

Abstract
Genetic alterations can drive oncogenic events and cancer development. However, this is only half of the story. It is now evident that tumor progression only occurs if powerful stress signaling pathways and microenvironmental signals are overcome. Two recent Nature Cell Biology papers study how niche signals of primary and target organ barriers have to be overridden for oncogenes to allow for colorectal cancer (CRC) initiation and metastasis (Urosevic et al, 2014; Whissell et al, 2014).
See also: G Whissell et al (July 2014) and J Urosevic et al (July 2014)
Whissell et al elegantly define the signals controlling stem cell programs in colon cancer stem cells (CSC) during tumor initiation. They show that deletion of GATA6 in an APC−/− background decreased proliferation, self-renewal and tumorigenesis of Lrg5+ CSCs by increasing bone morphogenetic protein-4 (BMP4) signaling. The authors also showed that GATA6 binds to the promoter regions of BMP4 outcompeting β-catenin/TCF4 complexes and repressing BMP4 expression by modulating histone-H3 post-translational modifications (PTMs) (Fig1A). These changes conspired to create CSCs and microenvironments permissive for CRC development. These findings suggest that BMP4 in the microenvironment is a rate-limiting signal for CRC development and that the use of H3-PTM modulators that restore BMP4 expression might limit CRC progression. These data are in line with work showing that BMPs signaling avoids proliferation and expansion of normal adult stem cells by maintaining a quiescent state (i.e. hair follicle SC, hematopoietic SC) (van der Flier & Clevers, 2009; Whissell et al, 2014). Accordingly, GREM1, a BMP inhibitor, is commonly secreted from the normal stroma, and duplication of this gene predisposes patients to CRC (Whissell et al and refs within) (Fig1A). Thus, gain of GREM1 and GATA6 may increase the chances of CRC development. Importantly, these findings suggest that even when oncogenic signals are activated, restoration of niche-derived inhibitory signals (like BMPs) limits tumorigenesis. Niche cues (including BMPs) and PTEN activity in hair follicle stem cells can also maintain quiescence of adult stem cells and “insulate” these from oncogenic K-RasG12D signaling (White et al, 2014). These data raise key questions. Could the microenvironment maintain for long periods dominance over powerful oncogenic signals present in pre-malignant cells? Whissell et al tested BMP4 expression in CRC precursors, but could stroma-derived BMP4 be the first line defense in limiting mutant CSC expansion? Finally is GATA6 required for CRC tumor maintenance? Questions that have important therapeutic implications.
Figure 1. Mechanisms of colorectal cancer initiation and dissemination.
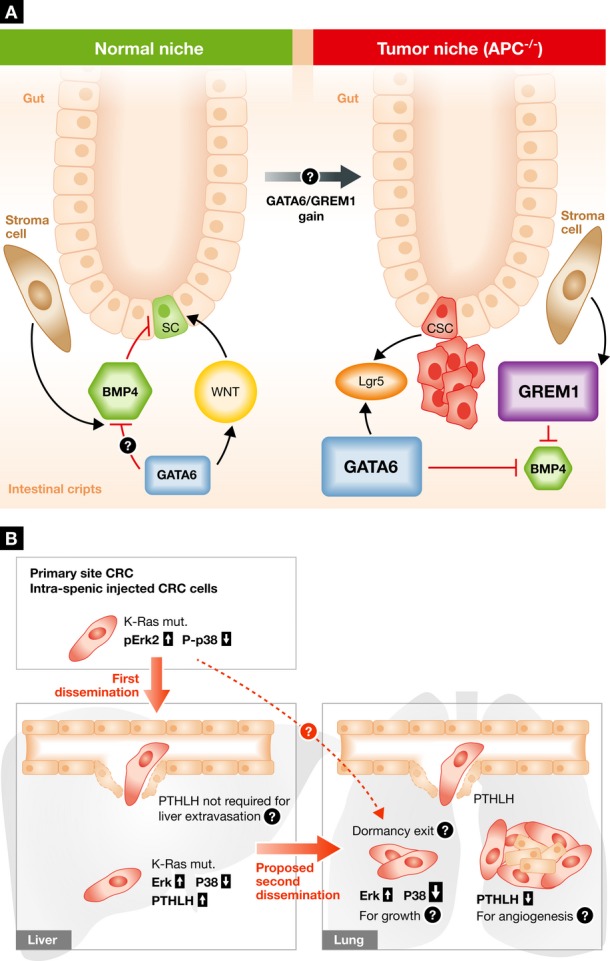
(A) The normal intestinal crypt niche is maintained by a continuous balance between growth-promoting (i.e., WNT) and inhibitory signals (i.e., BMPs). Batlle and co-workers show that in an APC, null background GATA6 signals promote tumorigenesis via inhibition of BMP4 expression and function in cancer stem cells (CSC). This effect is achieved by a competition of GATA6 for the same binding elements of β-catenin/TCF4 factors and by modifying the epigenetic landscape of the BMP4 promoter. Importantly, in patients, germ line gene duplication of GREM1, a BMP inhibitor, is linked to increased predisposition to CRC development. (B) High phospho-ERK2 is required for CRC metastatic seeding to the liver from the spleen or primary site, whereas metastatic seeding of the lung from the liver requires a strong downregulation of p38 activation. Loss of P-p38 also induces PTHLH secretion that facilitates cancer cell dissemination in the lung by killing the endothelial cells. It remains to be elucidated how PTHLH is not required for liver extravasation, how primary tumor endothelium and that associated with metastasis become insensitive to PTHLH, and whether P-ERK2 and P-p38 levels also affect dormancy and reactivation of disseminated tumor cells in this model.
In another story, Urosevic et al investigate metastatic dissemination in CRC to gain further understanding of a phenomenon called: metastasis from metastasis. While liver is the predominant site of CRC metastasis, a smaller proportion of patients carrying mutant K-Ras primary CRC also develop lung metastasis, which also show enrichment for the K-Ras mutation (Tie et al, 2011). The authors selected, à la Fiedler, a more powerful liver metastatic variant of mutant K-Ras SW620 CRC cells (originally derived from a lymph node metastasis). When injected in the spleen, to more easily obtain liver colonization, the resulting SW620 variant (LiM2) spontaneously grew in the liver and, albeit less powerful, spread into the lung. Expression profiling and pathway analysis identified high ERK1/2 activity and low levels of p38 phosphorylation in metastatic variants. Importantly, even lower p38 activity was required for LiM2 cell lung metastasis (Fig1B). It appeared that ERK2—rather than ERK1—was more critically required for LiM2 liver metastasis (though not for lung spreading). Importantly, a low ERK/p38 signal ratio was determined in primary tumor biopsies of CRC patients with high liver metastatic activity as compared to patients carrying no metastases. Since K-Ras is a strong activator of ERK1/2, its role in LiM2 cells is in line with previous literature. Similarly, a balance between ERK and p38 signaling where ERK signaling favors disseminated tumor cell (DTC) colonization and growth is in concordance with previous findings from others laboratories, including ours (Bragado et al, 2013 and refs within). An intriguing twist proposed by the new data is that p38 inhibition is exclusively required for LiM2 cells in lung colonization. The authors propose that loss of p38 activity induces the cytokine PTHLH, in turn essential for lung (but not liver) metastases. In vitro induction of endothelial cell apoptosis by PTHLH is taken as cell-biological evidence to explain the extravasation into the lung.
The Urosevic study thus raises intriguing questions: Based on abundant patient data, Hölzel et al had excluded the formation of metastases from established metastases, rather tracing these back to primary tumor spread before surgery (Hölzel et al, 2010). In contrast, the Urosevic study suggests that lung metastasis came from liver lesions. Thus, more precise fate mapping tools and single-cell resolution imaging as used in CRC tumor initiation analysis (van der Flier & Clevers, 2009) and/or genetic analysis of clonal lesions (Klein, 2013) would be required to settle this question. Another question is whether p38 inhibition contributes to lung metastasis by other means as well; could activation of ERK be involved? Given that SW620 cells colonize the lung efficiently (Ma et al, 2008), have the emerging LiM2 clones regained sensitivity to dormancy signals via p38 activation (Fig1B)? This would be in accordance with recent findings showing that TGF-β2 and BMPs induce dormancy post-extravasation by establishing a low ERK/p38 signaling ratio (Kobayashi et al, 2011; Bragado et al, 2013). Finally, how does PTHLH selectively kill lung—and not liver—or primary tumor endothelial cells? How does the lung endothelium become resistant to PTHLH after extravasation when in fact endothelial cells support further metastatic growth? These issues inspire further studies on metastases biology and support key findings that the balance between mitogenic and stress signaling pathways (Bragado et al, 2013) is a key in determining DTC fate.
Circling back to the studies by Whissell et al, the striking result that BMPs, TGF-β2 and other regulators (Shiozawa et al, 2011) of adult stem cell quiescence and pluripotency, might antagonize metastasis development is thought provoking. It implies that solitary, though aggressive DTCs that managed to lodge in target organs retain their sensitivity to microenvironmental cues that originally restricted their expansion. Understanding the underlying mechanisms and the genetic traits driving this phenomenon holds the promise to develop specific therapies to successfully modulate key microenvironmental inputs to eradicate both, primary tumor progression and DTC reactivation to form metastases.
References
- Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, Schewe DM, Aguirre-Ghiso JA. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol. 2013;15:1351–1361. doi: 10.1038/ncb2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–260. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- Hölzel D, Eckel R, Emeny RT, Engel J. Distant metastases do not metastasize. Cancer Metastasis Rev. 2010;29:737–750. doi: 10.1007/s10555-010-9260-1. [DOI] [PubMed] [Google Scholar]
- Klein CA. Selection and adaptation during metastatic cancer progression. Nature. 2013;501:365–372. doi: 10.1038/nature12628. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, Pai SK, Liu W, Fukuda K, Chambers C, Wilber A, Watabe K. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med. 2011;208:2641–2655. doi: 10.1084/jem.20110840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Rong Y, Radiloff DR, Datto MB, Centeno B, Bao S, Cheng AW, Lin F, Jiang S, Yeatman TJ, Wang XF. Extracellular matrix protein betaig-h3/TGFBI promotes metastasis of colon cancer by enhancing cell extravasation. Genes Dev. 2008;22:308–321. doi: 10.1101/gad.1632008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, Kim JK, Patel LR, Ying C, Ziegler AM, Pienta MJ, Song J, Wang J, Loberg RD, Krebsbach PH, Pienta KJ, Taichman RS. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121:1298–1312. doi: 10.1172/JCI43414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie J, Lipton L, Desai J, Gibbs P, Jorissen RN, Christie M, Drummond KJ, Thomson BN, Usatoff V, Evans PM, Pick AW, Knight S, Carne PW, Berry R, Polglase A, McMurrick P, Zhao Q, Busam D, Strausberg RL, Domingo E. KRAS mutation is associated with lung metastasis in patients with curatively resected colorectal cancer. Clin Cancer Res. 2011;17:1122–1130. doi: 10.1158/1078-0432.CCR-10-1720. [DOI] [PubMed] [Google Scholar]
- Urosevic J, Garcia-Albéniz X, Planet E, Real S, Céspedes MV, Guiu M, Fernandez E, Bellmunt A, Gawrzak S, Pavlovic M, Mangues R, Dolado I, Barriga FM, Nadal C, Kemeny N, Batlle E, Nebreda AR, Gomis RR. Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH. Nat Cell Biol. 2014;16:685–694. doi: 10.1038/ncb2977. [DOI] [PubMed] [Google Scholar]
- Whissell G, Montagni E, Martinelli P, Hernando-Momblona X, Sevillano M, Jung P, Cortina C, Calon A, Abuli A, Castells A, Castellvi-Bel S, Nacht AS, Sancho E, Stephan-Otto Attolini C, Vicent GP, Real FX, Batlle E. The transcription factor GATA6 allows self-renewal of colon adenoma stem cells by repressing BMP gene expression. Nat Cell Biol. 2014;16:695–707. doi: 10.1038/ncb2992. [DOI] [PubMed] [Google Scholar]
- White AC, Khuu JK, Dang CY, Hu J, Tran KV, Liu A, Gomez S, Zhang Z, Yi R, Scumpia P, Grigorian M, Lowry WE. Stem cell quiescence acts as a tumour suppressor in squamous tumours. Nat Cell Biol. 2014;16:99–107. doi: 10.1038/ncb2889. [DOI] [PMC free article] [PubMed] [Google Scholar]