori-Sato C, Parmely TJ, Yao T, et al. The mammalian Mediator complex. FEBS Lett. 2005;579:904–8. doi: 10.1016/j.febslet.2004.11.031. [DOI] [PubMed] [Google Scholar]
- 54.Timmers HT, Tora L. SAGA unveiled. Trends Biochem Sci. 2005;30:7–10. doi: 10.1016/j.tibs.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 55.Sanders SL, Jennings J, Canutescu A, Link AJ, Weil PA. Proteomics of the eukaryotic transcription machinery: identification of proteins associated with components of yeast TFIID by multidimensional mass spectrometry. Mol Cell Biol. 2002;22:4723–38. doi: 10.1128/MCB.22.13.4723-4738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McMahon SJ, Pray-Grant MG, Schieltz D, Yates JR, 3rd, Grant PA. Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SAGA and SLIK histone acetyltransferase activity. Proc Natl Acad Sci U S A. 2005;102:8478–82. doi: 10.1073/pnas.0503493102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Helmlinger D, Hardy S, Sasorith S, Klein F, Robert F, Weber C, et al. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet. 2004;13:1257–65. doi: 10.1093/hmg/ddh139. [DOI] [PubMed] [Google Scholar]
- 58.Palhan VB, Chen S, Peng GH, Tjernberg A, Gamper AM, Fan Y, et al. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci U S A. 2005;102:8472–7. doi: 10.1073/pnas.0503505102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martinez E, Palhan VB, Tjernberg A, Lymar ES, Gamper AM, Kundu TK, et al. Human STAGA complex is a chromatin-acetylating transcription coactivator that interacts with pre-mRNA splicing and DNA damage-binding factors in vivo. Mol Cell Biol. 2001;21:6782–95. doi: 10.1128/MCB.21.20.6782-6795.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Helmlinger D, Hardy S, Abou-Sleymane G, Eberlin A, Bowman AB, Gansmuller A, et al. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol. 2006;4:e67. doi: 10.1371/journal.pbio.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strom AL, Forsgren L, Holmberg M. A role for both wild-type and expanded ataxin-7 in transcriptional regulation. Neurobiol Dis. 2005;20:646–55. doi: 10.1016/j.nbd.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 62.Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, et al. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–43. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 63.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–8. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 64.Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 65.Zhang S, Xu L, Lee J, Xu T. Drosophila atrophin homolog functions as a transcriptional corepressor in multiple developmental processes. Cell. 2002;108:45–56. doi: 10.1016/s0092-8674(01)00630-4. [DOI] [PubMed] [Google Scholar]
- 66.Okazawa H, Rich T, Chang A, Lin X, Waragai M, Kajikawa M, et al. Interaction between mutant ataxin-1 and PQBP-1 affects transcription and cell death. Neuron. 2002;34:701–13. doi: 10.1016/s0896-6273(02)00697-9. [DOI] [PubMed] [Google Scholar]
- 67.Tsai CC, Kao HY, Mitzutani A, Banayo E, Rajan H, McKeown M, et al. Ataxin 1, a SCA1 neurodegenerative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc Natl Acad Sci U S A. 2004;101:4047–52. doi: 10.1073/pnas.0400615101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li F, Macfarlan T, Pittman RN, Chakravarti D. Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities. J Biol Chem. 2002;277:45004–12. doi: 10.1074/jbc.M205259200. [DOI] [PubMed] [Google Scholar]
- 69.Evert BO, Araujo J, Vieira-Saecker AM, de Vos RA, Harendza S, Klockgether T, et al. Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J Neurosci. 2006;26:11474–86. doi: 10.1523/JNEUROSCI.2053-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lin X, Antalffy B, Kang D, Orr HT, Zoghbi HY. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci. 2000;3:157–63. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- 71.Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, et al. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9:1259–71. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- 72.La Spada AR, Taylor JP. Polyglutamines placed into context. Neuron. 2003;38:681–4. doi: 10.1016/s0896-6273(03)00328-3. [DOI] [PubMed] [Google Scholar]
- 73.Hughes RE, Lo RS, Davis C, Strand AD, Neal CL, Olson JM, et al. Altered transcription in yeast expressing expanded polyglutamine. Proc Natl Acad Sci U S A. 2001;98:13201–6. doi: 10.1073/pnas.191498198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yvert G, Lindenberg KS, Picaud S, Landwehrmeyer GB, Sahel JA, Mandel JL. Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum Mol Genet. 2000;9:2491–506. doi: 10.1093/hmg/9.17.2491. [DOI] [PubMed] [Google Scholar]
- 75.Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, et al. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. Embo J. 1996;15:1255–64. [PMC free article] [PubMed] [Google Scholar]
- 76.Koyano S, Iwabuchi K, Yagishita S, Kuroiwa Y, Uchihara T. Paradoxical absence of nuclear inclusion in cerebellar Purkinje cells of hereditary ataxias linked to CAG expansion. J Neurol Neurosurg Psychiatry. 2002;73:450–2. doi: 10.1136/jnnp.73.4.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Strahlendorf JC, Brandon T, Miles R, Strahlendorf HK. AMPA receptor-mediated alterations of intracellular calcium homeostasis in rat cerebellar Purkinje cells in vitro: correlates to dark cell degeneration. Neurochem Res. 1998;23:1355–62. doi: 10.1023/a:1020742404945. [DOI] [PubMed] [Google Scholar]
- 78.Brorson JR, Manzolillo PA, Gibbons SJ, Miller RJ. AMPA receptor desensitization predicts the selective vulnerability of cerebellar Purkinje cells to excitotoxicity. J Neurosci. 1995;15:4515–24. doi: 10.1523/JNEUROSCI.15-06-04515.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–7. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 80.Xia H, Mao Q, Eliason SL, Harper SQ, Martins IH, Orr HT, et al. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med. 2004;10:816–20. doi: 10.1038/nm1076. [DOI] [PubMed] [Google Scholar]
- 81.Mount HT, Dean DO, Alberch J, Dreyfus CF, Black IB. Glial cell line-derived neurotrophic factor promotes the survival and morphologic differentiation of Purkinje cells. Proc Natl Acad Sci U S A. 1995;92:9092–6. doi: 10.1073/pnas.92.20.9092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Springer JE, Mu X, Bergmann LW, Trojanowski JQ. Expression of GDNF mRNA in rat and human nervous tissue. Exp Neurol. 1994;127:167–70. doi: 10.1006/exnr.1994.1091. [DOI] [PubMed] [Google Scholar]
- 83.Alberch J, Perez-Navarro E, Canals JM. Neuroprotection by neurotrophins and GDNF family members in the excitotoxic model of Huntington's disease. Brain Res Bull. 2002;57:817–22. doi: 10.1016/s0361-9230(01)00775-4. [DOI] [PubMed] [Google Scholar]
- 84.Bonde C, Kristensen BW, Blaabjerg M, Johansen TE, Zimmer J, Meyer M. GDNF and neublastin protect against NMDA-induced excitotoxicity in hippocampal slice cultures. Neuroreport. 2000;11:4069–73. doi: 10.1097/00001756-200012180-00032. [DOI] [PubMed] [Google Scholar]
- 85.Cheng H, Fu YS, Guo JW. Ability of GDNF to diminish free radical production leads to protection against kainate-induced excitotoxicity in hippocampus. Hippocampus. 2004;14:77–86. doi: 10.1002/hipo.10145. [DOI] [PubMed] [Google Scholar]
- 86.Gratacos E, Perez-Navarro E, Tolosa E, Arenas E, Alberch J. Neuroprotection of striatal neurons against kainate excitotoxicity by neurotrophins and GDNF family members. J Neurochem. 2001;78:1287–96. doi: 10.1046/j.1471-4159.2001.00538.x. [DOI] [PubMed] [Google Scholar]
- 87.Tolbert DL, Clark BR. GDNF and IGF-I trophic factors delay hereditary Purkinje cell degeneration and the progression of gait ataxia. Exp Neurol. 2003;183:205–19. doi: 10.1016/s0014-4886(03)00172-9. [DOI] [PubMed] [Google Scholar]
- 88.Tolbert DL, Bradley MW, Tolod EG, Torres-Aleman I, Clark BR. Chronic intraventricular infusion of glial cell line-derived neurotrophic factor (GDNF) rescues some cerebellar Purkinje cells from heredodegeneration. Exp Neurol. 2001;170:375–9. doi: 10.1006/exnr.2001.7718. [DOI] [PubMed] [Google Scholar]
- 89.Nieto-Bona MP, Garcia-Segura LM, Torres-Aleman I. Orthograde transport and release of insulin-like growth factor I from the inferior olive to the cerebellum. J Neurosci Res. 1993;36:520–7. doi: 10.1002/jnr.490360504. [DOI] [PubMed] [Google Scholar]
- 90.Aguado F, Sanchez-Franco F, Cacidedo L, Fernandez T, Rodrigo J, Martinez-Murillo R. Subcellular localization of insulin-like growth factor I (IGF-I) in Purkinje cells of the adult rat: an immunocytochemical study. Neurosci Lett. 1992;135:171–4. doi: 10.1016/0304-3940(92)90429-b. [DOI] [PubMed] [Google Scholar]
- 91.Digicaylioglu M, Garden G, Timberlake S, Fletcher L, Lipton SA. Acute neuroprotective synergy of erythropoietin and insulin-like growth factor I. Proc Natl Acad Sci U S A. 2004;101:9855–60. doi: 10.1073/pnas.0403172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Escartin C, Boyer F, Bemelmans AP, Hantraye P, Brouillet E. Insulin growth factor-1 protects against excitotoxicity in the rat striatum. Neuroreport. 2004;15:2251–2254. doi: 10.1097/00001756-200410050-00022. [DOI] [PubMed] [Google Scholar]
- 93.Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–42. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- 94.Nakao N, Odin P, Lindvall O, Brundin P. Differential trophic effects of basic fibroblast growth factor, insulin-like growth factor-1, and neurotrophin-3 on striatal neurons in culture. Exp Neurol. 1996;138:144–57. doi: 10.1006/exnr.1996.0053. [DOI] [PubMed] [Google Scholar]
- 95.Gamboa C, Ortega A. Insulin-like growth factor-1 increases activity and surface levels of the GLAST subtype of glutamate transporter. Neurochem Int. 2002;40:397–403. doi: 10.1016/s0197-0186(01)00106-1. [DOI] [PubMed] [Google Scholar]
- 96.Fernandez AM, Gonzalez de la Vega AG, Planas B, Torres-Aleman I. Neuroprotective actions of peripherally administered insulin-like growth factor I in the injured olivo-cerebellar pathway. Eur J Neurosci. 1999;11:2019–30. doi: 10.1046/j.1460-9568.1999.00623.x. [DOI] [PubMed] [Google Scholar]