

Abstract
Intracellular protein degradation is an essential process in all life domains. While in all eukaryotes regulated protein degradation involves ubiquitin tagging and the 26S-proteasome, bacterial prokaryotic ubiquitin-like protein (Pup) tagging and proteasomes are conserved only in species belonging to the phyla Actinobacteria and Nitrospira. In Mycobacterium tuberculosis, the Pup-proteasome system (PPS) is important for virulence, yet its physiological role in non-pathogenic species has remained an enigma. We now report, using Mycobacterium smegmatis as a model organism, that the PPS is essential for survival under starvation. Upon nitrogen limitation, PPS activity is induced, leading to accelerated tagging and degradation of many cytoplasmic proteins. We suggest a model in which the PPS functions to recycle amino acids under nitrogen starvation, thereby enabling the cell to maintain basal metabolic activities. We also find that the PPS auto-regulates its own activity via pupylation and degradation of its components in a manner that promotes the oscillatory expression of PPS components. As such, the destructive activity of the PPS is carefully balanced to maintain cellular functions during starvation.
Keywords: mycobacteria, nitrogen limitation, proteasome, proteolysis, pupylation
Introduction
The eukaryotic ubiquitin-proteasome system participates in a large variety of cellular processes and maintains homeostasis of protein folding through the degradation of regulatory and misfolded proteins (Schmidt & Finley, 2014). Proteasomal degradation also serves nutritional roles under starvation conditions through the recycling of amino acids (Vabulas & Hartl, 2005; Suraweera et al, 2012). In bacteria, proteasomes are only found in species belonging to the phyla Actinobacteria and Nitrospira, where they are strictly conserved (Lupas et al, 1997; De Mot, 2007; Valas & Bourne, 2008); other bacterial species rely on smaller and less complex proteases for regulated protein degradation (Gur et al, 2011). In Mycobacterium tuberculosis, an actinobacterial species, it was demonstrated that the bacterial proteasome degrades protein targets conjugated to a prokaryotic ubiquitin-like protein (Pup) (Fig1) (Pearce et al, 2008; Burns et al, 2009). Pup is a natively unfolded 64 residue-long protein, and, in contrast to ubiquitylation where poly-ubiquitin chains are added to target proteins, no evidence for poly-Pup chains has been presented yet (Pearce et al, 2008; Chen et al, 2009; Liao et al, 2009; Festa et al, 2010). The enzyme responsible for attaching Pup to protein substrates, proteasomal accessory factor A (PafA), catalyzes the ATP-dependent formation of isopeptide bonds between the Pup C-terminal glutamate γ-carboxylate and the side chain of protein substrates lysine residues (Fig1) (Pearce et al, 2008; Guth et al, 2011). However, in many proteasome-containing species, Pup is translated with a C-terminal glutamine, rather than a glutamate. As such, a deamidase of Pup (Dop) converts this terminal glutamine into glutamate, thus allowing PafA-mediated Pup conjugation to protein substrates (i.e., pupylation; Striebel et al, 2009). Not only does Dop catalyze the deamidation reaction to promote substrate pupylation, it can also depupylate substrates, namely detach Pup from proteins already pupylated (Fig1) (Burns et al, 2010b; Imkamp et al, 2010b).
Figure 1. The Pup-proteasome system.
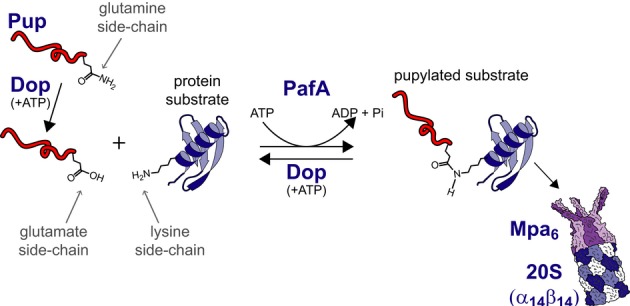
Protein degradation by the PPS starts with deamidation of Pup by Dop. Deamidated Pup can be conjugated to lysine side chains of protein substrates by PafA at the expense of one ATP molecule per conjugation. A pupylated protein can either be degraded by the proteasome or depupylated by Dop.
Pup recognition by the proteasome regulatory subunit, termed Mpa in mycobacteria, results in its translocation, beginning at the Pup N-terminal end, into the proteolytic chamber of the 20S core proteasomal particle (Pearce et al, 2008; Sutter et al, 2009; Striebel et al, 2010; Wang et al, 2010). Bacterial proteasomes are simpler than their eukaryotic counterparts, containing only one or two types of α and β subunits in the 20S core particle and relying on a simpler regulatory particle comprising a homo-hexameric AAA+ protein (Wolf et al, 1998; Darwin et al, 2005; Hu et al, 2006). Mpa itself is a pupylation substrate, and it was demonstrated in vitro that proteasome function can be negatively regulated by Mpa pupylation (Delley et al, 2012).
The physiological role of bacterial proteasomes has been unclear since their discovery, almost two decades ago (Tamura et al, 1995). In M. tuberculosis, the Pup-proteasome system (PPS) is essential for full virulence and persistence in the host (Darwin et al, 2003; Darwin 2009; Cerda-Maira et al, 2010; Gandotra et al, 2010). However, as the vast majority of PPS-containing bacteria are non-pathogenic, it would seem that the PPS plays a fundamental role in bacterial physiology, rather than serving a direct function in virulence. Nonetheless, no clear explanation for the PPS physiological role was presented thus far. On the one hand, whereas growth defects of M. tuberculosis PPS mutants were reported by Gandotra et al (2010), other studies indicated that pupylation-deficient mutants multiply as rapidly as do wild-type cells (Darwin et al, 2003; Cerda-Maira et al, 2010; Imkamp et al, 2010a; Küberl et al, 2014). An additional layer of complexity was introduced when proteomics studies failed to reveal a clear substrate specificity of PafA or defining traits for pupylation substrates (Festa et al, 2010; Poulsen et al, 2010; Watrous et al, 2010; Cerda-Maira et al, 2011). Rather, hundreds of different protein species were found to be pupylated, with no obvious PafA recognition motif detected.
In this study, we report that the PPS is essential for the survival of M. smegmatis, a mycobacterial model organism, under conditions of nitrogen starvation and, to a lesser extent, under conditions of carbon starvation. Whereas a PPS-deficient M. smegmatis mutant fails to survive starvation, the wild-type strain remains viable and responds by a dramatic, yet delicately regulated, induction of pupylation and degradation of pupylated substrates. We also find that the proteasome mediates the degradation of PPS components. Such negative auto-regulation leads to oscillatory expression of the PPS in response to starvation. Overall, our findings point to a model by which under starvation conditions, the PPS facilitates amino acid recycling so as to generate protein building blocks and a carbon source, thereby allowing for the maintenance of basal metabolic activities and survival under prolonged nutrient limitation. In considering PPS substrates merely as sources for amino acids, this model provides a simple explanation for the broad substrate specificity of PafA, the Pup ligase.
Results
Pupylation dynamics during M. smegmatis growth
To understand the physiological role of the PPS, we considered whether the level of pupylated proteins in M. smegmatis changes during growth. As such, stationary-phase M. smegmatis cultures were diluted into fresh liquid media, and growth was assessed over time by measuring turbidity until the cultures once again reached stationary phase (Fig2A). Aliquots were collected at various intervals for Western blot analysis using anti-Pup antibodies (Fig2B, upper panel). In addition, the level of pupylated proteins in each sample was quantified (Fig 2A and Supplementary Fig S1A) as a percentage of the total protein mass using, as standard, a purified pupylome, namely the isolated pool of pupylated proteins, generated as described in Materials and Methods. The results indicated that the level of pupylated proteins changed in correlation with changes in growth phase. Specifically, the level of pupylated proteins at the beginning of the experiment, while the cells were still adjusting to growth in liquid media, accounted for approximately 2% (w/w) of the total protein population (Fig2A). This level, however, eventually dropped to approximately 0.3% as the cultures resumed exponential growth (Fig2A). Notably, as the bacteria re-entered stationary phase following 36 h of growth from the point of dilution, a sharp increase in the level of pupylated proteins, followed by an additional reduction in their level, was noted as the cells remained at stationary phase (Fig2A and B). The increased level of pupylated proteins appearing at stationary phase correlated with an increase in PafA levels (Fig2B, lower panel), suggesting an elevated pupylation rate at stationary phase. Loading controls for Fig2B, as well as for other Western blot analyses in this study, are presented in Supplementary Fig S2.
Figure 2. Elevated pupylation is observed at stationary phase.
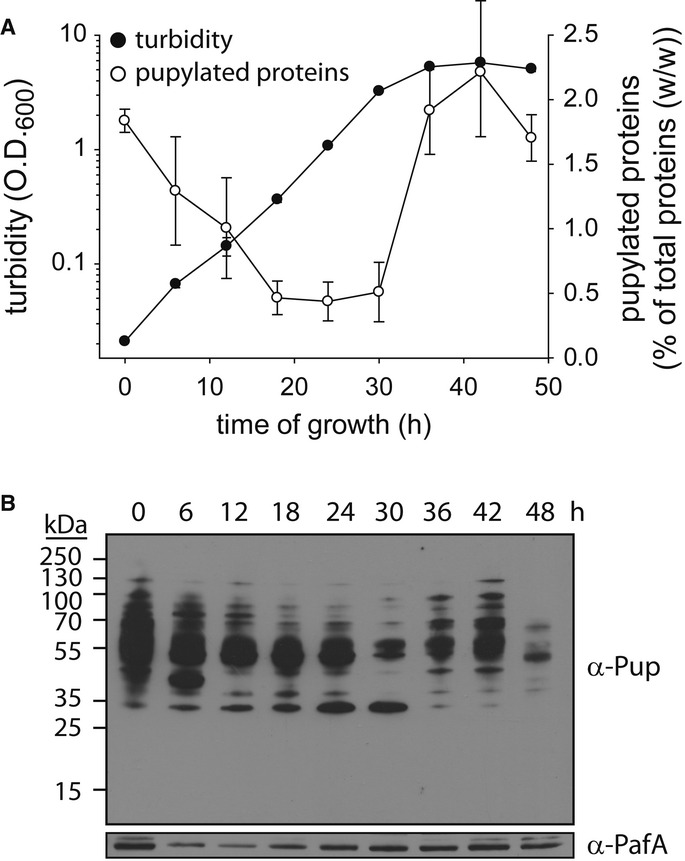
- Three stationary-phase Mycobacterium smegmatis cultures were diluted into fresh media, and growth at 30°C was monitored by measuring turbidity. In addition, culture samples were collected every 6 h for quantification of pupylated proteins as described in Materials and Methods and in the legend to Supplementary Fig S1A. Data are presented as mean ± SD; n = 3.
- Western blot analysis of aliquots removed at the indicated intervals was carried out using anti-Pup and anti-PafA antibodies.
To compare the pupylomes of exponential and stationary-phase M. smegmatis cultures, we adopted the methodology described by Festa et al (2010) for pupylome purification. In short, simultaneously polyhistidine- and Strep-tagged Pup was expressed in M. smegmatis, and pupylomes were purified from exponential- and stationary-phase cultures via tandem purification procedures using Ni2+-NTA and Strep-Tactin affinity chromatography (Supplementary Fig S1B and C). As controls, cultures not expressing the dually tagged Pup were used. Proteins purified from all cultures were identified by mass spectrometry, and a protein was considered to be part of the pupylome only if it was detected in extracts prepared from cultures that expressed the dually tagged version of Pup and not in the control culture. We detected 42 and 93 protein species (excluding Pup itself) in the pupylomes of exponential- and stationary-phase cultures, respectively (Fig3 and Supplementary Tables S1 and S2). Of these, 19 were detected in both pupylomes. Comparison of protein levels in each pupylome indicated that the protein concentration in the stationary-phase pupylome was much higher than that of the exponential-phase pupylome (Supplementary Fig S1B). Therefore, many more copies of each identified protein species were pupylated during stationary phase. There is a concern that some of the proteins detected were not pupylated, but rather associated with the pupylome. Nonetheless, many proteins in our list were also identified in previous M. smegmatis pupylome analyses (Poulsen et al, 2010; Watrous et al, 2010), and proteins homologous to those included in our list were also identified in the M. tuberculosis pupylome (Festa et al, 2010). For instance, both model pupylation substrates PanB and InoI (Pearce et al, 2006; Burns et al, 2010b; Ofer et al, 2013) were detected in our analysis. Importantly, although more pupylated proteins were detected in stationary phase, and although some of these proteins merely associated with the pupylome rather than undergoing pupylation, the markedly broad specificity of PafA, a hallmark of protein pupylation, manifested itself at both growth stages (Fig3).
Figure 3. Pupylome analysis.
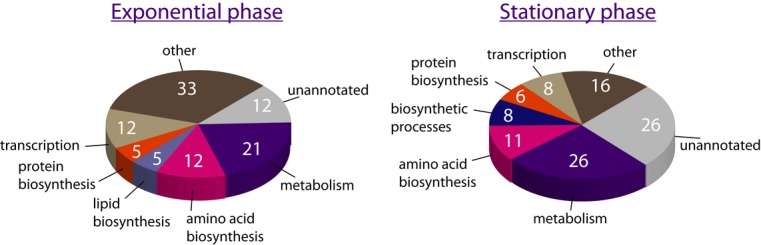
Classification of the exponential- and stationary-phase pupylomes according to protein function. The numbers are the percentage of each functional group out of the total pupylome (42 proteins at exponential phase and 93 at stationary phase). Functional groups comprising less than 3% of the pupylomes were included in the group designated as “other”.
Phenotypic examination of a pupylation-deficient mutant
We next considered the possibility that the PPS plays an important role in M. smegmatis physiology during stationary phase. To test this hypothesis, the survival of a PPS-deficient mutant (ΔprcSBA) following arrival at stationary phase was examined. As prcS encodes Pup and prcBA encodes the 20S β and α subunits, respectively (Fig4A), the ΔprcSBA mutant cannot pupylate proteins and lacks intact proteasomes (Shenkerman et al, 2013 and Fig4B). Indeed, when Western blot analysis was performed using antibodies against the 20S α subunit, no bands were detected in the mutant strain (Fig4B, lower panel). When similar analysis was conducted using anti-Pup antibodies, multiple bands representing pupylated proteins were detected in the wild-type strain, whereas only two bands were detected in the mutant (Fig4B, upper panel). Through the use of a ΔpafA mutant (Supplementary Fig S3A–C), we found that these two bands represent antibody-cross-reactive proteins, rather than corresponding to pupylated proteins. Despite the absence of PafA, known to be essential for pupylation (Pearce et al, 2008), the two proteins in questions were nonetheless detected by Western blot analysis using anti-Pup antibodies (Supplementary Fig S3D). Furthermore, expression and tandem purification of polyhistidine- and FLAG-tagged Pup in this mutant resulted in the elution of the dually tagged Pup while the cross-reacting proteins were washed away during the purification steps (Supplementary Fig S3E and F). As such, it can be concluded the ΔprcSBA mutant completely lacks pupylated proteins. Expression of the prcSBA operon from a chromosomally integrated plasmid fully complemented the pupylation and proteasome deficiencies of this mutant (Fig4B). Both the wild-type and the mutant grew equally well at 30°C during the exponential phase, with a generation time of about 4 h (Fig4C). These findings are consistent with previous reports showing that PPS-deficient mutants of M. tuberculosis and M. smegmatis multiply as fast as do the parental wild-type strains (Darwin et al, 2003; Cerda-Maira et al, 2010; Imkamp et al, 2010a). By contrast, after prolonged incubation at stationary phase (17 days after inoculation), 10-fold fewer ΔprcSBA cells were alive, in comparison with wild-type cells (Fig4D and E). As both strains reached a similar cell density upon entrance into stationary phase (Fig4C), this phenotype reflects the reduced survival of the PPS-deficient mutant at stationary phase. This phenotype was fully complemented upon expression of the prcSBA operon from a chromosomally integrated plasmid, indicating that the reduced survival of the mutant results from the lack of intact PPS rather than because of a polar effect of the deletion mutation (Fig4D and E). Moreover, integration of an empty vector into either the wild-type or the mutant chromosome did not affect the ability of these cells to survive during stationary phase. Collectively, the data presented thus far indicate that the elevated levels of pupylated proteins noted in stationary-phase cultures correlate with the reduced survival of a PPS-deficient mutant after prolonged incubation at stationary phase.
Figure 4. Starvation sensitivity of a PPS-deficient mutant.
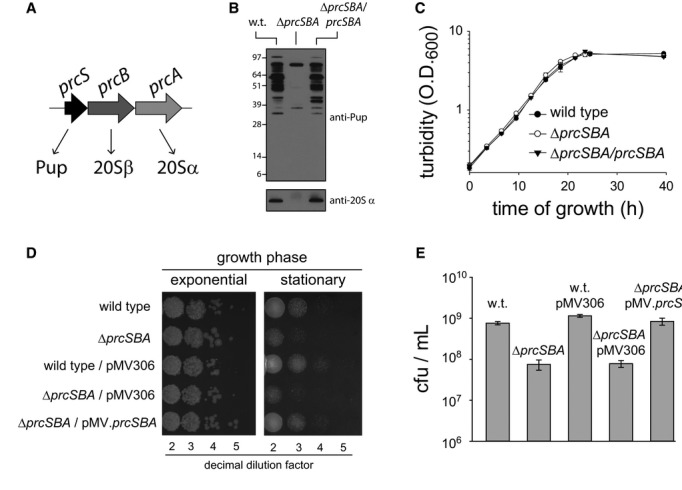
- The prcSBA operon.
- Western blot analysis using anti-Pup and anti-20Sα antibodies, as indicated.
- Exponentially growing cultures were diluted into fresh media, and turbidity was measured (n = 3) at the indicated time points.
- Exponentially growing cultures were diluted into fresh media (day 0), and samples were collected during the exponential phase at day 18. All samples were diluted to an OD600 of 0.5, serial dilutions were carried out, and 10 μl of the indicated dilutions were plated on solid media.
- The concentration of live cells in 17-day-old liquid cultures was determined via colony counts (n = 3) following sample plating on solid media.
Carbon versus nitrogen limitation
Cells enter stationary phase when nutrients become limiting (Blackman, 1905). In the standard M. smegmatis growth medium (7H9 containing 0.05% (v/v) Tween-80 and 0.4% (v/v) glycerol), such as used in the experiments described in Figs2, 3 and 4, the carbon source, glycerol, is a growth-limiting factor (Supplementary Fig S4). At higher glycerol concentrations, however, the amount of available nitrogen becomes limiting (Supplementary Fig S4). Hence, to examine the importance of the PPS at stationary phase in more depth, the survival of wild-type and ΔprcSBA strains was monitored under conditions of either carbon or nitrogen limitation. To this end, cultures that grew exponentially in defined minimal medium were centrifuged and resuspended in a similar medium lacking either carbon or nitrogen sources. These cultures were kept at 30°C with aeration, and cell viability was monitored over the course of 15 days (Fig5A). To account for changes in cell density during the prolonged period of incubation, turbidity measurements were carried out and viability was normalized accordingly (Fig5B and C). At the first time point considered after either carbon or nitrogen starvation, we detected an increase in the number of cells for both the wild-type and the mutant strains, probably due to consumption of internal nutrient reservoirs. At the same time, the culture turbidity under carbon starvation decreased mildly, possibly reflecting changes in cell size under starvation. Importantly, in response to carbon or nitrogen limitation, the PPS-deficient mutant presented significant survival defects in comparison with the wild-type strain. Under carbon limitation, a approximately 10-fold decrease in the viability of the mutant cells was measured following 15 days, whereas that of the wild type was hardly compromised. These findings are consistent with the observations reported in Fig4D and E showing a 10-fold decrease in survival of the mutant following prolonged incubation in stationary phase. Strikingly, under nitrogen limitation, the differences between the wild type and the mutant culminated up to approximately 150-fold. It seems, therefore, that the PPS-deficient mutant is hypersensitive to nitrogen limitation, and, to a lesser extent, to carbon limitation. Expression of the prcSBA operon from a chromosomally integrated plasmid fully complemented the mutant phenotype in the face of nitrogen limitation (Fig5D), indicating that the PPS deficiency is indeed responsible for the acute phenotype observed under these conditions. Interestingly, whereas prcBA expression in the mutant did not restore survival of the mutant under nitrogen limitation, expression of prcS alone resulted in partial complementation of the mutant phenotype (Fig5D). Indeed, whereas the mutant presents an approximately 150-fold decrease in survival following nitrogen starvation, the prcS-complemented strain is only approximately 10-fold less viable than either the wild-type or the fully complemented strain. These findings indicate that either pupylation alone, without degradation, plays an important role under nitrogen starvation or pupylated proteins can be degraded in the absence of the 20S complex, albeit less efficiently than in the presence of this particle. Results presented below support the latter possibility.
Figure 5. Hypersensitivity of a PPS-deficient mutant to nitrogen starvation.
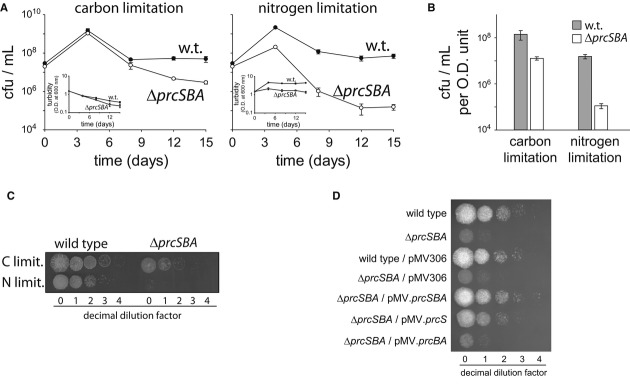
A, B Exponentially growing cultures (OD600 = 0.5) were centrifuged and resuspended in media lacking either carbon (left) or nitrogen (right) sources. Following resuspension (time = 0), aliquots were collected at the indicated time points for determination of live cell concentration (n = 3).
C Spot test of the cultures shown in (A) following 15 days of nutrient limitation. Cell density of all cultures was normalized to OD600 of 0.5 prior to serial dilution.
D Spot test of the indicated cultures following 11 days of nutrient limitation. Cell density of all cultures was normalized to OD600 of 0.5 prior to serial dilution.
PPS induction in response to nitrogen deprivation
Since the phenotype of a PPS-deficient mutant is considerably more severe under conditions of nitrogen deprivation, as compared to carbon deprivation, we focused our attention on the response of the PPS in the face of nitrogen limitation. To follow the dynamics of pupylation and PPS induction in response to restricted access to nitrogen, samples were collected from a wild-type culture at time points from the onset of nitrogen starvation and subjected to Western blot analysis using antibodies against Pup. Such analysis showed that the level of pupylated proteins increased 30 min following the onset of nitrogen limitation, continued to increase throughout additional 1 h and a half, and remained high for at least 24 h (Fig6A, upper panel). Next, in the time frame spanning days 1–6, most of the pupylated proteins disappeared. The temporally correlated dramatic increase in the level of the 20S α subunit (hereafter, 20Sα), as observed via Western blot analysis using antibodies against this protein (Fig6A, lower panel), suggested that the disappearance of the pupylated proteins resulted from proteasomal degradation. This was indeed supported by a comparison of the level of pupylated proteins in proteasome-deficient and proteasome-expressing strains. In both cases, pupylated proteins accumulated during the first day following nitrogen starvation (Fig6B). However, these proteins almost completely disappeared at later time points in the wild-type strain, while their levels remained high in the proteasome-deficient strain (Fig6B). This is the first indication of a robust proteasome-dependent degradation of the entire pupylome in response to an external stimulus. We did, however, notice some reduction in the level of pupylated proteins on day 13, even in that strain lacking a 20S particle. Since equal protein amounts were loaded in each lane of the gel used for the Western blot analysis (see loading control in Supplementary Fig S2), this reduction in the levels of pupylated proteins can be interpreted as either reflecting depupylation or 20S-independent degradation. We also noticed that pupylated proteins accumulated to a much higher level in the 20S-deficient strain as early as the first day of nitrogen starvation (Fig6B). We considered whether the reduced accumulation in the wild type could be the result of 20S-dependent degradation of pupylated proteins at early stages of nitrogen starvation. Addressing this issue is somewhat complex, as the levels of pupylated proteins are affected at each time point by three distinct processes, namely pupylation, depupylation, and degradation. To analyze the degradation of pupylated proteins at the early stages of nitrogen starvation, we designed an experimental system that is unaffected by either pupylation or depupylation. To this end, we relied on the use of a previously described Pup-Zur chimeric protein (Burns et al, 2010a). Msm Zur lacks lysines and, therefore, is not a PafA substrate. Moreover, Pup-Zur presents Pup as a N-terminal fusion to Zur, rather than isopeptide-bonded to a lysine side chain. As such, Pup-Zur, and in general, Pup fusions, cannot serve as substrates for depupylation by Dop (Imkamp et al, 2010b), although they serve as proteasome substrates (Burns et al, 2010a; Striebel et al, 2010). Conveniently, Zur presents five consecutive histidines, allowing its detection using anti-polyhistidine antibodies. Nonetheless, to facilitate sensitive detection, we cloned a genetic fusion that encodes six histidines at the C-terminus of Pup-Zur. In our experimental system, prcS-zur is cloned into plasmid pJV53 (Van Kessel et al, 2008) under the transcriptional control of the acetamidase promoter. As such, chimera expression can be induced upon addition of acetamide to the growth medium. To examine degradation of Pup-Zur following nitrogen starvation, the fusion protein was initially expressed in exponentially growing wild-type and ΔprcSBA cells. We found expression by the acetamide promoter to be tightly controlled, as no Pup-Zur was detected prior to the induction step, as determined by Western blot analysis using anti-polyhistidine antibodies (Fig6C). Next, the cells were starved for nitrogen via multiple steps of centrifugation and resuspension in nitrogen- and acetamide-depleted media. Control cultures were resuspended in medium depleted of acetamide yet containing nitrogen. Rifampicin was added to ensure complete arrest of Pup-Zur expression, and aliquots were removed over time for Western blot analysis using anti-polyhistidine antibodies. We found that Pup-Zur disappeared in the wild-type strain as early as 5 h following starvation, whereas Pup-Zur concentration remained largely constant in the ΔprcSBA mutant (Fig6C), indicating that the chimeric protein is indeed degraded by the proteasome. Importantly, degradation was much faster in the wild-type following nitrogen starvation than in non-starved cells (Fig6C). This degradation was Pup dependent, as Zur, when not fused to Pup, was not proteolyzed under similar conditions (Fig6C, lower panel). These results indicate that proteasomal degradation of pupylated proteins is accelerated in M. smegmatis following nitrogen starvation. Altogether, our findings indicate that both tagging and degradation of proteins by the PPS are induced in response to nitrogen starvation.
Figure 6. PPS induction under nitrogen starvation.
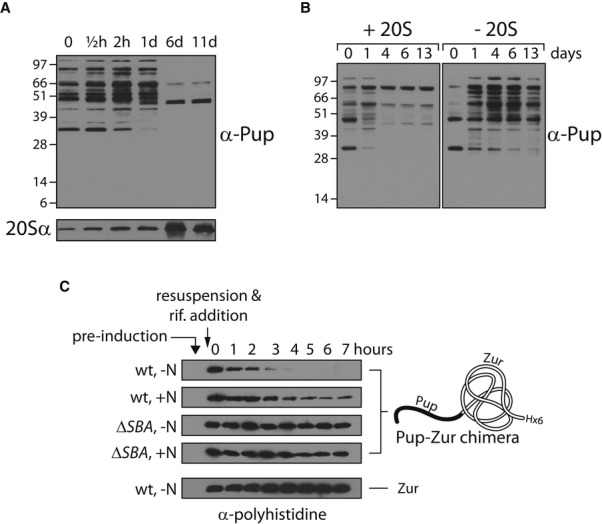
- Exponentially growing cultures (OD600 = 0.5) of wild-type Mycobacterium smegmatis were starved for nitrogen, and samples were collected at the indicated time points from the onset of starvation (t = 0) for Western blot analysis
- As in (A), except that in addition to the wild-type strain (designated +20S), a ΔprcSBA mutant that expresses the prcS gene from a chromosomally integrated plasmid was used (designated -20S).
- M. smegmatis cultures carrying a plasmid encoding either Zur or a prcS-zur fusion, as indicated, under the transcriptional control of the acetamidase promoter were grown to exponential phase (OD600 = 0.5) before 0.2% (w/v) acetamide was added. Three hours later, the cultures were centrifuged and resuspended in media lacking acetamide and either lacking or containing a nitrogen source, as indicated. Rifampicin (200 μg/ml) was added to the cultures, and samples were collected at the indicated time points for Western blot analysis using antibodies against a polyhistidine tag.
Data information: Loading controls for all the blots in this figure are presented in Supplementary Fig S2.
PPS-negative auto-regulation
Induced 20S expression, as reflected in Fig6A, was accompanied not only by a disappearance of pupylated proteins but also by a reduction in Mpa, PafA, and Dop levels (Fig7A). Mpa is known to be a pupylation target and a proteasome substrate (Delley et al, 2012; Forer et al, 2013). As such, the loss of Mpa in response to 20S induction is expected. By contrast, the disappearance of PafA and Dop came as a surprise, as pupylation of PafA and Dop has not been previously documented. Nevertheless, like Mpa, PafA and Dop were identified in the M. smegmatis pupylome (Supplementary Tables S1 and S2), suggesting that these PPS components are pupylated prior to their degradation by the proteasome. To obtain more direct evidence for PafA pupylation, an in vitro assay was conducted by mixing purified PafA in pupylation buffer together with a purified Pup variant presenting glutamate at its C-terminus (PupE). Following addition of ATP, pupylation products accumulated over time (Fig7B, upper panel). No pupylation was observed in the absence of either ATP or PupE (Fig7C, upper panel). Strikingly, PafA underwent multiple pupylation events in a manner reminiscent of the pattern observed upon poly-ubiquitylation. Due to its depupylase activity, a similar assay could not be performed for Dop.
Figure 7. PPS-negative auto-regulation.
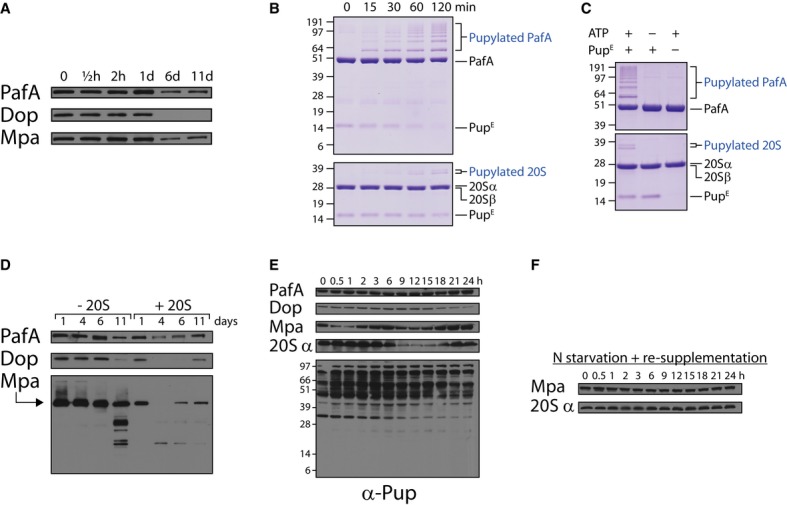
- As in Fig6A, except that antibodies against the indicated proteins were used in the Western blot analysis.
- 20 μM PupE were incubated in pupylation buffer at 30°C together with 5 μM PafA (upper panel) or with 1 μM PafA and 5 μM 20S (lower panel). The first sample was removed (t = 0), ATP (2 mM) was added to start the reactions, and additional samples were removed at the indicated time points for SDS–PAGE followed by Coomassie Brilliant Blue staining.
- Reactions were mixed as in (B) with or without ATP or PupE, as indicated. Following a 2-h incubation at 30°C, aliquots were collected and subjected to SDS–PAGE followed by Coomassie Brilliant Blue staining.
- As in (A), except that a ΔprcSBA mutant was used that expresses from a chromosomally integrated plasmid either the prcS gene (designated -20S) or the prcSBA operon (designated +20S).
- Western blot analysis using antibodies against the indicated proteins was performed on samples collected at the indicated time points during a nitrogen starvation experiment that was performed as described in Fig6A.
- As in (D), except nitrogen source was re-supplemented following an hour of starvation.
Next, PafA, Mpa, and Dop levels were compared in 20S-positive and 20S-negative cells. Following the first few days of nitrogen limitation, PafA and Dop levels, as well as those of Mpa, decreased in proteasome-expressing cells, yet remained stable in the proteasome-deficient strain (Fig7D and Supplementary Fig S5A). After 11 days of starvation, however, the levels of Mpa, Dop, and PafA decreased even in 20S-deficient cells (Fig7D), as already noted for pupylated proteins (Fig6B). In the case of Mpa, what appear to be degradation intermediates were observed at day 11 of nitrogen starvation (Fig7D). It is possible, therefore, that Mpa, PafA, and Dop are degraded by an alternative route in the absence of the 20S, albeit much less efficiently than in the presence of the particle.
Following degradation of PPS components in 20S-positive cells, we noticed that these often reappear at later time points. For instance, whereas both Dop and Mpa disappeared by the fourth day following starvation, Dop was detected again at day 11, while Mpa already reappeared by the sixth day of starvation. A similar tendency was observed for PafA, albeit less dramatically and less consistently (Fig7D and Supplementary Fig S5A). These findings raise the possibility that PPS proteins oscillate during starvation. This was found to indeed be the case when the levels of PPS proteins were monitored at short intervals over the course of the first day following starvation (Fig7E). Whereas PafA levels hardly changed, remarkable oscillations were observed in the levels of Mpa and 20Sα. Dop levels oscillated as well, albeit with an amplitude that appeared to be lower than those noted for Mpa and the 20S α subunit. Moreover, these proteins oscillated with different periods. Whereas Mpa levels peaked 0, 6, and 24 h following starvation, Dop levels peaked only once, 6 h following starvation, during the 24 h of the experiment. The 20Sα was maintained at a stable level at the beginning of starvation and started to oscillate only 6 h later. 20Sα levels then initially decreased for 12 h, at which point they again increased throughout the rest of the experiment. The finding that the 20Sα oscillates indicates that this protein, like other PPS components, is proteolyzed in vivo. In an agreement, 20S subunits were previously identified in the M. smegmatis pupylome (Poulsen et al, 2010), and we could show in an in vitro pupylation assay similar to the one performed for PafA that the 20S is indeed a pupylation substrate (Fig7B and C, lower panels). Despite the oscillatory expression of PPS components, no dramatic oscillations in the levels of pupylated proteins were detected (Fig7E).
Given our ability to detect oscillations of PPS components following nitrogen starvation, it follows that starved cells should be synchronized as otherwise oscillations would not be observable, considering that our measurements detect the average level of each examined component. Yet, a week following starvation, oscillations of PPS components were much less obvious over the course of the next 24 h (Supplementary Fig S5B). Presumably, cells in the starved cultures gradually desynchronize from the moment of starvation and, concomitantly, synchronization.
Finally, we wondered whether the oscillatory expression of PPS components is triggered by nitrogen starvation or whether oscillations occur constantly yet were only detected in our experiments after cell synchronization. To differentiate between these two possibilities, we starved cells for nitrogen for an hour and then replenished culture nitrogen sources. After an hour of starvation, the cells should be synchronized, as in the absence of any source of nitrogen, Mpa oscillations were already detected (Fig7E). By contrast, following addition of a nitrogen source, the levels of both Mpa and the 20Sα, those two proteins for which oscillations were most obvious in our experiments, appeared stable over the course of the subsequent 24 h from the moment of nitrogen re-supplementation (Fig7F). These findings thus indicate that nitrogen starvation induces oscillatory expression of Mpa, Dop, and the 20S particle.
Discussion
The work presented here provides the first link between the PPS and bacterial physiology. It also provides the first evidence for a PPS response to external stimuli. The data indicate that the PPS plays a crucial role in M. smegmatis physiology under starvation. This conclusion is supported both by phenotypic analysis of a PPS-deficient mutant and by analysis of the PPS response to nutrient limitation. We find the phenotype of a PPS-deficient mutant, the broad substrate specificity, and the delicate regulation of the PPS under nitrogen limitation to be consistent with the model illustrated in Fig8. This model proposes that the PPS acts as an amino acid recycling machinery for the generation of protein building blocks and a carbon source under conditions in which de novo synthesis of amino acids is restricted. Amino acid recycling under starvation is not a novel concept (Lilly et al, 1991; Vabulas & Hartl, 2005; Suraweera et al, 2012). Up until now, however, it was not conceived as a role played necessarily by the PPS.
Figure 8. Protein recycling by the PPS in response to nutrient starvation.
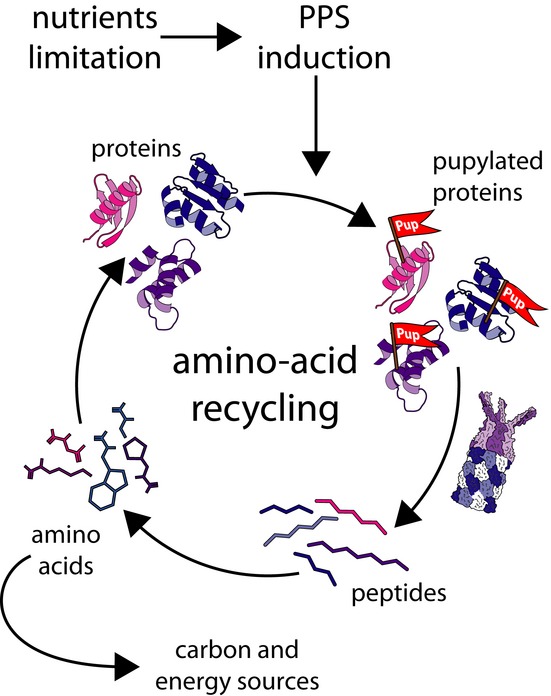
In response to nutrient limitation, PPS induction leads to accelerated protein pupylation and degradation. The resulting protein building blocks can be used to remodel the proteome in response to changing environmental conditions.
An amino acid recycling machinery
The response of M. smegmatis to nitrogen limitation was recently characterized at the genomic and transcriptomic levels. Such analyses found that, among mycobacterial species examined thus far, M. smegmatis carries the highest number of genes involved in nitrogen metabolism (Amon et al, 2009; Jenkins et al, 2013; Williams et al, 2014). As such, M. smegmatis is well equipped to withstand nitrogen stress. As the nitrogen source becomes a growth-limiting factor for M. smegmatis, a reduction in metabolic activity is accompanied by a global adaptation at the level of gene expression. More than 100 genes responsible for nitrogen scavenging, accelerated nitrogen transport into the cell, and efficient assimilation of various nitrogen sources are induced in the first few hours of nitrogen limitation (Jenkins et al, 2013; Williams et al, 2013). However, these mechanisms are only efficient up to a certain point. Below a minimum of extracellular nitrogen concentration, M. smegmatis must rely on internal nitrogen sources to maintain minimal metabolic activity. We propose that the PPS answers this requirement. By degrading proteins, the PPS generates protein building blocks that cannot support an increase in biomass, yet allow adaptation to environmental changes via the ad hoc synthesis of proteins as required by the organism in light of these changes. For example, as a saprophytic soil bacterium, M. smegmatis experiences periodic changes, such as the temperature fluctuations that accompany day/night cycles, in its natural habitat. Adaptation to such fluctuations often requires flexibility at the proteome level. The PPS could provide M. smegmatis the flexibility to remodel its proteome even in the absence of an external nitrogen source. In view of the role suggested here for the PPS, the extreme broad specificity of PafA makes perfect sense. Indeed, if the PPS functions primarily to generate protein building blocks, substrate specificity is of little importance. We find that Pup expression in a ΔprcSBA mutant partially complements the survival deficiencies of this mutant under conditions of nitrogen starvation. Does this mean that pupylation alone plays an important role in starved M. smegmatis cells or that pupylated proteins are degraded to some extent even in the absence of the 20S? While not excluding the former possibility, our data reinforce the latter, as we observe modest disappearance of pupylated proteins in 20S-deficient cells (Fig6B). This disappearance could be attributed to depupylation, rather than degradation, of the pupylated proteins. However, PafA, Mpa, and Dop were found to be degraded in these cells with the same timing as the disappearance of pupylated proteins in 20S-deficient cells, the former events albeit occurring much less efficiently than in wild-type cells. As PafA, Mpa, and apparently Dop are pupylation substrates, their slow degradation, together with the simultaneous slow disappearance of other pupylated proteins in the absence of the 20S particle, suggests that pupylated proteins can be degraded, inefficiently as it may be, by an alternative route. This possibility indeed explains why expression of Pup alone can partially complement the nitrogen sensitivity of the ΔprcSBA mutant. It is noteworthy that alternative degradation routes were reported in parallel proteolytic systems (Kanemori et al, 1997; Lies & Maurizi, 2008). For instance, SsrA-tagged proteins are degraded in Escherichia coli primarily by ClpXP, yet can also be degraded less efficiently by other cytoplasmic proteases (Lies & Maurizi, 2008).
PPS dynamics and negative auto-regulation
Due to their destructive nature, intracellular proteolytic systems are carefully regulated so as to prevent unnecessary degradation of cellular proteins (Gur et al, 2011). Our data indicate that this principle also applies to the PPS. During exponential growth, both pupylation and proteasomal degradation are not induced, resulting in slow pupylation and degradation rates, respectively. In response to nitrogen starvation, PPS activity is induced, leading to accelerated pupylation and degradation of pupylated proteins. Intriguingly, whereas pupylated proteins accumulated during the early stages of stress, these proteins almost completely disappeared at later stages following starvation. These findings indicate that during the early stages of starvation, pupylation dominates over degradation, as if the cell “prepares the setting” for degradation (together with inducing nitrogen-scavenging mechanisms), while accelerating the irreversible destructive step of proteasome-mediated degradation only later during starvation, as a last resort.
The proteasome degrades Mpa, PafA, and Dop, and seemingly also the 20S α subunit, thereby reducing the rates of both protein pupylation and degradation. In other words, the PPS negatively auto-regulates its own activity via self-degradation of its components. As a result, a negative feedback loop is formed in which accelerated pupylation facilitates degradation of PPS components, which, in turn, reduces the pupylation rate and, concomitantly, proteolysis of PPS components (Fig9A and B). As such, this negative feedback loop prevents uncontrolled pupylation and degradation of cellular proteins. Such negative auto-regulation, rather than ensuring stable steady-state levels of the PPS components, promotes the oscillatory expression of these proteins in response to nitrogen starvation. Indeed, negative feedback loops are often essential components of oscillatory systems, alongside proper time-scale balancing, as dictated by the kinetics of those enzymes involved and their rates of synthesis (Novák & Tyson, 2008). It appears that these kinetic requirements are satisfied in the case of the PPS following nitrogen starvation, when both pupylation and proteasomal degradation are accelerated. At present, however, in-depth understanding of the factors that promote PPS oscillations is limited by the availability of information regarding the regulatory mechanisms that control PPS gene expression and challenges to describing the kinetics of PPS activity in vivo. Despite the oscillatory expression of PPS components, modest fluctuations in the levels of pupylated proteins were observed. This outcome reflects how the levels of pupylated proteins are determined by three parallel processes at every time point considered (i.e., pupylation, depupylation, and degradation). As such, an increased pupylation rate, for instance, would not lead to an increase in pupylated protein levels if balanced by a similar increase in the rate of degradation.
Figure 9. PPS auto-regulation through negative feedback loops.
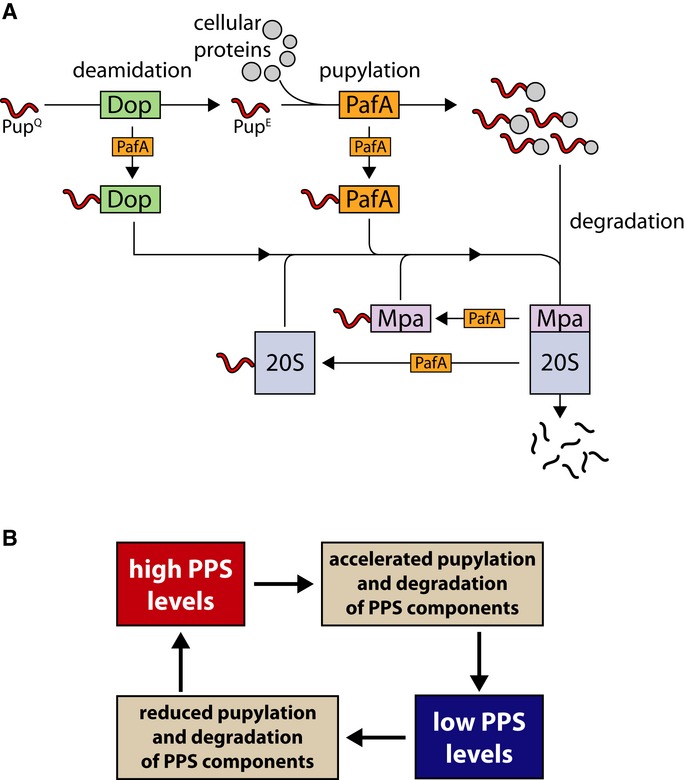
- Like many other cellular proteins, PPS components are subjected to pupylation and proteasomal degradation. For simplicity, depupylation is ignored in the illustration presented.
- As a result of the interactions illustrated in (A), a negative feedback loop is formed that prevents uncontrolled tagging and degradation of cellular proteins.
What purpose could such oscillations of PPS components serve? One reasonable answer is the fine-tuning of amino acid recycling so as to balance nutritional requirements under starvation with the need to prevent unnecessary proteolysis. By acting in pulses, the PPS generates a transient pool of amino acids that allows the cell to reassess its nutritional needs before another cycle of degradation is activated. By analogy, the PPS can be considered to act as an oscillating thermostat that avoids over-heating the system via controlled release of short pulses of heat, all the while measuring the temperature. Still, how nitrogen starvation is sensed by the PPS remains an open question.
General implications
The PPS is conserved in Actinobacteria and Nitrospira, two bacterial phyla that together comprise thousands of species. As many of these species are, like M. smegmatis, saprophytic soil microorganisms encountering fluctuating environmental conditions, it is highly likely that protein recycling under starvation conditions is a general role of the PPS. Still, there exist actinobacterial species, such as Corynebacterium glutamicum, that encode an incomplete PPS in their genomes. Specifically, they lack the 20S-coding genes. In these species, protein pupylation may thus play a regulatory role, rather than functioning as a protein recycling machinery. Alternatively, pupylated proteins may be degraded by a non-proteasomal protease in these species. Finally, it remains to be determined to what extent our model applies to parasitic mycobacteria, such as M. tuberculosis. Undoubtedly, such species encounter conditions and stresses that differ markedly from those encountered by saprophytic species. Accordingly, the PPS in such bacteria may have evolved to respond to the unique conditions encountered in their hosts. It appears, however, that a necessity for the PPS under starvation conditions is conserved between M. smegmatis and M. tuberculosis. Indeed, a proteasome-deficient M. tuberculosis mutant presents compromised survival at stationary phase and reduced persistence in host tissues (Gandotra et al, 2010). These observations, therefore, suggest that our findings represent a general property of the PPS that applies both to saprophytic species and to mycobacterial pathogens, such as M. tuberculosis. As a rule, bacterial pathogens must acquire nutrients within their hosts in order to persist and replicate (Appelberg, 2006; Rohmer et al, 2011). Here, an enthralling comparison can be made between M. tuberculosis and Legionella pneumophila (both are intracellular pathogens). Whereas the latter depends on host proteasome-mediated protein degradation to replenish its nitrogen and carbon supply (Price et al, 2011), M. tuberculosis relies on its own proteasome during infection, probably for the same reason.
Materials and Methods
Strains, plasmids, and growth conditions
Unless stated otherwise, M. smegmatis MC2155 (wild-type and mutant) cultures were grown at 30°C in Middlebrook 7H9 broth containing 0.05% (v/v) Tween-80 and 0.4% (v/v) glycerol. For nitrogen and carbon starvation experiments, cultures were grown in defined minimal medium adapted from Davis and Mingioli (1950) containing 40 mM K2HPO4, 22 mM KH2PO4, 15 mM (NH4)2SO4, 1.7 mM sodium citrate, 0.4 mM MgSO4, 0.4% glycerol (v/v), and 0.05% Tween-80 (v/v). Exponentially growing cultures were harvested, centrifuged, washed thrice, and resuspended in similar media lacking either (NH4)2SO4 (to induce nitrogen starvation) or glycerol (to induce carbon starvation). Solid media were prepared using Middlebrook 7H10 supplemented with 0.4% glycerol. E. coli ER2566 (New England Biolabs) was routinely used for all cloning procedures and was grown using typical procedures on LB broth and plates at 37°C. Plasmid pMV306 (Stover, 1991) was used in complementation assays to integrate a cloned prcSBA operon into the chromosome of a M. smegmatis ΔprcSBA mutant. Plasmid pMV206 (Stover, 1991) was used for cloning and expression of PafA. A polyhistidine-strep-pup fusion and zur variants were cloned into plasmid pJV53 (Van Kessel et al, 2008) instead of genes Che9c 60-61, under the transcriptional control of the acetamidase promoter (Parish et al, 1997). Plasmid pET11a was used for cloning and expression of prcBA.
Protein purification
For purification of the M. smegmatis pupylome, expression of a polyhistidine-strep-pup fusion was induced in M. smegmatis cells, in mid-exponential phase at 37°C by addition of 0.2% (w/v) acetamide to the growth medium. The cultures were harvested 3 and 24 h later, and pupylome purification was carried out as previously described (Festa et al, 2010). Cultures that did not express the dually tagged Pup served as controls.
PafA and Pup were purified as previously described (Ofer et al, 2013). For 20S purification, E. coli BL21 cells carrying plasmid pET.prcBA were grown in LB medium containing ampicillin (100 μg/ml) at 37°C. At OD600 = 0.7, 20S expression was induced for 18 h by the addition of IPTG (0.5 mM). The cells were harvested and resuspended in 25 mM Tris–HCl, pH 8.0, 1 mM DTT (hereafter, Tris-DTT). Following sonication, the lysate was centrifuged (12,000 g, 4°C, 20 min) and the clear supernatant was loaded onto a HiTrap QFF column (GE Healthcare) pre-equilibrated with Tris-DTT buffer. Elution was performed using a 0–1 M linear NaCl gradient. 20S-rich fractions were pooled, concentrated, and loaded on a Superose 6 (GE Healthcare) size-exclusion column, pre-equilibrated with Tris-DTT buffer containing 100 mM NaCl. Following elution, 20S-containing fractions were pooled and loaded onto a Mono Q column (GE Healthcare) pre-equilibrated with Tris-DTT buffer containing 200 mM NaCl and elution was performed using a linear NaCl gradient (200–800 mM). 20S-containing fractions were pooled and loaded on a Superose 6 column, pre-equilibrated with Tris-DTT buffer containing 600 mM NaCl. Following elution, 20S-containing fractions were pooled, and the buffer was exchanged for 50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM DTT and 10% glycerol. The purified protein was stored in aliquots at −80°C.
Western blot analysis of M. smegmatis lysates
M. smegmatis lysates were prepared by sonication of cell pellets in microcentrifuge tubes containing 0.5 ml of 1 mM Tris–HCl, pH 8.0, 1 mM EDTA. Cell debris was removed by centrifugation (20,000 g, 4°C). After determining protein content in each sample by Bradford assay, equal protein amounts were loaded onto SDS–PAGE for electrophoretic separation, followed by transfer onto PVDF membranes and immuno-detection using standard procedures. As a final step after completion of immunodetection, probed membranes were stained by Coomassie Brilliant Blue to verify the equal loading and transfer of proteins in each lane.
Polyclonal antibodies against Pup, Mpa, Dop, and the 20S core particle were produced by Covance, while anti-PafA antibodies were produced by Adar Biotech. Full-length proteins served as antigens for immunization, except in the case of Dop, where a synthetic peptide was used. All antibodies were affinity purified.
Deletion of the paf operon
DNA fragments flanking the Msm paf operon were amplified by PCR and cloned into the suicide plasmid, pYUB854 (Bardarov et al, 2002), upstream and downstream of the hygromycin resistance cassette. Wild-type Msm cells were then transformed with the cloned plasmid and spread onto plates containing hygromycin (150 μg/ml). To identify paf deletion mutants, colonies were doubly screened by Western blotting using anti-Pup and anti-PafA antibodies. For complementation assays, the Msm pafA open reading frame and a region 207 bp upstream of the translation start site were cloned into the mycobacterial shuttle vector, pMV206.
Southern blot analysis
Chromosomal DNA was purified from wild-type and ΔpafA strains using the Qiagen RNeasy Mini kit. DNA probes were labeled with a PCR DIG Probe Synthesis kit (Roche), and band detection was carried out with an anti-DIG alkaline phosphatase antibody (Roche cat# 1093274) and a CDP-star substrate solution (Roche cat# 1759051). Hygromycin-specific probe preparation was performed with primers 5′-ccctgttacttctcgacc-3′/5′-cagcagttccgggaaga-3′, whereas preparation of the external probe was performed with the primer pairs 5′-cgtcgacgaagtagtgct-3′/5′- tcaacgaactcggtctca-3′.
In vitro pupylation
Pupylation assays were carried out at 30°C in a buffer containing 50 mM HEPES, pH 7.5, 100 mM KCl, 20 mM MgCl2, and 10% glycerol (v/v). Following preparation of reaction mixtures as described in the text, pupylation was initiated by the addition of ATP at a final concentration of 2 mM.
Pupylated protein quantification
The M. smegmatis lysates to be examined were prepared at a total protein concentration of 1 μg/ml. As a blank, lysates from a pupylation-deficient mutant were similarly prepared, whereas a purified pupylome, diluted to a final concentration of 0.1 μg/ml and added to Pup-lacking lysates at a total protein concentration of 1 μg/ml, provided standards. Serial dilutions were prepared from each sample, and dot blot analysis was carried out using anti-Pup antibodies (Supplementary Fig S1A). Dot intensities were measured using ImageJ software (NIH), and a standard curve was constructed to calculate the concentration of pupylated proteins in each sample.
LC/MS analysis
MS analysis was performed using an Eksigent nano-HPLC connected to the LTQ Orbitrap XL (Thermo Fisher Scientific). Reverse-phase chromatography of peptides was performed using an homemade C-18 column (15 cm long, 75 μm ID) packed with Jupiter C18, 300 Å, 5 μm beads (Phenomenex). Peptides were separated by a 70-min linear gradient, starting with 100% buffer A (5% acetonitrile, 0.1% formic acid) and ending with 80% buffer B (80% acetonitrile, 0.1% formic acid), at a flow rate of 300 nl/min. A full scan, acquired at 60,000 resolution, was followed by CID MS/MS analysis performed for the five most abundant peaks, in the data-dependent mode. Fragmentation (with minimum signal trigger threshold set at 1,000) and detection of fragments were carried out in the linear ion trap. Maximum ion fill time settings were 300 ms for the high-resolution full scan in the Orbitrap analyzer and 100 ms for MS/MS analysis in the ion trap. The AGC settings were 5′105 and 1′104 for Orbitrap and linear ion trap analyzers, respectively.
Bioinformatics
Following acquisition of MS data, proteins were identified on the basis of their precursor mass and the sequence information included in their fragmentation spectra, by using the Proteome Discoverer 1.4 software package (Thermo Fisher Scientific). The acquired spectra were searched against NCBI-derived M. smegmatis protein database, using the SEQUEST search engine. The following search parameters were used: enzyme specificity is trypsin; maximum two missed cleavage sites; maximum 10 ppm or 0.8 Da error tolerance for the full scan and MS/MS analysis, respectively; static modification of cysteine carbamidomethylation; and dynamic modification of methionine oxidation and lysine pup-modification (GGE). Threshold criteria for protein identification were defined as having at least two peptides of high confidence as calculated by the SEQUEST algorithm and a false discovery rate (FDR) P-value < 0.01 acquired by the Percolator node. The reverse M. smegmatis database was chosen as a target decoy. The biological function of each identified protein was extracted from the UniProt-SwissProt protein database.
Acknowledgments
We thank Ekaterina Eeremenko and Guy Adler (Ben-Gurion University) for assistance with the dot blot procedure. We also thank Graham Hatfull (University of Pittsburgh) for kindly providing us with plasmid pJV53 and Torin Weisbrod (Albert Einstein College of Medicine) for generously providing us with plasmids pMV206 and pMV306. Special thanks to Anat Ben-Zvi, Nir Hecht, Jerry Eichler, Amir Epstein, Michael Meijler, Boaz Shaanan, Maayan Korman (Ben-Gurion University), and to Uri Gophna (Tel-Aviv University) for helpful comments and fruitful discussions.
Author contributions
YE performed most of the experiments described, analyzed the data, and helped writing the manuscript. ZR performed mass spectrometry analysis and helped writing the manuscript. IH constructed the pafA deletion strain and analyzed pupylation in stationary-phase M. smegmatis cells. AM analyzed pupylation in stationary-phase M. smegmatis cells. GP analyzed the cross-reactivity of anti-Pup antibodies. YS constructed the prcSBA deletion strain and performed experiments. MV helped to construct deletion strains and purified proteins. IK performed mass spectrometry analysis, and EG performed experiments, analyzed the data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Amon J, Titgemeyer F, Burkovski A. A genomic view on nitrogen metabolism and nitrogen control in mycobacteria. J Mol Microbiol Biotechnol. 2009;17:20–29. doi: 10.1159/000159195. [DOI] [PubMed] [Google Scholar]
- Appelberg R. Macrophage nutriprive antimicrobial mechanisms. J Leukoc Biol. 2006;79:1117–1128. doi: 10.1189/jlb.0206079. [DOI] [PubMed] [Google Scholar]
- Bardarov S, Bardarov S, Jr, Pavelka MS, Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR., Jr Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis M. bovis BCG and M. smegmatis. Microbiology. 2002;148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- Blackman FF. Optima and limiting factors. Ann Bot. 1905;19:282–295. [Google Scholar]
- Burns KE, Liu WT, Boshoff HI, Dorrestein PC, Barry CE., III Proteasomal protein degradation in Mycobacteria is dependent upon a prokaryotic ubiquitin-like protein. J Biol Chem. 2009;284:3069–3075. doi: 10.1074/jbc.M808032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KE, Pearce MJ, Darwin KH. Prokaryotic ubiquitin-like protein provides a two-part degron to Mycobacterium proteasome substrates. J Bacteriol. 2010a;192:2933–2935. doi: 10.1128/JB.01639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KE, Cerda-Maira FA, Wang T, Li H, Bishai WR, Darwin KH. “Depupylation” of prokaryotic ubiquitin-like protein from mycobacterial proteasome substrates. Mol Cell. 2010b;39:821–827. doi: 10.1016/j.molcel.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerda-Maira FA, Pearce MJ, Fuortes M, Bishai WR, Hubbard SR, Darwin KH. Molecular analysis of the prokaryotic ubiquitin-like protein (Pup) conjugation pathway in Mycobacterium tuberculosis. Mol Microbiol. 2010;77:1123–1135. doi: 10.1111/j.1365-2958.2010.07276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerda-Maira FA, McAllister F, Bode NJ, Burns KE, Gygi SP, Darwin KH. Reconstitution of the Mycobacterium tuberculosis pupylation pathway in Escherichia coli. EMBO Rep. 2011;12:863–870. doi: 10.1038/embor.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Solomon WC, Kang Y, Cerda-Maira F, Darwin KH, Walters KJ. Prokaryotic ubiquitin-like protein pup is intrinsically disordered. J Mol Biol. 2009;392:208–217. doi: 10.1016/j.jmb.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- Darwin KH, Lin G, Chen Z, Li H, Nathan CF. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol Microbiol. 2005;55:561–571. doi: 10.1111/j.1365-2958.2004.04403.x. [DOI] [PubMed] [Google Scholar]
- Darwin KH. Prokaryotic ubiquitin-like protein (Pup) proteasomes and pathogenesis. Nat Rev Microbiol. 2009;7:485–491. doi: 10.1038/nrmicro2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BD, Mingioli ES. Mutants of Escherichia coli requiring methionine or vitamin B12. J Bacteriol. 1950;60:17–28. doi: 10.1128/jb.60.1.17-28.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mot R. Actinomycete-like proteasomes in a Gram-negative bacterium. Trends Microbiol. 2007;15:335–338. doi: 10.1016/j.tim.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Delley CL, Striebel F, Heydenreich FM, Özcelik D, Weber-Ban E. Activity of the mycobacterial proteasomal ATPase Mpa is reversibly regulated by pupylation. J Biol Chem. 2012;287:7907–7914. doi: 10.1074/jbc.M111.331124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festa RA, McAllister F, Pearce MJ, Mintseris J, Burns KE, Gygi SP, Darwin KH. Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis. PLoS ONE. 2010;5:e8589. doi: 10.1371/journal.pone.0008589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forer N, Korman M, Elharar Y, Vishkautzan M, Gur E. Bacterial proteasome and PafA, the Pup ligase, interact to form a modular protein tagging and degradation machine. Biochemistry. 2013;52:9029–9035. doi: 10.1021/bi401017b. [DOI] [PubMed] [Google Scholar]
- Gandotra S, Lebron MB, Ehrt S. The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLoS Pathog. 2010;6:e1001040. doi: 10.1371/journal.ppat.1001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur E, Biran D, Ron EZ. Regulated proteolysis in Gram-negative bacteria-how and when? Nat Rev Microbiol. 2011;9:839–848. doi: 10.1038/nrmicro2669. [DOI] [PubMed] [Google Scholar]
- Guth E, Thommen M, Weber-Ban E. Mycobacterial ubiquitin-like protein ligase PafA follows a two-step reaction pathway with a phosphorylated pup intermediate. J Biol Chem. 2011;286:4412–4419. doi: 10.1074/jbc.M110.189282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Lin G, Wang M, Dick L, Xu RM, Nathan C, Li H. Structure of the Mycobacterium tuberculosis proteasome mechanism of inhibition by a peptidyl boronate. Mol Microbiol. 2006;59:1417–1428. doi: 10.1111/j.1365-2958.2005.05036.x. [DOI] [PubMed] [Google Scholar]
- Imkamp F, Rosenberger T, Striebel F, Keller PM, Amstutz B, Ser P, Weber-Ban E. Deletion of dop in Mycobacterium smegmatis abolishes pupylation of protein substrates in vivo. Mol Microbiol. 2010a;75:744–754. doi: 10.1111/j.1365-2958.2009.07013.x. [DOI] [PubMed] [Google Scholar]
- Imkamp F, Striebel F, Sutter M, Ozcelik D, Zimmermann N, Ser P, Weber-Ban E. Dop functions as a depupylase in the prokaryotic ubiquitin-like modification pathway. EMBO Rep. 2010b;11:791–797. doi: 10.1038/embor.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins VA, Barton GR, Robertson BD, Williams KJ. Genome wide analysis of the complete GlnR nitrogen-response regulon in Mycobacterium smegmatis. BMC Genomics. 2013;14:301. doi: 10.1186/1471-2164-14-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemori M, Nishihara K, Yanagi H, Yura T. Synergistic roles of HslVU and other ATP-dependent proteases in controlling in vivo turnover of sigma32 and abnormal proteins in Escherichia coli. J Bacteriol. 1997;179:7219–7225. doi: 10.1128/jb.179.23.7219-7225.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küberl A, Fränzel B, Eggeling L, Polen T, Wolters DA, Bott M. Pupylated proteins in Corynebacterium glutamicum revealed by MudPIT analysis. Proteomics. 2014;14:1531–1542. doi: 10.1002/pmic.201300531. [DOI] [PubMed] [Google Scholar]
- Liao S, Shang Q, Zhang X, Zhang J, Xu C, Tu X. Pup a prokaryotic ubiquitin-like protein is an intrinsically disordered protein. Biochem J. 2009;422:207–215. doi: 10.1042/BJ20090738. [DOI] [PubMed] [Google Scholar]
- Lies M, Maurizi MR. Turnover of endogenous SsrA-tagged proteins mediated by ATP-dependent proteases in Escherichia coli. J Biol Chem. 2008;283:22918–2229. doi: 10.1074/jbc.M801692200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly WW, Wallweber GJ, Higgins SM. Proteolysis and amino acid recycling during nitrogen deprivation in Schizophyllum commune. Curr Microbiol. 1991;23:27–32. [Google Scholar]
- Lupas A, Zühl F, Tamura T, Wolf S, Nagy I, De Mot R, Baumeister W. Eubacterial proteasomes. Mol Biol Rep. 1997;24:125–131. doi: 10.1023/a:1006803512761. [DOI] [PubMed] [Google Scholar]
- Novák B, Tyson JJ. Design principles of biochemical oscillators. Nat Rev Mol Cell Biol. 2008;9:981–991. doi: 10.1038/nrm2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofer N, Forer N, Korman M, Vishkautzan M, Khalaila I, Gur E. Allosteric transitions direct protein tagging by PafA the prokaryotic ubiquitin-like protein (Pup) ligase. J Biol Chem. 2013;288:11287–11293. doi: 10.1074/jbc.M112.435842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish T, Mahenthiralingam E, Draper P, Davis EO, Colston MJ. Regulation of the inducible acetamidase gene of Mycobacterium smegmatis. Microbiology. 1997;143:2267–2276. doi: 10.1099/00221287-143-7-2267. [DOI] [PubMed] [Google Scholar]
- Pearce MJ, Arora P, Festa RA, Butler-Wu SM, Gokhale RS, Darwin KH. Identification of substrates of the Mycobacterium tuberculosis proteasome. EMBO J. 2006;25:5423–5432. doi: 10.1038/sj.emboj.7601405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science. 2008;322:1104–1107. doi: 10.1126/science.1163885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen C, Akhter Y, Jeon AH, Schmitt-Ulms G, Meyer HE, Stefanski A, Stühler K, Wilmanns M, Song YH. Proteome-wide identification of mycobacterial pupylation targets. Mol Syst Biol. 2010;6:386. doi: 10.1038/msb.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price CT, Al-Quadan T, Santic M, Rosenshine I, Abu Kwaik Y. Host proteasomal degradation generates amino acids essential for intracellular bacterial growth. Science. 2011;334:1553–1557. doi: 10.1126/science.1212868. [DOI] [PubMed] [Google Scholar]
- Rohmer L, Hocquet D, Miller SI. Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol. 2011;19:341–348. doi: 10.1016/j.tim.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M1, Finley D. Regulation of proteasome activity in health and disease. Biochim Biophys Acta. 2014;1843:13–25. doi: 10.1016/j.bbamcr.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkerman Y, Elharar Y, Vishkautzan M, Gur E. Efficient and simple generation of unmarked gene deletions in Mycobacterium smegmatis. Gene. 2013;533:374–378. doi: 10.1016/j.gene.2013.09.082. [DOI] [PubMed] [Google Scholar]
- Stover CK, et al. New use of BCG for recombinant vaccines. Nature. 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- Striebel F, Imkamp F, Sutter M, Steiner M, Mamedov A, Weber-Ban E. Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat Struct Mol Biol. 2009;16:647–651. doi: 10.1038/nsmb.1597. [DOI] [PubMed] [Google Scholar]
- Striebel F, Hunkeler M, Summer H, Weber-Ban E. The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup's N-terminus. EMBO J. 2010;29:1262–1271. doi: 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraweera A, Münch C, Hanssum A, Bertolotti A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell. 2012;48:242–253. doi: 10.1016/j.molcel.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter M, Striebel F, Damberger FF, Allain FH, Weber-Ban E. A distinct structural region of the prokaryotic ubiquitin-like protein (Pup) is recognized by the N-terminal domain of the proteasomal ATPase Mpa. FEBS Lett. 2009;583:3151–3157. doi: 10.1016/j.febslet.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Tamura T, Nagy I, Lupas A, Lottspeich F, Cejka Z, Schoofs G, Tanaka K, De Mot R, Baumeister W. The first characterization of a eubacterial proteasome: the 20S complex of Rhodococcus. Curr Biol. 1995;5:766–774. doi: 10.1016/s0960-9822(95)00153-9. [DOI] [PubMed] [Google Scholar]
- Vabulas RM, Hartl FU. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science. 2005;310:1960–1963. doi: 10.1126/science.1121925. [DOI] [PubMed] [Google Scholar]
- Valas RE, Bourne PE. Rethinking proteasome evolution: two novel bacterial proteasomes. J Mol Evol. 2008;66:494–504. doi: 10.1007/s00239-008-9075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kessel JC, Marinelli LJ, Hatfull GF. Recombineering mycobacteria and their phages. Nat Rev Microbiol. 2008;6:851–857. doi: 10.1038/nrmicro2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Darwin KH, Li H. Binding-induced folding of prokaryotic ubiquitin-like protein on the Mycobacterium proteasomal ATPase targets substrates for degradation. Nat Struct Mol Biol. 2010;7:1352–1357. doi: 10.1038/nsmb.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrous J, Burns K, Liu WT, Patel A, Hook V, Bafna V, Barry CE, III, Bark S, Dorrestein PC. Expansion of the mycobacterial “PUPylome”. Mol BioSyst. 2010;6:376–385. doi: 10.1039/b916104j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KJ, Bryant WA, Jenkins VA, Barton GR, Witney AA, Pinney JW, Robertson BD. Deciphering the response of Mycobacterium smegmatis to nitrogen stress using bipartite active modules. BMC Genomics. 2013;14:436. doi: 10.1186/1471-2164-14-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf S, Nagy I, Lupas A, Pfeifer G, Cejka Z, Müller SA, Engel A, De Mot R, Baumeister W. Characterization of ARC a divergent member of the AAA ATPase family from Rhodococcus erythropolis. J Mol Biol. 1998;277:13–25. doi: 10.1006/jmbi.1997.1589. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.