

Abstract
In flowering plants, the somatic-to-reproductive cell fate transition is marked by the specification of spore mother cells (SMCs) in floral organs of the adult plant. The female SMC (megaspore mother cell, MMC) differentiates in the ovule primordium and undergoes meiosis. The selected haploid megaspore then undergoes mitosis to form the multicellular female gametophyte, which will give rise to the gametes, the egg cell and central cell, together with accessory cells. The limited accessibility of the MMC, meiocyte and female gametophyte inside the ovule is technically challenging for cytological and cytogenetic analyses at single cell level. Particularly, direct or indirect immunodetection of cellular or nuclear epitopes is impaired by poor penetration of the reagents inside the plant cell and single-cell imaging is demised by the lack of optical clarity in whole-mount tissues.
Thus, we developed an efficient method to analyze the nuclear organization and chromatin modification at high resolution of single cell in whole-mount embedded Arabidopsis ovules. It is based on dissection and embedding of fixed ovules in a thin layer of acrylamide gel on a microscopic slide. The embedded ovules are subjected to chemical and enzymatic treatments aiming at improving tissue clarity and permeability to the immunostaining reagents. Those treatments preserve cellular and chromatin organization, DNA and protein epitopes. The samples can be used for different downstream cytological analyses, including chromatin immunostaining, fluorescence in situ hybridization (FISH), and DNA staining for heterochromatin analysis. Confocal laser scanning microscopy (CLSM) imaging, with high resolution, followed by 3D reconstruction allows for quantitative measurements at single-cell resolution.
Keywords: Plant Biology, Issue 88, Arabidopsis thaliana, ovule, chromatin modification, nuclear architecture, immunostaining, Fluorescence in situ Hybridization, FISH, DNA staining, Heterochromatin
Introduction
In flowering plants, the establishment of reproductive lineages begins with the differentiation of SMCs, female MMC and male microspore mother cell. The MMC develops from a sub-epidermal nucellar cell at the distal tip of the ovule primordium, and the microspore mother cell develops from sporogenous tissue in the anther locule, which are located deep inside the floral organs1. SMCs undergo meiosis to produce haploid spores, which then give rise to the gametophytes upon mitosis. The female gametophyte, or embryo sac, consists of one egg cell, one central cell, two synergids and three antipodals. The male gametophyte, or pollen, is composed of one vegetative cell and two sperm cells. While the male gametophyte remains a relatively accessible object-of-study, the female gametophyte is embedded inside the ovule, itself enclosed in the flower carpel, and thus poses specific challenges to molecular and cytological analyses. Recently, however, laser-assisted microdissection offered an elegant solution allowing transcriptomic analyses in the MMC and female gametophytic cells2-4. In addition to candidate gene expression analyses, using e.g. RNA in situ hybridization or reporter gene assays, cytological analyses allows investigating the dynamics of endogenous cellular components using specific direct cellular staining or indirect immunostaining. Particularly, cytogenetic staining using FISH and DNA staining, together with immunostaining of chromatin modifications or chromatin components are central approaches to elucidate chromatin dynamics and nuclear organization in Arabidopsis5. Typically, meiosis entails specific chromosome dynamics which has been well investigated in plant male meiocytes6,7; further large-scale, cell-specific chromatin reorganization, likely reflecting dynamic epigenetic reprogramming has been described during pollen development8-10. By contrast, due to the relative inaccessibility of the female meiocyte and gametophyte, these investigations remain technically difficult to apply, and often require sectioning or manual dissection and enzymatic digestion (see below). In addition, the prevalent lack of optical clarity in whole-mount is an obstacle to high-resolution imaging of reproductive cells in intact ovules.
A classical method for cytological analysis of chromosome organization in whole-mount ovules uses Feulgen’s staining11-13. It involves acid hydrolysis (using hypochlorous acid) of the DNA which results in protein denaturation and thus causes destruction of the chromatin structure. Alternatively, chromosome organization in female meiocytes and gametophytic cells can be observed using DAPI staining and immunostaining on semi-thin sections or dissected embryo sacs and MMC (for instance see14-18). Clearly, however, manual dissection and sectioning can be labor intensive and impedes on the qualitative and quantitative analysis of a large number of chromatin epitopes.
Here we provide an efficient protocol to prepare a large number of Arabidopsis ovules suitable for a variety of downstream cytological staining in whole-mount. In brief, flower buds are incubated in a fixative solution, rows of ovules are dissected from the carpel and embedded in acrylamide on slide as done for pollen meiocytes19,20. The embedded ovules are further cleared and fixed in methanol, ethanol, and xylene before cell wall digestion and permeabilization. Possible variations of these steps are discussed. The samples can then be used for DNA staining, immunostaining, and FISH. The preparation mode is efficient and allows for parallel experimental set-up (up to 16 slides can be prepared in a day for different downstream analysis). The treatments described enable homogeneous signals in whole-mount and well preserved histological, cellular, and nuclear organization in reproductive cells and surrounding nucellar cells which benefit qualitative and quantitative comparisons between cell types. Calibrated, CLSM-based high-resolution imaging followed by 3-dimensional reconstruction enables meaningful quantitative measurements of fluorescent signals. We successfully used this procedure to analyze chromatin dynamics in the differentiating MMC21 and developing female gametophyte22; we present here representative results of heterochromatin analysis, chromatin immunostaining, GFP immunostaining and FISH in whole-mount ovules. We further believe that our protocol will be suitable for other plant tissues and species.
Protocol
The procedure is described in the workflow in Figure 1, and the setup for dissection and embedding of tissues are presented in Figure 2.
1. Tissue Fixation
Collect 20-30 carpels in a microfuge tube containing freshly made BVO fixative buffer on ice.
Fix the tissue 30 min with gentle shaking at room temperature.
Spin the tubes containing the carpels in fixative in a benchtop microcentrifuge 1 min at 400 x g.
Remove carefully the fixative buffer and add 1 ml of PBT, place the tubes on ice.
2. Dissection and Embedding
Prepare five Eppendorf tubes with each 200 μl of a freshly made, 5% acrylamide mix.
Prepare five Superfrost slides pre-cleaned with 70% ethanol and labeled with a pencil.
Thaw one aliquot of 20% APS and 20% NaPS each, on ice.
Take 4-5 carpels with a cut-end tip, place them on a clean slide, remove the excess of liquid.
Make longitudinal cuts with a fine needle and detach the carpel walls to release rows of ovules as shown Figure 2, avoid drying by covering with PBS (not more than 10 μl).
Quickly add and mix 12 μl NaPS, 12 μl APS with an aliquot of 200 μl acrylamide mix.
Add 30 μl of the activated acrylamide onto the dissected ovules.
Cover with a 20 x 20 mm coverslip, let polymerize at room temperature, 45-60 min.
Remove the coverslip using a razor blade. At this stage, the samples can be kept overnight at 4 °C in a Coplin jar containing PBS.
3. Tissue Processing
NOTE: All steps except 3.2.1, 3.2.3, 3.3.2 and 3.4.3 are carried out in Coplin jars with 80 ml solution under the chemical hood at room temperature. Slides are transferred with a flat-tip forceps.
- Tissue clarification and fixation:
- Incubate 5 min in methanol.
- Incubate 5 min in ethanol.
- Incubate 30 min in ethanol:xylene (1:1).
- Incubate 5 min in ethanol.
- Incubate 5 min in methanol.
- Incubate 15 min in methanol and PBT (1:1), complemented with 2.5% formaldehyde.
- Rinse 2 x 10 min in PBT. At this stage, slides can be kept overnight at 4 °C.
- Cell wall digestion:
- Thaw an aliquot of the cell wall digestion mix on ice.
- Take a slide from the Coplin jar, drain the excess of liquid by placing it vertically on a paper towel.
- Add 100 μl of cell wall digestion mix over the acrylamide pad and cover with a 23 x 46 mm coverslip. Repeat for the other slides. Incubate for 2 hr at 37 °C in a moist chamber (described in Materials).
- Wash the slides 2 x 5 min in PBT.
- RNase A treatment:
- Take a slide from the Coplin jar, drain the excess of liquid as before.
- Incubate each slide with 100 μl of RNAseA at 100 μg/ml in PBS with 1% Tween-20 for 1 hr at 37 °C in a moist chamber.
- Wash the slides for 2 x 5 min in PBT.
- Post-fixation and permeabilization:
- Post-fix for 20 min in freshly made PBT-F.
- Rinse the slides for 10 min in PBT.
- Permeabilize for 2 hr in PBS with 2% Tween-20 at 4 °C.
- Rinse the slides for 2 x 5 min in PBT.
4. Immunostaining
NOTE: For this step, the optimal concentration of the primary antibody has to be tested by using different dilutions (1:200, 1:500, 1:1000) of the antibodies.
Incubate each slide with 100 μl of primary antibody diluted in PBS with 0.2% Tween-20 for 12-24 hr at 4 °C.
Wash the slides in PBT for 2-4 hr at room temperature under gentle shaking.
Apply the secondary antibody 1:200 in PBS + 0.2% Tween-20 for 24 hr at 4 °C.
Wash slides in PBT for 1 hr at room temperature under gentle shaking.
Counterstain with 10 μg/ml propidium iodide in PBS for 15 min, then rinse 15 min in PBS under gentle shaking, at room temperature.
Mount in anti-fading liquid mountant supplemented with 10 μg/ml propidium iodide. Let the mounting medium harden for 1 hr before acquiring images by CLSM.
5. Quantitative Imaging
- Image acquisition:
- Acquire high resolution images using CLSM, ideally using a resonance scanning mode, which allows better preservation of fluorescent signals over prolonged imaging23, and a 63X glycerol immersion lens.
- Test the acquisition parameters such as laser intensity, gain, pinhole, voxel size and zoom factor at the beginning of the experiment to define a standard acquisition procedure to strictly follow throughout all slides for consistent quantitative measurements.
- Verify the absence of cross-talk between fluorochromes. If present, set up a sequential scan. Acquire transmission images separately and not simultaneously.
- Perform serial, three-dimensional image acquisition with highest possible resolution in the x and y dimensions and with 2x oversampling in the z dimension (Nyquist’s rule).
- Image processing:
- Reconstruct serial images in three-dimensions using commercial or open source software.
- Define contour surfaces around each nucleus (or cell) of interest in 3D.
- Quantify fluorescence in each channel as the sum of pixel intensities in each object.
- Export the data to Excel for statistical analyses. Normalize antibody signals against e.g. DNA staining signals.
Representative Results
We provide a robust protocol for large-scale preparation and processing of Arabidopsis ovules suitable for cytological staining in whole-mount. Thanks to the embedding, the ovules retain a 3-dimensional structure (Figure 3). Furthermore, the tissue processing including optical clarification enables imaging subcellular structures at high-resolution. Figure 4 shows DNA staining in whole-mount ovule primordia where heterochromatin appears as bright, well defined conspicuous foci (no deconvolution was used for this picture). These images were used for analyzing heterochromatin content in the MMC and nucellus (Figure 4)21.
In addition, we successfully used this protocol to quantitatively analyze chromatin dynamics by immunostaining in megaspore mother cells, functional megaspore, developing female gametophytes and early embryo21, 22-24. In Figure 5, we show representative results of whole-mount immunostaining on Arabidopsis ovules. Figure 5A shows an example of immunodetection in ovule primordia, including the megaspore mother cell, of a euchromatin-associated permissive mark (H3K4me3) and a heterochromatin-associated repressive mark (H3K27me1). Figure 5B shows an example of GFP immunodetection in a mature ovule, including the embryo sac (in this case, the protocol was slightly modified for using the GFP booster antibody (see discussion). We also detected native chromatin proteins such as H3 and H121 showing that the procedure preserves chromatin protein epitopes. The procedure also allows for reproducible quantifications enabling comparison between cell types (e.g. reproductive vs. somatic, surrounding cells)21.
Finally, we also successfully applied this procedure to carry out FISH analyses on whole-mount ovule primordia. An example is shown Figure 6 showing FISH signals using a probe against 45S rDNA repeats defining the nucleolar organizing regions25. The DNA probe was directly labeled with Alexa 488 using FISH-Tag26, hybridization was done essentially as described27 with minor modifications, while DNA counterstaining was done as described in our protocol.
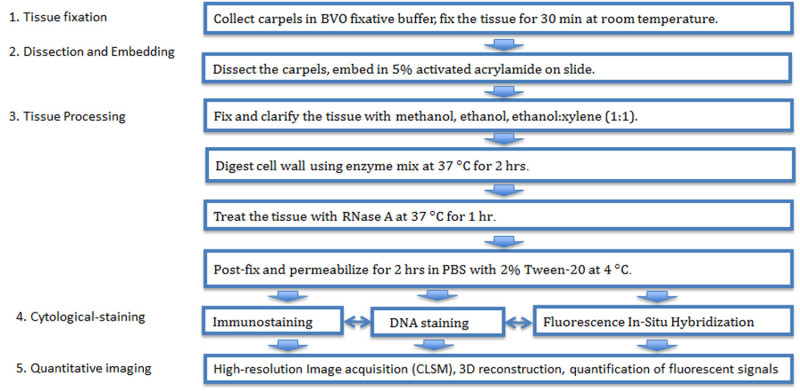 Figure 1. Workflow of immunostaining, DNA staining and Fluorescence in situ Hybridization in Arabidopsis ovules.
Please click here to view a larger version of this figure.
Figure 1. Workflow of immunostaining, DNA staining and Fluorescence in situ Hybridization in Arabidopsis ovules.
Please click here to view a larger version of this figure.
 Figure 2. Setup for dissection and embedding of carpels on slide. The carpel wall is removed and the carpel is dissected on the slide to release rows of ovules (see close up of dissected ovules in step 3), and then the dissected carpel is embedded in activated acrylamide mix, covered by 20 x 20 mm coverslip. Please click here to view a larger version of this figure.
Figure 2. Setup for dissection and embedding of carpels on slide. The carpel wall is removed and the carpel is dissected on the slide to release rows of ovules (see close up of dissected ovules in step 3), and then the dissected carpel is embedded in activated acrylamide mix, covered by 20 x 20 mm coverslip. Please click here to view a larger version of this figure.
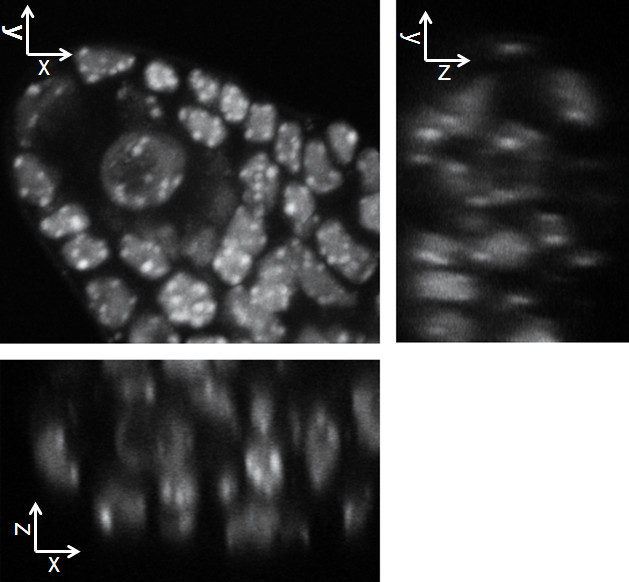 Figure 3. The protocol enables preserving the 3-dimensional structure while allowing optical clarity and homogenous staining. The images shows a split section view of the 3D image in xy, xz and yz axis as indicated. The image data have been acquired by confocal laser scanning microscopy and reconstructed in 3 dimensions using the Imaris software. Please click here to view a larger version of this figure.
Figure 3. The protocol enables preserving the 3-dimensional structure while allowing optical clarity and homogenous staining. The images shows a split section view of the 3D image in xy, xz and yz axis as indicated. The image data have been acquired by confocal laser scanning microscopy and reconstructed in 3 dimensions using the Imaris software. Please click here to view a larger version of this figure.
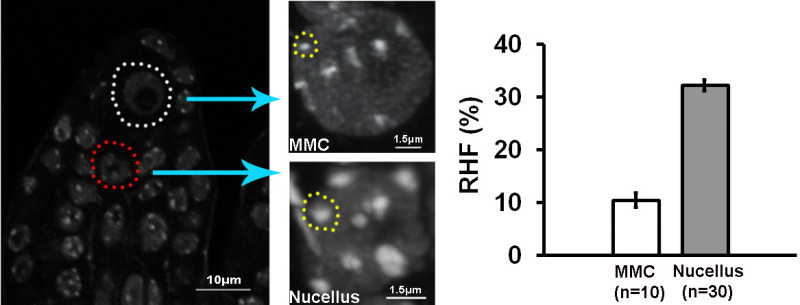 Figure 4. Whole-mount DNA staining by propidium iodide in ovule primodia allows for precise heterochromatin quantification. The image on the left shows whole-mount DNA staining throughout an ovule primordia. An MMC nucleus is marked by a white contour and a nucellar nucleus in red. Projections of 3D-reconstructed nuclei are shown on the right. The clarity of the tissue enables high-resolution imaging of the heterochromatin foci marked by a yellow contour and quantification of the fluorescent signals therein. The graphs show the relative heterochromatin fraction21. Please click here to view a larger version of this figure.
Figure 4. Whole-mount DNA staining by propidium iodide in ovule primodia allows for precise heterochromatin quantification. The image on the left shows whole-mount DNA staining throughout an ovule primordia. An MMC nucleus is marked by a white contour and a nucellar nucleus in red. Projections of 3D-reconstructed nuclei are shown on the right. The clarity of the tissue enables high-resolution imaging of the heterochromatin foci marked by a yellow contour and quantification of the fluorescent signals therein. The graphs show the relative heterochromatin fraction21. Please click here to view a larger version of this figure.
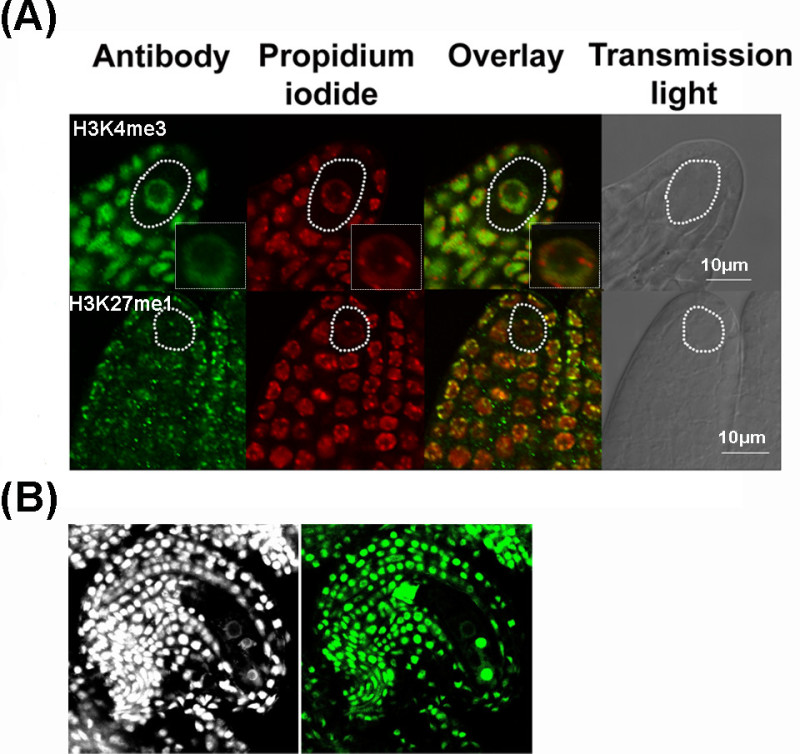 Figure 5. Representative results of whole-mount immunostaining in Arabidopsis ovules. A) Immunostaining of chromatin modifications in young ovule primordia detecting euchromatin (H3K4me3) and heterochromatin (H3K27me1). The antibody signal is green, the DNA counterstained by propidium iodide in red. An overlay of fluorescent signals is shown together with a picture in transmission light using differential interference contrast (grey). MMCs are indicated by white contours. A close-up of the MMC nucleus is shown as inset in the upper panel. The images are single confocal section. B) Immunodetection of GFP in a mature ovule. The GFP was immunostained using GFP-booster antibody and the ovule was counterstained with DAPI. Please click here to view a larger version of this figure.
Figure 5. Representative results of whole-mount immunostaining in Arabidopsis ovules. A) Immunostaining of chromatin modifications in young ovule primordia detecting euchromatin (H3K4me3) and heterochromatin (H3K27me1). The antibody signal is green, the DNA counterstained by propidium iodide in red. An overlay of fluorescent signals is shown together with a picture in transmission light using differential interference contrast (grey). MMCs are indicated by white contours. A close-up of the MMC nucleus is shown as inset in the upper panel. The images are single confocal section. B) Immunodetection of GFP in a mature ovule. The GFP was immunostained using GFP-booster antibody and the ovule was counterstained with DAPI. Please click here to view a larger version of this figure.
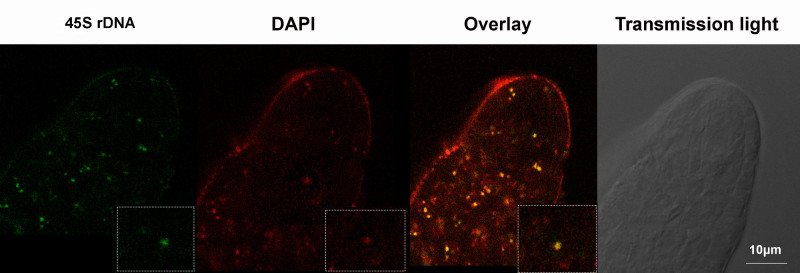 Figure 6. Whole-mount Fluorescence in situ Hybridization in Arabidopsis ovules. The ovule primordium was hybridized with a DNA probe specific to 45S rDNA repeat loci and labeled with Alexa 488 using the FISH-Tag technology, and counterstained with DAPI26. The overlay of 45S rDNA with DAPI and image acquired in the transmission light channel (grey) are also shown. A close-up of the MMC nucleus is shown as inset. Please click here to view a larger version of this figure.
Figure 6. Whole-mount Fluorescence in situ Hybridization in Arabidopsis ovules. The ovule primordium was hybridized with a DNA probe specific to 45S rDNA repeat loci and labeled with Alexa 488 using the FISH-Tag technology, and counterstained with DAPI26. The overlay of 45S rDNA with DAPI and image acquired in the transmission light channel (grey) are also shown. A close-up of the MMC nucleus is shown as inset. Please click here to view a larger version of this figure.
 Figure 7. Influence of the fixation, digestion and staining procedure on DNA signals in whole-mount ovules. A) Mature ovules were fixed 30 min either with 4% paraformaldehyde or BVO fixative and processed 30 min or 1 hr with cell wall digest enzyme mix before DNA staining with propidium iodide. For a given batch, longer incubation affects more negatively on the DNA staining ovules that were fixed with paraformaldehyde than with BVO. B) Whole-mount DNA staining using the Feulgen reagent following a required acid-hydrolysis28 or using propidium iodide following the non-denaturing protocol described in the text. The upper panel shows a single plane section through the embryo sac, the lower panel presents a magnification over the central cell nucleus showing clearly alteration of chromatin organization in Feulgen-stained ovules. ccn, central cell nucleus, ecn, egg cell nucleus, syn, synergid nucleus. Please click here to view a larger version of this figure.
Figure 7. Influence of the fixation, digestion and staining procedure on DNA signals in whole-mount ovules. A) Mature ovules were fixed 30 min either with 4% paraformaldehyde or BVO fixative and processed 30 min or 1 hr with cell wall digest enzyme mix before DNA staining with propidium iodide. For a given batch, longer incubation affects more negatively on the DNA staining ovules that were fixed with paraformaldehyde than with BVO. B) Whole-mount DNA staining using the Feulgen reagent following a required acid-hydrolysis28 or using propidium iodide following the non-denaturing protocol described in the text. The upper panel shows a single plane section through the embryo sac, the lower panel presents a magnification over the central cell nucleus showing clearly alteration of chromatin organization in Feulgen-stained ovules. ccn, central cell nucleus, ecn, egg cell nucleus, syn, synergid nucleus. Please click here to view a larger version of this figure.
Discussion
In flowering plants, the female reproductive lineage is surrounded by several cell layers including the nucellus and the ovule teguments, thus rendering cytological staining in whole-mount technically challenging. Here we present an efficient protocol enabling the preparation and processing of a large number of ovules suitable for cytological staining such as immunostaining, DNA staining and fluorescence in situ hybridization in whole-mount. We successfully used it for the analysis of the female reproductive germline in Arabidopsis21,22. This method is highly efficient as several slides can be treated in parallel for different staining. It is also robust and gives homogeneous signal distribution and allows for reproducible quantitative analyses. By contrast to classical method such as Feulgen staining which involves the denaturation of the chromatin structure, our protocol preserves chromatin organization and nuclear epitopes. In addition, tissue clarification enables imaging the signals at high resolution at the single-cell level.
This protocol can be expedited by omitting the steps described in 3.1 if the flowers have been fixed in 4% paraformaldehyde (in PBS + 1% Tween) instead of BVO buffer (1.1). While this shorter procedure proved functional for several immunostaining22-24, we found that it attenuates the robustness of the staining across samples, antibody and batch of cell wall digestion enzyme mix (see below). Furthermore we were not successful for FISH hybridization with this short procedure. For immunodetection of GFP’s using the booster molecules as shown Figure 5B, a similar protocol as described here was used with slight modifications at step 1 and 3: carpels were fixed in 2.5% Formaldehyde for 45 min (1.1), the first step of tissue processing was shortened to 5-10 min methanol treatment (3.1), a blocking step was introduced (30 min in 2% BSA in PBS) prior to antibody application overnight as described. No secondary antibody is necessary with the booster.
We discuss below some critical steps:
Tissue Fixation, dissection and embedding.
Immunostaining and DNA staining signals were consistently more robust and homogenously distributed when the tissue was sampled from plants less than 5 weeks old (following transfer of the seedling in soil). Possibly, in our growth conditions, a prolonged period of cultivation may be accompanied by changes in the biochemical composition of the cell wall, influencing in turn the efficiency of tissue processing. Thus we recommend sampling tissue from relatively young plants. In addition, the tissue should be prevented from drying during dissection (leading otherwise to histological alteration and absence of staining signals) while an excess of PBS challenges the manipulation; a gentle draining of the excess of solution with the tip around the tissue deposited on slide is thus recommended. Furthermore, bubbles should be avoided in the acrylamide mixture and while covering with a coverslip. Finally, Superfrost Plus slides are strongly recommended for adequate acrylamide adhesion (standard quality lead to fragile and unstable pads in our hands), as they appear superior to others for tissue and acrylamide adhesion.
Fixation, permeabilisation and cell wall digestion.
Cell wall digestion is a critical step of tissue processing. It is thought that this step facilitates a good penetration of the staining reagents homogenously throughout the plant tissue. We experienced variability in staining homogeneity (ranging from no signal, signal in only part of the tissue, to 100% tissue staining) depending on the digestion time and the enzymatic activity (batch-specific, described by the provider). It is recommended to produce a large amount of stock solution of the enzyme mix (e.g. 100 ml) and keep 1 ml aliquots at -20 °C. Each stock solution should first be tested on 1-2 slides before using at large scale. Furthermore, we experienced that the type of fixative influences the efficiency of DNA staining in combination with different processing time for cell wall digest: ovules fixed in 4% paraformaldehyde were negatively affected by prolonged incubation with the cell wall digestion mix, while tissue fixed with the BVO solution were tolerant to longer digestion times and allowed better DNA staining (Figure 7A). In addition, an RNAse (DNase free) treatment is strictly necessary if the tissue is counterstained with propidium iodide as it also binds to RNA molecules. We advise against using 4',6-diamidino-2-phenylindole (DAPI) due to its broad fluorescent emission spectra overlapping with other fluorophores but also to achromatic aberrations that necessitate channel shift corrections post-acquisition. We also recommend against Feulgen staining which requires an acid-hydrolysis during tissue processing28 leading to chromatin denaturation particularly in the embryo sac (Figure 7B). Alternative DNA dyes may also be used29, but their efficiency has not been tested here.
Immunostaining.
For immunostaining of chromatin modifications, we recommend to verify the specificity of the primary antibody in the open source database http://compbio.med.harvard.edu/antibodies/projects/130, as some commercially available antibodies showed cross-reactions for other modifications. For downstream quantification analyses, it is important to calibrate the antibodies concentration and incubation time to measure signals in a linear relationship with the epitope. We recommend to calibrate the antibody dilution and detection time using an antibody dilution series of 1:200, 1:500, 1:1,000 and from 12-24 hr incubation, respectively, to identify the conditions giving robust and homogenous signals. The highest dilution and shortest incubation time allowing reproducible signals should be used for quantitative analyses. Controls without primary antibody should be performed to test for the specificity. If immunostaining produces unspecific staining, it is advised to block with 5% BSA + 0.1% Tween in PBS for 2 hr at 4 °C before applying the primary antibody. Antibody signals can be checked on the slide before DNA counterstaining to verify the success of the experiment. In our hands, washing in PBT for 2-4 hr after incubation with the primary antibody, and 1 hr after the secondary antibody allows for low background signals even without blocking.
Finally, this protocol also is likely applicable to other plant tissues (e.g. root, leaf fragment, floral meristem) and probably to other plant species, providing some adjustment on the dissection, cell wall digest and permeabilization.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Ueli Grossniklaus (University of Zürich) for technical and financial support. We are thankful to Valeria Gagliardini, Christof Eichenberger, Arturo Bolanos and Peter Kopf for general lab support. This research was funded by the University of Zürich, grants from the Swiss National Foundation to CB (31003A_130722) and Ueli Grossniklaus (31003A_141245 and 31003AB-126006), and the Agence Nationale de la Recherche to DG (Programme ANR-BLANC-2012).
References
- Maheshwari P. An introduction to the embryology of angiosperms. New York: McGraw-Hill; 1950. [Google Scholar]
- Schmidt A, et al. Transcriptome analysis of the Arabidopsis megaspore mother cell uncovers the importance of RNA helicases for plant germline development. PLoS Biol. 2011;9(9) doi: 10.1371/journal.pbio.1001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MW, et al. A powerful method for transcriptional profiling of specific cell types in eukaryotes: laser-assisted microdissection and RNA Sequencing. PLoS One. 2012;7(1) doi: 10.1371/journal.pone.0029685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuest SE, et al. Arabidopsis female gametophyte gene expression map reveals similarities between plant and animal gametes. Curr Biol. 2010;20(6):506–512. doi: 10.1016/j.cub.2010.01.051. [DOI] [PubMed] [Google Scholar]
- Koornneef M, et al. Cytogenetic tools for Arabidopsis thaliana. Chromosome Res. 2003;11(3):183–194. doi: 10.1023/a:1022827624082. [DOI] [PubMed] [Google Scholar]
- Oliver C, et al. The dynamics of histone H3 modifications is species-specific in plant meiosis. Planta. 2013;238(1):23–33. doi: 10.1007/s00425-013-1885-1. [DOI] [PubMed] [Google Scholar]
- Ravi M, et al. Meiosis-specific loading of the centromere-specific histone CENH3 in Arabidopsis thaliana. PLoS Genet. 2011;7(6) doi: 10.1371/journal.pgen.1002121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges F, et al. Reprogramming the epigenome in Arabidopsis pollen. Cold Spring Harb Symp Quant Biol. 2012;77:1–5. doi: 10.1101/sqb.2013.77.014969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey P, et al. Chromatin alterations during pollen development in Hordeum vulgare. Cytogenet Genome Res. 2013;141(1):50–57. doi: 10.1159/000351211. [DOI] [PubMed] [Google Scholar]
- Schoft VK, et al. Induction of RNA-directed DNA methylation upon decondensation of constitutive heterochromatin. EMBO Rep. 2009;10(9):1015–1021. doi: 10.1038/embor.2009.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrell PJ, Grossniklaus U. Confocal microscopy of whole ovules for analysis of reproductive development: the elongate1 mutant affects meiosis II. Plant J. 2005;43(2):309–320. doi: 10.1111/j.1365-313X.2005.02456.x. [DOI] [PubMed] [Google Scholar]
- Braselton JP, et al. Feulgen staining of intact plant tissues for confocal microscopy. Biotech. Histochem. 1996;71:84–87. doi: 10.3109/10520299609117139. [DOI] [PubMed] [Google Scholar]
- Scott RJ, et al. Parent-of-origin effects on seed development in Arabidopsis thaliana. Development. 1998;125:3329–3341. doi: 10.1242/dev.125.17.3329. [DOI] [PubMed] [Google Scholar]
- Armstrong SJ, Jones GH. Female meiosis in wild-type Arabidopsis thaliana and in two meiotic mutants. Sex Plant Reprod. 2001;13:177–183. [Google Scholar]
- Grimanelli D, et al. Heterochronic expression of sexual reproductive programs during apomictic development in Tripsacum. Genetics. 2003;165:1521–1531. doi: 10.1093/genetics/165.3.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Marcos JF, et al. Maternal gametophytic baseless1 is required for development of the central cell and early endosperm patterning in maize (Zea mays) Genetics. 2006;174(1):317–329. doi: 10.1534/genetics.106.059709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedojadło K, et al. Ribosomal RNA of Hyacinthus orientalis L. female gametophyte cells before and after fertilization. Planta. 2012;236:171–184. doi: 10.1007/s00425-012-1618-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JH, Friedman WE. The four-celled female gametophyte of Illicium (Illiciaceae; Austrobaileyales): Implications for understanding the origin and early evolution of monocots, eumagnoliids, and eudicots. Am J Bot. 2004;91(3):332–351. doi: 10.3732/ajb.91.3.332. [DOI] [PubMed] [Google Scholar]
- Bass HW, et al. Telomeres cluster de novo before the initiation of synapsis: a three-dimensional spatial analysis of telomere positions before and during meiotic prophase. J Cell Biol. 1997;137(1):5–18. doi: 10.1083/jcb.137.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe ES, et al. Three-dimensional acrylamide fluorescence in situ hybridization for plant cells. Methods Mol Biol. 2013;990:53–66. doi: 10.1007/978-1-62703-333-6_6. [DOI] [PubMed] [Google Scholar]
- She W, et al. Chromatin reprogramming during the somatic-to-reproductive cell fate transition in plants. Development. 2013;140(19):4008–4019. doi: 10.1242/dev.095034. [DOI] [PubMed] [Google Scholar]
- Pillot M, et al. Embryo and endosperm inherit distinct chromatin and transcriptional states from the female gametes in Arabidopsis. Plant Cell. 2010;22(2):307–320. doi: 10.1105/tpc.109.071647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlinghaus RT. MRT letter: high speed scanning has the potential to increase fluorescence yield and to reduce photobleaching. Microsc Res Tech. 2006;69(9):689–692. doi: 10.1002/jemt.20363. [DOI] [PubMed] [Google Scholar]
- Autran D, et al. Maternal epigenetic pathways control parental contributions to Arabidopsis early embryogenesis. Cell. 2011;145(5):707–719. doi: 10.1016/j.cell.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Fransz P, et al. Interphase chromosomes in Arabidopsis are organized as well defined chromocenters from which euchromatin loops emanate. Proc Natl Acad Sci USA. 2002;99(22):14584–14589. doi: 10.1073/pnas.212325299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox WG, Singer VL. Fluorescent DNA hybridization probe preparation using amine modification and reactive dye coupling. BioTechniques. 2004;36(1):114–122. doi: 10.2144/04361RR02. [DOI] [PubMed] [Google Scholar]
- Lysak M, et al. In: Cytogenetic analyses of Arabidopsis In Arabidopsis Protocols: Methods in Molecular Biology, 2nd ed. Salinas J, Sanchez-Serrano JJ, editors. Totowa, NJ: Humana Press; 2006. pp. 173–186. [DOI] [PubMed] [Google Scholar]
- Chieco P, Derenzini M. The Feulgen reaction 75 years on. Histochem Cell Biol. 1999;111(5):345–358. doi: 10.1007/s004180050367. [DOI] [PubMed] [Google Scholar]
- Suzuki T, et al. DNA staining for fluorescence and laser confocal microscopy. J Histochem Cytochem. 1997;45(1):49–53. doi: 10.1177/002215549704500107. [DOI] [PubMed] [Google Scholar]
- Egelhofer TA, et al. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 2011;18(1):91–93. doi: 10.1038/nsmb.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauwens S, Oostveldt PV. Whole mount fluorescence in situ hybridization (FISH) of repetitive DNA sequences on interphase nuclei of the small cruciferous plant Arabidopsis thaliana. Procedures for In Situ Hybridization to Chromosomes, Cells, and Tissue Sections. Nonradioactive in situ hybridization application manual. 1996. pp. 165–171.