

Abstract
The formation of neuronal synapses and the dynamic regulation of their efficacy depend on the assembly of the postsynaptic neurotransmitter receptor apparatus. Receptor recruitment to inhibitory GABAergic and glycinergic synapses is controlled by the scaffold protein gephyrin and the adaptor protein collybistin. We derived new insights into the structure of collybistin and used these to design biochemical, cell biological, and genetic analyses of collybistin function. Our data define a collybistin-based protein interaction network that controls the gephyrin content of inhibitory postsynapses. Within this network, collybistin can adopt open/active and closed/inactive conformations to act as a switchable adaptor that links gephyrin to plasma membrane phosphoinositides. This function of collybistin is regulated by binding of the adhesion protein neuroligin-2, which stabilizes the open/active conformation of collybistin at the postsynaptic plasma membrane by competing with an intramolecular interaction in collybistin that favors the closed/inactive conformation. By linking trans-synaptic neuroligin-dependent adhesion and phosphoinositide signaling with gephyrin recruitment, the collybistin-based regulatory switch mechanism represents an integrating regulatory node in the formation and function of inhibitory postsynapses.
Keywords: autoinhibition, conformational activation, gephyrin, inhibitory synapse, neuroligin-2
Introduction
Synaptogenesis, the generation of synapses between nerve cells, is a fundamental cell biological determinant of neuronal network formation in the developing brain. During synaptogenesis, contact sites of pre- and postsynaptic neurons undergo spatially and temporally controlled changes in molecular content and morphology to form fully functional synapses. This differentiation process is coordinated by synaptic adhesion proteins and involves the specific accumulation of synaptic vesicles and active zone components at the presynaptic transmitter release site, in precise apposition to the postsynaptic membrane, where synapse-type-specific neurotransmitter receptors are clustered via scaffolding proteins.
Gephyrin is the main scaffolding protein of inhibitory glycinergic and GABAergic postsynapses in mammals and is largely responsible for clustering glycine and GABAA receptors apposed to corresponding transmitter release sites (Kneussel & Betz, 2000; Moss & Smart, 2001). Gephyrin directly interacts with glycine and GABAA receptors (Meyer et al, 1995; Tretter et al, 2008, 2011; Maric et al, 2011; Mukherjee et al, 2011), thus regulating their cell surface dynamics (Meier et al, 2001; Jacob et al, 2005), and the amount of gephyrin recruited from the cytosol to the postsynaptic membrane determines the receptor content and sensitivity of inhibitory synapses (Specht et al, 2013). Loss of gephyrin in mice leads to severe defects in inhibitory synapse function and death after birth due to a lack of motor neuron inhibition and consequent muscle stiffness, and gephyrin dysfunction may be causally involved in certain forms of epilepsy in human patients (Tretter et al, 2012).
In many brain regions, the recruitment of gephyrin from intracellular deposits to postsynaptic membranes depends on the adaptor protein collybistin (CB) (Papadopoulos & Soykan, 2011). Loss of CB leads to a massive reduction of gephyrin and GABAA receptor clusters in several regions of the forebrain, altered synaptic plasticity, increased anxiety, and impaired spatial learning (Papadopoulos et al, 2007, 2008; Jedlicka et al, 2009), which indicates an essential role of CB in the initial assembly and maintenance of gephyrin-GABAA receptor clusters. The critical role of CB in the differentiation of inhibitory postsynapses is further illustrated by disease-associated CB mutations in humans, which cause hyperekplexia, epilepsy, and mental retardation (Harvey et al, 2004; Kalscheuer et al, 2009; Shimojima et al, 2011).
CB is a Dbl-family guanine nucleotide exchange factor (GEF). All known CB mRNA transcripts encode tandem Dbl-homology (DH) and pleckstrin-homology (PH) domains, and the vast majority encode an additional N-terminal src-homology 3 (SH3) domain (Harvey et al, 2004). Earlier studies indicated that CB-mediated gephyrin recruitment to and clustering at synapses is dependent upon binding of its PH domain to phosphatidylinositol 3-phosphate [PI(3)P] (Kalscheuer et al, 2009), whereas the GEF activity of its DH domain is dispensable (Reddy-Alla et al, 2010). Structural data on CB are currently restricted to an NMR solution structure of the SH3 domain (pdb entry 2YSQ), which shows a typical SH3 fold, and to a crystal structure of the Cdc42–CB2SH3− complex (Xiang et al, 2006; pdb code 2DFK), which reveals a novel conformation of the switch I region of Cdc42 as well as conformational changes in the orientation of the PH domain relative to the DH domain.
Importantly, the gephyrin-clustering activity of CB appears to be inhibited by its N-terminal SH3 domain. A CB variant lacking this domain (CB2SH3−) has the intrinsic capacity to mediate gephyrin clustering at the plasma membrane of non-neuronal cells. In contrast, CB variants that carry the SH3 domain (e.g. CB2SH3+ or CB3SH3+) are not targeted to the plasma membrane but remain colocalized with intracellular gephyrin deposits (Kins et al, 2000; Harvey et al, 2004). These variants require additional neuronal factors, such as neuroligin-2 (NL2), neuroligin-4 (NL4), or the GABAA receptor α2 subunit, which all bind to the SH3 domain, thereby activating CB, and promoting its plasma membrane targeting (Poulopoulos et al, 2009; Saiepour et al, 2010; Hoon et al, 2011). Correspondingly, mice lacking any one of these CB activators exhibit a brain layer-specific loss of gephyrin clusters at inhibitory synapses and impairments in GABAergic and/or glycinergic transmission in various regions of the central nervous system (Poulopoulos et al, 2009; Hoon et al, 2011; Jedlicka et al, 2011; Panzanelli et al, 2011).
The aim of the present study was to elucidate the molecular mechanism of CB autoinhibition and of its activation through NL2 binding. We present a low-resolution crystal structure of an SH3 domain-containing CB, which shows that the SH3 domain of CB forms an intramolecular interface with the DH and PH domains, resulting in a compact conformation. Based on the primary and tertiary structures of CB and their high similarity to the corresponding structures of the closest CB homologue Asef (Mitin et al, 2007), we identified interface-forming residues in the SH3 and DH domains and found that their substitution with alanine leads to conformational activation of CB, enhanced PI(3)P binding, and increased gephyrin-clustering activity in non-neuronal cells and neurons. Furthermore, we identified the NL2-binding site within the SH3 domain of CB and provide evidence for the critical role of NL2 in potentiating the gephyrin-clustering activity of CB in neurons. Combining our in vitro data with careful structure function analyses in neurons from different genetic backgrounds, we were able to formulate a molecular model of the mechanisms by which gephyrin scaffolds are assembled and maintained at inhibitory postsynapses.
Results
CB isoforms in mouse brain contain an N-terminal SH3 domain
The rodent CB (Arhgef9) gene gives rise to four differentially spliced mRNAs in the brain (Fig1A). Among these, three major variants encode CBs that contain an N-terminal SH3 domain (SH3+) but differ in their C-termini (CB1SH3+, CB2SH3+, CB3SH3+), while a fourth minor variant encodes a CB2 isoform (CB2SH3−) without an SH3 domain (Harvey et al, 2004). To determine the relative abundances of SH3+ and SH3− CB isoforms in mouse brain, we used a novel polyclonal antiserum that was raised against recombinantly expressed full-length CB2SH3+ and can detect all four CB isoforms. SDS–PAGE and Western blot analysis of whole brain lysates from 8-week-old wild-type (WT) mice using the new antiserum revealed two bands of 55–60 kDa (Fig1B). An SH3 domain-specific monoclonal anti-CB antibody detected the same two bands, indicating that both CB isoforms contain an SH3 domain. The two bands likely correspond to CB3SH3+ and CB2SH3+ (60.99 and 55.94 kDa calculated molecular masses, respectively), whose mRNAs are the most abundant CB mRNAs in rodent spinal cord and brain (Harvey et al, 2004). The new polyclonal anti-CB antiserum detected recombinantly expressed myc-CB2SH3−, but no endogenous CB2SH3− (49.17 kDa calculated molecular mass) in mouse brain. A comparison of brains from different developmental stages showed that CBSH3+ isoforms dominate at all developmental stages and that CB2SH3− levels remain below our detection limit throughout development (Fig1B and Supplementary Fig S1). These data show that the vast majority of CB variants in mouse brain contain an N-terminal SH3 domain.
Figure 1. The structure of autoinhibited CBSH3+.

- Domain architecture of CB isoforms and Asef. The different C-termini of CB1–3 are indicated. Mutations used in this study are marked in CB2SH3+. The C-terminally truncated CB used for crystallography is shown as CBSH3+. Sequence homology to Asef is indicated for each domain.
- Specificity of rabbit anti-CB antiserum and developmental expression profile of CB as determined by Western blotting of brain homogenates of adult WT mice, adult CB−/− mice, and WT CB mice at different developmental stages, and of lysates of myc-CB2SH3− and myc-CB2SH3+-expressing HEK293 cells (upper panel). The same samples were tested with a mouse antibody directed against the CB SH3 domain (lower panel). Green arrows indicate CB-specific bands, which likely represent endogenous CB3SH3+ and CB2SH3+. Red stars indicate unspecific bands.
- SIGMAA weighted 2Fo–Fc electron density of CBSH3+ after refinement contoured at an RMSD of 1.0 (gray). The DH and PH domains are shown as C-alpha traces in magenta and blue, respectively. Fo–Fc difference map for the SH3 domain (rainbow coloring from the N- to the C-terminus) is shown in green at an RMSD of 2.5 prior to inclusion of this domain in the model.
- Surface representation of CBSH3+ in two different orientations differing by a 90° rotation around the vertical axis (as indicated by the arrow). The SH3 domain is located in proximity to both the DH and PH domains. Domains are color coded as in (A).
- Comparison of different conformations of CB and the Asef structure. The top panel shows a superposition of CBSH3+ (colored according to its domains) and Asef (gray). In the bottom panel, the closed conformation of CBSH3+ has been superimposed onto the open conformations of CB2SH3− as observed in the Cdc42-CB2SH3− complex (chain A, gray; chain C, light orange) and of CB1SH3− (cyan) on the basis of their DH domains. Leu416 is marked in red in all structures to illustrate different conformations.
- Close-up view of the interface between the SH3 domain and the DH/PH tandem domain of CBSH3+ and Asef. Trp203 and Glu436 in Asef are crucial for the intramolecular SH3–DH/PH interaction and correspond to Trp24 and Glu262 in CBSH3+ (residues are shown in stick representation and domains are color coded as above, with the Asef structure in faded colors).
- Binding of CB2SH3− to SH3. MST measurements reveal an impairment of the interaction in the W24A (P < 0.001) and E202A mutants (P < 0.001) as indicated by an increase in the KD values compared to the WT proteins.
Data information: Data and error bars indicate mean ± standard error of the mean.
Closed structure of autoinhibited CB
Since CBSH3+ isoforms dominate in mouse brain, we sought to determine the structure of an SH3 domain-containing CB. To overcome limitations during expression and purification, we analyzed a C-terminally truncated form of CB, referred to as CBSH3+ (Fig1A). CBSH3+ was crystallized in the cubic space group I213, and we were able to solve the structure at a resolution of 5.5 Å by molecular replacement, with the individual DH, PH, and SH3 domains as search models (Supplementary Table S1, PDB code: 4MT6). Only a single copy of CBSH3+ was detected in the asymmetric unit, resulting in a Mathew's coefficient of 7.92 Å3/Da (85% solvent content), possibly explaining the poor diffraction. The structure was refined to an R-factor of 0.237 (Rfree = 0.282) with good stereochemical parameters (Supplementary Table S1). Omitting the SH3 domain from the final model increased the R-factor to 0.321 and Rfree to 0.349, thus demonstrating that the position of the SH3 domain is correct. This conclusion is further supported by unbiased electron density maps (Fig1C) and by a map-to-model correlation coefficient of 0.74 for the SH3 domain (Supplementary Fig S2).
In the crystal, CBSH3+ adopted a closed conformation, where the SH3 domain primarily engages in contacts with the DH but also with the PH domain (Fig1D). The overall conformation of CBSH3+ is strikingly similar to the closed conformation of the related GEF Asef (PDB code 2PZ1) (Mitin et al, 2007), as reflected by a low root mean square deviation (RMSD) of 2.0 Å for the main chain atoms. In contrast, the relative orientation of the DH and PH domains is fundamentally different between the CBSH3+ and previous CB2SH3- structures (Fig1E) (Xiang et al, 2006). CBSH3+ and Asef display high levels of sequence identity in their SH3, DH, and PH domains (Figs1A and 5A and Supplementary Fig S2B), which extends to the residues involved in inter-domain interactions in Asef, indicating a similar mode of domain interactions, although these cannot be unambiguously deduced due to the low resolution of the CBSH3+ crystal structure. In Asef, Trp203, Arg249, and Glu436 are most important for keeping the protein in a closed conformation (Mitin et al, 2007). These residues are identical in CB and correspond to Trp24, Arg70, and Glu262, respectively (Fig1F).
We also crystallized CB1SH3+ (space group C2221); however, SDS–PAGE of the crystals showed a truncated protein (Supplementary Fig S2D). We solved and refined the structure at a resolution of 3.5 Å (Supplementary Table S1, PDB code 4MT7) and found that the SH3 domain and a part of the C-terminus had been lost during crystallization and solely the DH/PH tandem was present, which we refer to as CB1SH3−. The degradation of the SH3 domain indicates that the closed state of CB is not as tight as the crystal structure of CBSH3+ implies and that subtle alterations may affect the equilibrium between the closed and open states of CB. Compared to the CBSH3+ structure, the apo CB1SH3− structure adopted a more extended conformation similar to both copies of CB2SH3− in the asymmetric unit of the Cdc42-CB2SH3− cocrystal (Xiang et al, 2006). While the position of the PH domain relative to the DH domain was different in all three CB molecules, the RMSDs for the individual DH and PH domains ranged from 0.34 to 1.52 Å, indicating no major intra-domain conformational changes (Fig1E and Supplementary Table S2). The radii of gyration (Rg) as calculated with Pymol indicated that the CB1SH3− structure is the most extended (Rg = 28.3 Å) whereas the CBSH3+ structure is the most compact model (Rg = 26.3 Å including the SH3 domain), with the CB2SH3− structures adopting intermediate conformations with Rg values of 27.4 and 26.8 Å. Critical for domain movement is the long flexible helix connecting the DH and PH domains, which was present in differently bent conformations: (1) almost straight as in CB1SH3−, (2) kinked at its center near Ile291 (CBSH3+), and (3) kinked toward its C-terminal end near Asp302 (molecules A and C of the Cdc42-CB2SH3− cocrystals and second kink in CBSH3+; Supplementary Fig S2C). Additionally, rotations around this helix, ranging from 38 to 92°, led to fundamental changes in the orientation of the PH domain with respect to the DH domain (Supplementary Table S2).
Intramolecular interactions of SH3 and DH/PH domains in CB
To assess the intramolecular interaction between the SH3 and DH/PH domains of CB and the contribution of individual amino acid residues, we performed microscale thermophoresis (MST) with fluorescently labeled CB2SH3− and the SH3 domain as ligand. The SH3–CB2SH3− interaction was found to be rather weak with a KD of 273 ± 34 μM, in good agreement with a KD of 56 μM calculated with PISA based on the SH3–DH/PH interface. Alanine substitutions of Trp24 in the SH3 domain and Glu202 in the DH domain (corresponding to Glu262 in CBSH3+, Fig1F) impaired the interaction, resulting in KD values of 908 ± 216 μM and 888 ± 276 μM, respectively (Fig1G). For the Arg70 mutation, binding was too weak to be measured in our MST setup (Supplementary Fig S1B). Thus, residues Trp24, Arg70, and Glu262 contribute to the intramolecular interactions in CB and modulate the equilibrium between the closed and open conformations of CBSH3+.
We further analyzed the conformational transitions in CB in solution by small angle X-ray scattering (SAXS) and atomic force microscopy (AFM), with a focus on WT CBSH3+ and the E262A variant. Due to aggregation/degradation problems, the W24A/E262A double mutant analyzed below could not be utilized for structural studies. SAXS measurements confirmed a monomeric state of both variants (Supplementary Table S3). The particle distance distribution functions P(r) for CBSH3+, with a Dmax (maximal intramolecular distance) of 95 Å, and for CBSH3+/E262A, with a Dmax of 125 Å (Fig2A), clearly indicated a more extended conformation of the E262A mutant and a more compact state of the WT protein. The radii of gyration (Rg) were estimated as approximately 30 and 37 Å for the WT and the mutant, respectively (Supplementary Table S3). In comparison, CBSH3+ in the crystal structure is characterized by an Rg of 26 Å (see above) and a Dmax of 88 Å. Ab initio shape reconstructions confirmed the more compact shape of CB2SH3+ compared to a more elongated shape of CBSH3+/E262A. The resulting averaged model for the WT CBSH3+ superimposes very well with the crystal structure, where CBSH3+ adopts a compact conformation (Fig2B). Adding the missing residues (SH3–DH inter-domain linker and the N/C-termini) not visible in the crystal structure using BUNCH (Petoukhov and Svergun, 2005) resulted in a clear improvement of the fit against the experimental data as reflected by a reduction of the discrepancy from χ = 1.07 to χ = 0.72 (Fig2A). In contrast to the WT, rigid body modeling of the CBSH3+/E262A variant could not be accomplished with the CBSH3+ crystal structure (χ = 1.70; Fig2A). However, the data could be interpreted with the CB1SH3− structure and the SH3 domain placed adjacent to the DH domain, resulting in an overall elongated conformation (χ = 0.79, Fig2A), as also supported by superposition with ab initio shape reconstructions (Fig2C). Interestingly, the Kratky plot of CBSH3+/E262A showed a considerably less pronounced maximum (0.64/nm) than CBSH3+ WT (Supplementary Fig S3), indicative of a potentially higher flexibility of CBSH3+/E262A in solution.
Figure 2. Extended conformation of CBSH3+ upon mutation of the intramolecular SH3–DH/PH interface as assessed by SAXS.

- Experimental and calculated scattering curves. Experimental data are shown in black (with error bars in gray) and the calculated curves in red as follows: (1) CBSH3+ crystal structure versus the experimental data for CBSH3+ WT; (2) CBSH3+ crystal structure with modeled missing linkers versus the experimental data for the CBSH3+ WT; (3) CBSH3+ crystal structure versus the experimental data for CBSH3+/E262A; (4) model of a single conformation of CBSH3+/E262A versus the experimental data for CBSH3+/E262A; (5) ensemble of conformations for CBSH3+/E262A versus the experimental data for CBSH3+/E262A. Inset: Distance distribution P(r) functions for CBSH3+ WT (solid) and the E262A variant (dashed).
- Ab initio shape from SAXS superimposed onto the CBSH3+ crystal structure with the SH3, DH and PH domains in green magenta, and blue, respectively. The two views differ by a 180° rotation around the vertical axis.
- Ab initio shape superimposed onto the BUNCH model of the CBSH3+/E262A variant, color coded as in (B). Regions not resolved in the crystal structures are presented in ocher. Front and back views are displayed
- EOM derived models of the CBSH3+/E262A variant in surface representation with the DH/PH tandem in magenta/blue and the flexible SH3 domain in different colors (red, yellow, orange, brown, cyan, black with the connecting linkers rendered semi-transparent). The contribution (in %) of the individual conformations to the ensemble together with the corresponding overall structural parameters Rg and Dmax is shown on the right.
To assess the flexibility of CBSH3+/E262A, the ensemble optimization method (EOM 2.0, developed by G. Tria) was employed allowing for co-existence of multiple conformers in solution. The selected ensemble displayed Rg values ranging from 30.3 to 44.1 Å and Dmax values between 100.0 and 159.3 Å. These distributions indicated a pronounced heterogeneity of conformations simultaneously present in solution with three discernible maxima representing compact, intermediate, and elongated conformations (Supplementary Fig S3). The ensemble resulting in the best fit (Fig2A) contained six models and described how the SH3 domain was located in relation to the DH/PH tandem. Positions proximate as well as distant to the DH/PH tandem were adopted, assuming that the SH3 domain is moving freely in solution, resulting in an equilibrium of closed and open states for CBSH3+/E262A (Fig2D). The ensemble yielded a slightly better fit to the experimental data with χ = 0.765 than the best single model from the BUNCH analysis χ = 0.788. More importantly, the Rg and Dmax values of the single conformation failed to match the experimentally determined values (Supplementary Table S4), indicating that the ensemble representation indeed more faithfully reproduces the overall parameters extracted from the experimental data.
The single molecule technique AFM provides information on structural heterogeneity in a sample and on flexibility and conformational transitions in protein molecules (Sander et al, 2013). AFM images of monomeric CB samples showed mixtures of circular and elongated shapes (Fig3A and Supplementary Fig S4). To quantify the observed elongation in the proteins, we described the molecules by ellipses and calculated the ratios between their major versus their minor axes. Using these simplified structural models, we found three distinct species for WT CB as well as CBSH3+/E262A, which we classified as compact, intermediate, and elongated states characterized by axial ratios of 1.23 ± 0.22 or 1.21 ± 0.17 (compact), 1.48 ± 0.21 or 1.45 ± 0.21 (intermediate), and 1.73 ± 0.28 or 1.74 ± 0.30 (elongated) for WT or CBSH3+/E262A, respectively (Fig3B). The distributions indicate an equilibrium between (at least) three conformational states of the proteins. The relative frequencies of the three conformations, however, differed between the WT and the E262A variant with respective values of 0.41 ± 0.08, 0.43 ± 0.1 and 0.16 ± 0.06 (WT) versus 0.46 ± 0.06, 0.28 ± 0.08 and 0.25 ± 0.1 (E262A) for compact, intermediate, and elongated species (Fig3C). The results showed that while the fraction of compact particles was comparable for both proteins, significantly less intermediate and more elongated particles were detected for the CBSH3+/E262A variant as compared to the WT protein (Fig3C), consistent with a stabilization of the open (elongated) conformation in the mutant.
Figure 3. Extended conformation of CB as visualized by AFM and increased phosphoinositide binding of CB upon mutation of the intramolecular SH3–DH/PH interface.

- Topographic AFM images of the CBSH3+ WT and the E262A variant. Different particle shapes are indicated by filled arrow heads (compact), open arrow heads (intermediate), and arrows (elongated).
- Relative frequencies of occurrence of CB particle shapes as classified by the different major/minor axis ratios featuring compact (red), intermediate (green), and elongated (blue) states for the WT and E262A samples. One representative experiment is shown for each protein with n representing the number of particles per experiment.
- Quantification of frequencies of compact, intermediate, and elongated particles with P-values representing the statistical significance of the observed differences calculated by pairwise Student's t-test. Here n represents the number of independent experiments.
- Custom-made PI(3)P strips were used to compare different CB variants for PI(3)P binding. Left, purified GST-fusion proteins used in the assay. Middle, bound proteins were detected using an anti-GST antibody. A grayscale image is displayed using a color lookup table from ImageJ (Fire). GST-CB2SH3+/W24A-E262A showed stronger PI(3)P binding than GST-CB2SH3+. Right, relative binding of each CB variant is plotted for each PI(3)P spot by measuring the intensity of the chemiluminescence. A paired Student's t-test was performed for statistical analysis, n = 3 independent experiments.
Data information: Data and error bars indicate mean ± standard error of the mean.
The SH3 domain inhibits phosphoinositide binding of CB
Despite the evidence for an autoinhibitory role of the CB SH3 domain, it is not known which function of CB during inhibitory synapse formation is inhibited by the SH3 domain. DH domain mutations of CB that abolish its GEF activity have no effect on CB-mediated gephyrin clustering (Reddy-Alla et al, 2010), and the SH3 domain of CB does not obstruct gephyrin binding (Supplementary Fig S5A), indicating that the functional autoinhibition neither involves its enzymatic activity nor gephyrin binding. Since membrane clustering of gephyrin by CB is dependent on an interaction of its PH domain with PI(3)P (Reddy-Alla et al, 2010), we hypothesized that the SH3 domain may regulate phosphoinositide binding. Using protein-lipid overlay assays with commercially available PIP-strips, we found that a ΔSH3 mutant of CB2 (equivalent to CB2SH3-) displayed strong binding to PI(3)P in a PH domain-dependent manner, while full-length CB2SH3+ had no specific preference for a particular phosphoinositide but rather bound indiscriminately and with low affinity to all phosphoinositides tested (Supplementary Fig S5B). To compare PI(3)P binding of WT CB and its open conformation mutant, we developed a custom-made PI(3)P-strip assay, where the CB variants and mutants were tested for binding to increasing amounts of PI(3)P. These assays showed that CB2/ΔSH3 bound PI(3)P much more strongly than CB2SH3+ (Fig3D). In contrast, CB2SH3+ bound to PI(3)P only weakly, similar to the PI(3)P-binding-deficient CB2/ΔSH3 R303A-R304A mutant, in which the functionally critical Arg residues in the PH domain were mutated (Reddy-Alla et al, 2010). A CB2SH3+ mutant with two substitutions in the SH3–DH/PH interface (CB2SH3+/W24A-E262A) displayed an increased affinity for PI(3)P (Fig3D), indicating that autoinhibition of CB2SH3+ by its SH3 domain leads to a reduced affinity to phosphoinositides. Hence, a CB activation mechanism that interferes with the interaction between the SH3 domain and the DH/PH domains may allow for an increased tethering of CB to lipid membranes.
Perturbation of the SH3 and DH domain binding interface causes CB autoactivation
SH3 domain-containing CB isoforms fail to redistribute gephyrin to the plasma membrane in non-neuronal cells and instead remain coclustered with cytoplasmic gephyrin aggregates, while CB2SH3− (equivalent to CB2/ΔSH3) recruits gephyrin to the plasma membrane in the form of submembraneous microclusters (Kins et al, 2000; Harvey et al, 2004). Based on these observations, we employed a cell-based assay to test the activity of various CB mutants to target gephyrin to the plasma membrane. COS7 cells were transfected with myc-tagged CB constructs, GFP-Gephyrin, and HA-NL3, an NL isoform that cannot activate SH3 domain-containing CB isoforms but can be sequestered by CB2/ΔSH3-induced gephyrin microclusters at the plasma membrane due to its gephyrin-binding ability (Poulopoulos et al, 2009). In this assay, the correlation coefficients of GFP-stained and surface-stained HA-signals of GFP-Gephyrin and HA-NL3, respectively, were measured to quantify the relative levels of intrinsic activity of each CB mutant. Compared to CB2SH3+, the W24A, R70A, and E262A single mutants and the W24A-E262A double mutant displayed increased intrinsic activities, reflected by the more frequent occurrence of membrane-targeted gephyrin microclusters that colocalized with surface-stained NL3 (Fig4A and B). These results indicate that the intramolecular interactions between the SH3 domain and DH/PH tandem keep CB in a closed and inactive state and that the intrinsic gephyrin-clustering activity of CB is increased upon release of these intramolecular interactions.
Figure 4. Open conformation mutants of CB have a higher intrinsic gephyrin-clustering activity.
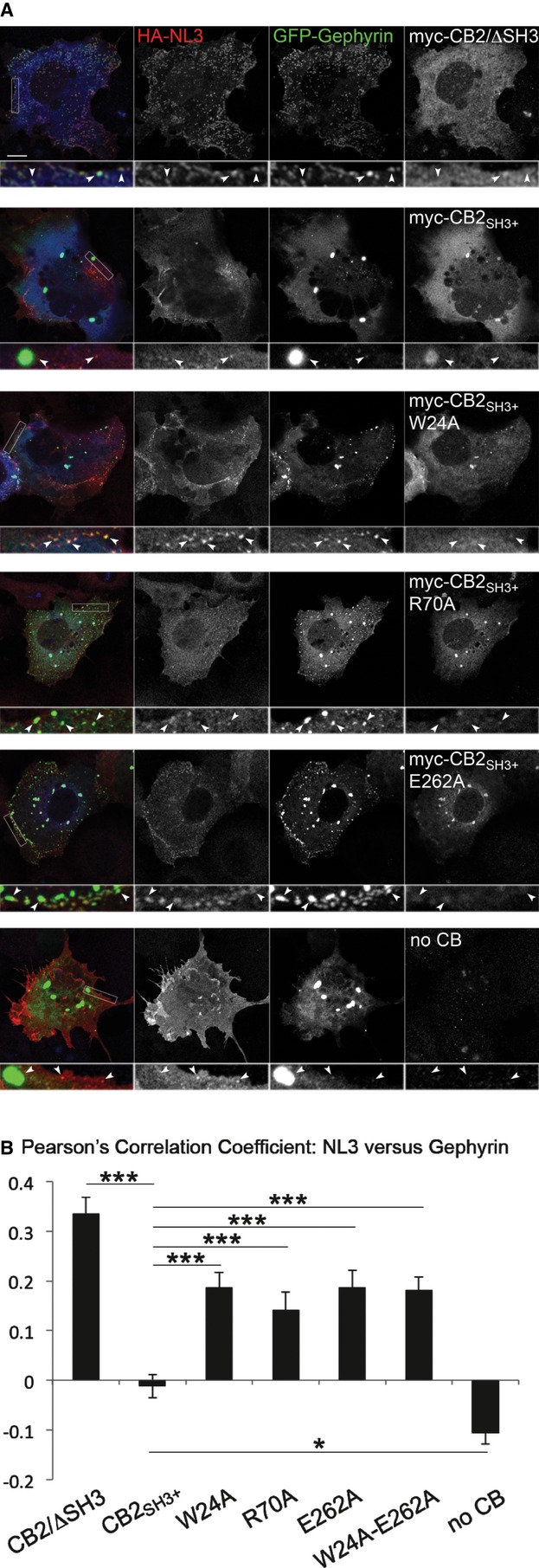
- COS7 cells were transfected with HA-NL3 (red), GFP-Gephyrin (green) and myc-CB2/ΔSH3, myc-CB2SH3+ or its point mutants (blue). Among the point mutants tested, myc-CB2SH3+/E262A showed the highest autoactivation and redistributed gephyrin to submembrane microclusters in the absence of NL2, unlike myc-CB2SH3+, which stays associated with intracytoplasmic gephyrin aggregates. Scale bar: 10 μm, main panels; 2.5 μm, insets.
- Pearson's correlation coefficients between green (GFP-Gephyrin) and red (surface-stained HA-NL3) labels were measured to quantify the relative levels of autoactivation of each CB variant. CB2SH3+ showed significantly reduced activity (−0.012 ± 0.024) as compared to mutants W24A (0.187 ± 0.029; P < 0.001), R70A (0.141 ± 0.036; P < 0.001), E262A (0.187 ± 0.034; P < 0.001), and W24A-E262A (0.181 ± 0.027; P < 0.001). N = 6 experiments, n = 33–50 cells per construct.
Data information: Data and error bars indicate mean ± standard error of the mean.
NL2 binding is essential for CB activation
The neuronal proteins that activate CB (NL2, NL4, and the GABAA receptor α2-subunit) have been shown to bind to the SH3 domain of CB, but the precise mechanism of CB activation has yet to be determined. To locate the NL2-binding site, we introduced point mutations on the CB SH3 domain to create NL2-binding-deficient CB mutants. An epilepsy-associated G62A mutation (G55A in human CB) in the SH3 domain of CB abolishes NL2 binding and CB activation (Poulopoulos et al, 2009). However, this mutation causes the formation of large non-synaptic CB/gephyrin aggregates and toxicity in transfected neurons (Harvey et al, 2004). This indicates that the G62A mutation disrupts the structure of the SH3 domain and might cause at least a local unfolding of the SH3 domain, and possibly a perturbation of NL2 binding without affecting autoinhibition and gephyrin-binding activity (Fig5C).
Figure 5. NL2–CB interaction and an intact PH domain are required for CB-mediated gephyrin clustering.

- Sequence alignment of SH3 domains from different mammalian proteins. Conserved residues are highlighted in green/black for structurally important amino acids and gray for residues involved in ligand binding. On top, β-strands and the 310 helix are indicated, along with the RT, N-Src, and distal loops.
- Left, cartoon representation of the CB SH3 domain (PDB: 2YSQ). Residues involved in ligand binding are depicted as gray sticks, while Gly62 is shown in black. Middle and right, surface representation of the CB SH3 domain with a proline-rich ligand modeled in the binding groove. After rotation by 90°, Gly62 is visible in black.
- GST-pulldown assay to test binding of GST-CB2SH3+, GST-CB2/ΔSH3, GST-CB2SH3+/G62A, GST-CB2SH3+/W52A, and GST-CB2SH3+/W63A to endogenously expressed gephyrin from HEK cells and exogenously expressed NL2 cytoplasmic domain. GST-CB2/ΔSH3 and all the point mutants of GST-CB2SH3+ were deficient in binding to NL2, while they all retained their ability to interact with gephyrin. GST-CB2SH3+ was shown previously to lack binding to the cytoplasmic domains of NL1 and NL3 (Hoon et al, 2011).
- Sequence alignment of intracellular domains of mouse NLs. Putative SH3 domain binding sites were identified with the SH3-Hunter software (Ferraro et al, 2007). Class I and II SH3 binding motifs are highlighted in blue and red, respectively (overlaps are marked in purple). The well-conserved gephyrin-binding and PDZ-binding regions are indicated.
- COS7 cells were transfected with HA-NL2 (red), GFP-Gephyrin (green) and myc-CB2/ΔSH3, myc-CB2SH3+ or its point mutants (blue). Myc-CB2SH3+ constructs bearing the W52A and W63A mutations were not activated by NL2 and did not redistribute gephyrin to submembranous microclusters. Scale bar: 10 μm, main panels; 2.5 μm, insets.
- Pearson's correlation coefficients between green (GFP-Gephyrin) and red (surface-stained HA-NL2) labels were determined to quantify the level of NL2-induced activation of each CB variant. Compared to CB2SH3+, the mutants W52A, G62A, and W63A showed a reduced response to NL2 activation (CB2SH3+: 0.241 ± 0.021; CB2SH3+/W52A: −0.015 ± 0.030, P < 0.001; G62A: −0.160 ± 0.035, P < 0.001; W63A: 0.065 ± 0.027, P < 0.001; no CB: −0.150 ± 0.031, P < 0.001; CB2SH3−: 0.398 ± 0.028, P < 0.001). N = 6 experiments, n = 38–51 cells per construct.
- COS7 cells were transfected with GFP-Gephyrin (green) and myc-CB2SH3+ or myc-CB2/ΔSH3 (blue), together with HA-NL2 or HA-NL2/P798stop (red). The P798stop mutant of NL2 did not activate CB2SH3+, although it retains the ability to cocluster with myc-CB2/ΔSH3-induced gephyrin microaggregates. Scale bar: 10 μm, main panels; 3.9 μm, insets.
- Pearson's correlation coefficients between green (GFP-Gephyrin) and red (surface-stained HA-NL2 or HA-NL2/P798stop) labels were determined to quantify the strength of NL2 and its P798stop mutant in activating CB. The P798stop mutation leads to reduced activation of CB2SH3+ (NL2: 0.346 ± 0.029; NL2/P798stop: −0.103 ± 0.056, P < 0.001), while it retained its capacity to cocluster with CB2SH3−-induced gephyrin microaggregates (NL2: 0.451 ± 0.050; NL2/P798stop: 0.444 ± 0.033, P = 0.901). N = 3 experiments, n = 15 cells per construct.
- COS7 cells were transfected with HA-NL2 (red), GFP-Gephyrin (green), myc-CB2SH3+ or its point mutants (blue). K358A-K359A and R363A-R364A mutants of CB2SH3+ did not induce the formation of gephyrin microaggregates. Scale bar: 10 μm, main panels; 2.5 μm, insets.
- Pearson's correlation coefficients between green (GFP-Gephyrin) and red (surface-stained HA-NL2) labels were determined to quantify the level of NL2-induced activation of PH domain mutants of CB. K358A-K359A and R363A-R364A mutants of CB2SH3+ have defects in inducing membrane microclusters of gephyrin (CB2SH3+: 0.451 ± 0.055; CB2SH3+/K358A-K359A: −0.253 ± 0.073, P < 0.001; CB2SH3+/R363A-R364A: −0.211 ± 0.052, P < 0.001). N = 3 experiments, n = 15 cells per construct.
Data information: Data and error bars indicate mean ± standard error of the mean.
We therefore attempted to design more specific mutations to perturb the NL2–CB interaction. Since NL2 and NL4 contain proline-rich stretches in their C-terminal tails that are lacking in the CB-binding-deficient NL1 and NL3 (Fig5D), we hypothesized that the interaction between CB and NL2 or NL4 might be a typical SH3/PXXP mode of interaction and assumed that residues mediating these interactions are not buried in the SH3–DH/PH interface. Alignment of a series of SH3 domains from various signaling and scaffolding molecules aided in the identification of the potential NL2-binding site within the CB SH3 domain (Fig5A and B). Based on this analysis, we generated several CB point mutants and tested them in GST-pulldown assays for NL2 binding. We identified Trp52 and Trp63 of CB as residues that mediate the NL2 interaction, since their mutation to alanine abolished binding to the intracellular domain of NL2 (NL2icd). In contrast, gephyrin binding was retained in these mutants (Fig5C). The location of Trp52 and Trp63 within the SH3 domain is on the opposite side of the SH3–DH/PH interface (Fig5B). Trp24, which we identified as a residue that contributes to the intramolecular interactions of CB (Fig1G), is not involved in binding to NL2 (Supplementary Fig S5C).
We further tested the SH3 domain mutant CB variants in cell-based NL2-activation assays, in which COS7 cells were transfected with myc-tagged CB constructs, GFP-Gephyrin, and HA-NL2. Like NL3 (Fig4A), NL2 can also be sequestered by submembraneous gephyrin microaggregates. However, in contrast to NL3, NL2 can relieve the autoinhibition of CB2SH3+, facilitate CB-mediated gephyrin microcluster formation, and become sequestered in these microclusters due to its binding to gephyrin (Poulopoulos et al, 2009). Therefore, calculating the level of colocalization between GFP- and HA-signals of gephyrin and NL2, respectively, provided a quantitative assessment of the response of each CB mutant to activation by NL2. Compared to CB2SH3+, the W52A and W63A mutants showed reduced responses to NL2 activation, reflected by the less frequent occurrence of membrane-targeted gephyrin microclusters that colocalized with NL2 (Fig5E and F), similar to the phenotype observed with the G62A mutant (Poulopoulos et al, 2009). These data indicate that perturbation of the NL2-binding site on the SH3 domain renders CB irresponsive to NL2-induced activation and that the interaction between NL2 and the SH3 domain is essential for the subsequent membrane tethering of the gephyrin/CB complex in the form of microaggregates. To further test the significance of NL2 binding for CB activation, we performed similar assays with an NL2 mutant with a Pro798stop mutation that truncates the NL2icd just upstream of the proline-rich stretch (Fig5D). This truncated NL2 mutant failed to activate CB2SH3+ and to induce gephyrin microcluster formation, although it was trapped by CB2/ΔSH3-induced gephyrin microaggregates due to its intact gephyrin-binding motif (Fig5G), indicating that the proline-rich stretch of NL2 might be essential for CB activation.
To test whether NL2 binding to the SH3 domain is sufficient for plasma membrane recruitment of CB, we generated alanine mutations of positively charged residues in the β3/β4 loop of the PH domain predicted to be involved in PI(3)P binding (Fig3D) (Reddy-Alla et al, 2010) and coexpressed these constructs in COS7 cells with NL2 and gephyrin. The K358A-K359A and R363A-R364A mutants of CB2SH3+ failed to redistribute gephyrin to the plasma membrane in the presence of NL2 and remained tightly coclustered with intracellular gephyrin aggregates (Fig5H). These data validate the significance of the basic PH domain residues in phosphoinositide binding and show that NL2-induced activation of CB and phosphoinositide binding of the PH domain are both required for gephyrin clustering.
NL2-induced CB activation drives neuronal gephyrin clustering
Our cell-based and biochemical assays showed that phosphoinositide-binding and gephyrin-clustering activities of CB are inhibited by its SH3 domain. CB is activated when a neuronal factor, such as NL2, binds to its SH3 domain, which likely leads to molecular rearrangements in the CB structure that allow for increased gephyrin clustering. To test whether this mechanism operates in neurons, we transfected cultured hippocampal neurons from CB−/− and NL2−/− mice with various CB constructs and measured the size and density of gephyrin clusters induced by each construct.
In CB−/− neurons, similar to published data on WT neurons (Chiou et al, 2011; Tyagarajan et al, 2011), expression of CB2/ΔSH3 led to gephyrin clusters that had a significantly larger size and higher fluorescence intensity and were around 50% more frequent than clusters induced by CB2SH3+ (Fig6A–D). Gephyrin clusters induced by the NL2-binding-deficient CB mutant CB2SH3+/W52A were smaller in size (Fig6B), had a reduced fluorescence intensity (Fig6C), and were fewer in number (Fig6D) as compared to CB2SH3+-induced clusters (see also Supplementary Fig S6A). In contrast, expression of the autoactive CB2SH3+/W24A-E262A mutant led to an increased number of gephyrin clusters (Fig6D) with a larger average size (Fig6B), as compared to WT CB2SH3+ (see also Supplementary Fig S6A). Presynaptic innervation of neurons (as indicated by VIAAT cluster density) and the degree of synaptic localization of gephyrin clusters were similar under all conditions tested (Fig6E and F), although the average size and the mean fluorescence intensity of VIAAT clusters were slightly increased in CB−/− neurons expressing either GFP or CB2/ΔSH3, as compared to CB−/− cells expressing WT CB2SH3+ (Supplementary Fig S6C and E). Assessment of the mean fluorescence intensity of CB signals revealed similar expression levels of the tested constructs (Fig6G). These data indicate that the density and size of gephyrin clusters are dependent upon the level of CB activity, which is regulated by NL2 binding to the SH3 domain of CB.
Figure 6. NL2-binding-deficient CB mutants have a reduced capacity to rescue gephyrin clustering in CB−/− neurons.

- Cultured hippocampal neurons from CB−/− mice were transfected with myc-CB2SH3+, myc-CB2SH3+/W52A, myc-CB2SH3+/W24A-E262A, myc-CB2/ΔSH3, or GFP (as a negative control) at DIV3–4, fixed at DIV14, and stained for gephyrin (green in overlay frame), VIAAT (red in overlay frame), and myc (blue in overlay frame). Scale bar: 10 μm, main panels; 3.33 μm, insets.
- Average size of perisomatic gephyrin clusters in μm2. CB2SH3+: 0.158 ± 0.005; CB2SH3+/W52A: 0.140 ± 0.07, P < 0.05; CB2SH3+/W24A-E262A: 0.194 ± 0.008, P < 0.001; CB2/ΔSH3: 0.274 ± 0.011, P < 0.001; GFP (no CB): 0.122 ± 0.016, P < 0.01.
- Mean intensity of gephyrin clusters, arbitrary units (a.u.). CB2SH3+: 44.6 ± 0.8; CB2SH3+/W52A: 41.8 ± 0.6, P < 0.01; CB2SH3+/W24A-E262A: 46.3 ± 0.6, n.s.; CB2/ΔSH3: 51.3 ± 0.8, P < 0.001; GFP (no CB): 39.3 ± 0.9, P < 0.001.
- Number of gephyrin clusters per μm2. CB2SH3+: 0.115 ± 0.008; CB2SH3+/W52A: 0.074 ± 0.007, P < 0.001; CB2SH3+/W24A-E262A: 0.155 ± 0.009, P < 0.01; CB2/ΔSH3: 0.168 ± 0.010, P < 0.001; GFP (no CB): 0.007 ± 0.002, P < 0.001.
- Number of VIAAT clusters per μm2. CB2SH3+: 0.155 ± 0.009; CB2SH3+/W52A: 0.146 ± 0.011, n.s.; CB2SH3+/W24A-E262A: 0.161 ± 0.009, n.s.; CB2/ΔSH3: 0.156 ± 0.009, n.s.; GFP (no CB): 0.129 ± 0.012, n.s.
- Percentage of gephyrin clusters apposed to VIAAT. CB2SH3+: 58.2 ± 2.7%; CB2SH3+/W52A: 61.2 ± 3.7%, n.s.; CB2SH3+/W24A-E262A: 59.8 ± 3.1%, n.s.; CB2/ΔSH3: 65.6 ± 2.8%, n.s.; GFP (no CB): 57.2 ± 9.7%, n.s.
- Mean intensity of CB signal, arbitrary units (a.u.). CB2SH3+: 73.6 ± 3.9; CB2SH3+/W52A: 69.7 ± 4.1, n.s.; CB2SH3+/W24A-E262A: 66.3 ± 3.8, n.s.; CB2/ΔSH3: 70.6 ± 3.4, n.s.
Data information: N = 3 experiments, n = 54 (CB2SH3+), 43 (CB2SH3+/W52A), 42 (CB2SH3+/W24A-E262A), 46 (CB2/ΔSH3), 42 (GFP) neurons in (B–G). Data and error bars indicate mean ± standard error of the mean.
The absence of NL2 leads to a decrease in perisomatic gephyrin and GABAAR clustering in various brain regions, including the hippocampus (Poulopoulos et al, 2009). We tested the degree of rescue of this NL2−/− phenotype by expressing CB2SH3+ and its W52A, W24A-E262A, and ΔSH3 mutants in hippocampal neurons of NL2−/− (Fig7A). Neurons transfected with the autoactive CB2SH3+/W24A-E262A construct displayed a significantly higher density of gephyrin clusters (Fig7D), which were slightly larger in size (Fig7B) but similar in fluorescence intensity (Fig7C), as compared to CB2SH3+-induced clusters (see also Supplementary Fig S6B). This indicates that in the absence of NL2, the autoactive CB mutant can more efficiently initiate gephyrin clustering, although these clusters were not as large and as bright as the clusters induced by the same construct in CB−/− neurons (Fig6B and C). Importantly, the density, the fluorescence intensity, and the average size of gephyrin clusters induced by CB2SH3+ and CB2SH3+/W52A in NL2−/− neurons were similar and equally small (Fig7B–D, see also Supplementary Fig S6B). This is strikingly different from the data on CB−/− neurons (i.e. in the presence of NL2) (Fig6A–D), where gephyrin clusters induced by CB2SH3+ were larger and more frequent than clusters induced by CB2SH3+/W52A, indicating that in the absence of NL2, the gephyrin-clustering activity of CB is compromised. Presynaptic innervation of neurons (as indicated by VIAAT cluster density) and the degree of synaptic localization of gephyrin clusters were similar under all conditions (Fig7E and F), although the mean fluorescence intensity (but not the average size) of VIAAT clusters was slightly increased in NL2−/− neurons expressing only GFP, as compared to NL2−/− cells expressing WT CB2SH3+ (Supplementary Fig S6D and F). Expression levels of the tested CB2 constructs were similar, as indicated by the mean intensity of CB signals (Fig7G). Altogether, the data acquired with CB−/− and NL2−/− neurons indicate that CB and NL2 act in synergy to determine the size and density of the perisomatic gephyrin clusters.
Figure 7. Open conformation mutants of CB have an increased capacity to rescue gephyrin clustering in NL2−/− neurons.

- Cultured hippocampal neurons from NL2−/− mice were transfected with myc-CB2SH3+, myc-CB2SH3+/W52A, myc-CB2SH3+/W24A-E262A, myc-CB2/ΔSH3, or GFP (as a negative control) at DIV3-4, fixed at DIV14, and stained for gephyrin (green in overlay frame), VIAAT (red in overlay frame), and myc (blue in overlay frame). Scale bar: 10 μm, main panels; 3.33 μm, insets.
- Average size of perisomatic gephyrin clusters in μm2. CB2SH3+: 0.133 ± 0.007; CB2SH3+/W52A: 0.138 ± 0.10, n.s.; CB2SH3+/W24A-E262A: 0.164 ± 0.010, P < 0.05; CB2/ΔSH3: 0.237 ± 0.012, P < 0.001; GFP (no CB): 0.141 ± 0.013, n.s.
- Mean intensity of gephyrin clusters, arbitrary units (a.u.). CB2SH3+: 40.8 ± 0.3; CB2SH3+/W52A: 40.0 ± 0.5, n.s.; CB2SH3+/W24A-E262A: 41.2 ± 0.3, n.s.; CB2/ΔSH3: 46.2 ± 0.6, P < 0.001; GFP (no CB): 40.7 ± 0.5, n.s.
- Number of gephyrin clusters per μm2. CB2SH3+: 0.075 ± 0.010; CB2SH3+/W52A: 0.059 ± 0.009, n.s.; CB2SH3+/W24A-E262A: 0.162 ± 0.016, P < 0.001; CB2/ΔSH3: 0.171 ± 0.015, P < 0.001; GFP (no CB): 0.067 ± 0.034, n.s.
- Number of VIAAT clusters per μm2. CB2SH3+: 0.130 ± 0.012; CB2SH3+/W52A: 0.146 ± 0.014, n.s.; CB2SH3+/W24A-E262A: 0.166 ± 0.015, n.s.; CB2/ΔSH3: 0.165 ± 0.013, n.s.; GFP (no CB): 0.131 ± 0.012, n.s.
- Percentage of gephyrin clusters apposed to VIAAT. CB2SH3+: 58.2 ± 5.1%; CB2SH3+/W52A: 61.1 ± 5.1%, n.s.; CB2SH3+/W24A-E262A: 55.2 ± 4.3%, n.s.; CB2/ΔSH3: 55.7 ± 4.4%, n.s.; GFP (no CB): 61.3 ± 5.6%, n.s.
- Mean intensity of CB signal, arbitrary units (a.u.). CB2SH3+: 72.1 ± 8.1; CB2SH3+/W52A: 61.8 ± 5.2, n.s.; CB2SH3+/W24A-E262A: 66.4 ± 6.5, n.s.; CB2/ΔSH3: 65.5 ± 5.9, n.s.
Data information: N = 3 experiments, n = 26 (CB2SH3+), 29 (CB2SH3+/W52A), 29 (CB2SH3+/W24A-E262A), 26 (CB2/ΔSH3), 29 (GFP) neurons in (B–G). Data and error bars indicate mean ± standard error of the mean.
Discussion
In the present study, we provide structural, biochemical, and cell biological data that together elucidate the mechanism of autoinhibition of CB and of its relief by synaptic proteins such as NL2 during the formation of postsynaptic gephyrin clusters at inhibitory synapses.
CB proteins with SH3 domains in mouse brain
CB1SH3+ and CB2SH3− were originally identified as gephyrin interactors (Kins et al, 2000) while additional SH3 domain-containing CB isoforms (CB2SH3+ and CB3SH3+) were discovered subsequently (Harvey et al, 2004). All SH3 domain-containing CB variants were shown to require additional neuronal factors in order to act as gephyrin adaptors at the plasma membrane. Various recent studies focused on the different roles of SH3− and SH3+ CB isoforms in gephyrin and GABAARs clustering and assigned new roles to the SH3− isoforms, for example, in gephyrin and Cdc42 binding or clustering of gephyrin and GABAARs (Chiou et al, 2011; Tyagarajan et al, 2011). However, it has remained unclear whether CB proteins lacking an SH3 domain actually exist in vivo.
Our Western blot analyses of mouse brain exclusively detected SH3 domain-containing CB variants, which likely represent CB3SH3+ and CB2SH3+ as judged by the known relative abundance of CB mRNAs (Harvey et al, 2004) and the calculated molecular weights. We failed to detect a protein band that would correspond to CB2SH3−. These results indicate that CB2SH3− might exist only in trace amounts and that the major CB proteins in the mouse brain contain the autoinhibitory SH3 domain and require neuronal activators such as NL2 (Poulopoulos et al, 2009), NL4 (Hoon et al, 2011), or GABAA-α2 (Saiepour et al, 2010).
Closed/inactive and open/active conformations of CB
The crystal structure of CBSH3+ displays a closed conformation, in which the SH3 domain is packed against the DH and PH domains, an arrangement that is very similar to the one seen in Asef (Mitin et al, 2007; Murayama et al, 2007). Although molecular details of the interface in CBSH3+ cannot be deduced due to the limited resolution of the crystal structure, PISA analyses of the interfaces indicate a fivefold higher theoretical KD in CBSH3+ (56 μM) as compared to Asef (11 μM). Furthermore, our structure of a CBSH3+ variant that lost its SH3 domain during crystallization, leaving only the DH/PH tandem (CB1SH3−), also indicates that the SH3 domain and DH/PH tandem interact with moderate affinity. This assessment is corroborated by our MST measurements, which detected a rather weak binding of the CB SH3 domain to CB2SH3− with a KD of 273 μM (Fig1G). Together, these findings indicate that CBSH3+ can rapidly interconvert between open and closed states.
There are important additional differences between CBSH3+ and Asef with regard to their activation and function, which may partly be related to their differential conformational heterogeneity. First, CB activation is accomplished by the binding of interaction partners to the CB SH3 domain, while Asef activation is triggered by the interaction of adenomatous polyposis coli protein (APC) with the Asef APC-binding region, which is located directly N-terminal of the Asef SH3 domain. Second, disinhibition of CB, for example, by removal of its SH3 domain, has only minor effects on its intrinsic GEF activity toward small GTPases (Cdc42 and TC10) in vitro or in transfected cells (Xiang et al, 2006; Tyagarajan et al, 2011; Mayer et al, 2013), whereas the GEF activity of Asef requires disinhibition, for example, by the removal of its SH3 domain or APC binding (Mitin et al, 2007; Zhang et al, 2012). Third, CB has a very low intrinsic GEF activity (Jaiswal et al, 2013) that is not required for its function in synaptic gephyrin clustering (Reddy-Alla et al, 2010), whereas Asef has a much higher GEF activity (Jaiswal et al, 2013) that is essential for its role as a tumor suppressor (Kawasaki et al, 2000, 2003).
Using SAXS and AFM, we probed the conformation of CB in the E262A variant, in which the interaction between the SH3 and DH/PH domains is perturbed. According to both techniques, the WT and E262A variants are monomeric. More importantly, SAXS and AFM revealed differences between mutant and WT CB that are consistent with a shift toward an open conformation in the mutant, as indicated by the increased overall Dmax and Rg values of the E262A variant as determined by SAXS and by the shifted equilibrium of the same mutant toward a more elongated state as demonstrated by AFM. Conformational variability in CB is not only limited to the SH3 domain, which either interacts with the DH/PH tandem or flexibly adopts various conformations as best illustrated by the EOM analysis, but also leads to changes in the relative arrangements of the DH and PH domains, as demonstrated by the crystal structures of the Cdc42–CB2SH3− complex (Xiang et al, 2006) and CB1SH3−.
Functional relevance of conformational changes in the CB structure
The intrinsic gephyrin-clustering activity of CB2SH3+ is increased in mutants that preferentially adopt an open conformation (Fig4), indicating that a switch in CB from a closed to an open conformation favors its synaptogenic activity. In contrast, mutations that perturb NL2 binding to the SH3 domain (Fig5E–G) or PH domain function (Fig5H) strongly inhibit gephyrin clustering by CB. These data indicate that NL2-dependent activation of CB, subsequent conformational changes in CB, and binding of the PH domain to lipids, but not the enzymatic activity of the DH domain toward small GTPases, are functionally interconnected.
A functional PH domain is essential for CB-induced synaptic targeting of gephyrin (Harvey et al, 2004) and likely acts by directly binding PI(3)P (Kalscheuer et al, 2009; Reddy-Alla et al, 2010). However, the physiological role of phosphoinositides during inhibitory postsynaptic differentiation is unknown (Papadopoulos & Soykan, 2011). Further confounding the issue, previous studies on the role of the PH domain mainly focused on the very rare CB isoform CB2SH3−. We tested PH domain mutants in CB2SH3+ for their gephyrin-clustering activity in NL2-expressing COS7 cells (Fig5G) and found that the NL2–CB interaction is not sufficient for retaining CB/gephyrin complexes at the plasma membrane. Rather, both the NL2–SH3 domain interaction and the PH domain–PI(3)P interaction seem to be essential for the formation of gephyrin microclusters by CB. In the structure of autoinhibited CB (Fig1C and D), the SH3 domain covers a part of the PH domain surface, where defined residues of the SH3 domain are in close proximity of the phosphoinositide-binding β3/β4 loop. In addition, a comparison of the structures of the Cdc42–CB2SH3− complex, CB1SH3−, and CBSH3+ (Fig1E) indicates that the PH domain can move substantially relative to the DH domain, hence rendering the phosphoinositide-binding surface of the PH domain fully accessible. We found that CB2/ΔSH3 has a strong affinity for PI(3)P, whereas CB2SH3+ binds only very weakly to PI(3)P. Furthermore, the open conformation mutant W24A-E262A displayed a stronger binding to PI(3)P, indicating that the SH3 domain directly inhibits the membrane-tethering function of the PH domain by affecting PI(3)P binding (Fig3D). The binding affinity of the CB2SH3+/W24A-E262A mutant for PI(3)P is still lower compared to the ΔSH3 variant, indicating that the SH3 domain exerts a partial autoinhibitory effect on the W24A-E262A mutant despite mutations of the interface-forming residues. The results described above confirm that the ability of CB to mediate gephyrin clustering depends on its ability to bind to membrane phosphoinositides and show in addition that the conformational activation of CB facilitates phosphoinositide binding and gephyrin clustering. It is likely that the assembly of the ternary NL2–gephyrin–CB complex causes molecular rearrangements in CB that lead to an increased accessibility of its PH domain and a stronger tethering to its target membranes.
The role of CB activation in neuronal synapse formation
The interaction between NL2 and CB is essential for CB activation as demonstrated by the reduced levels of gephyrin clustering in COS7 cells expressing non-interacting NL2 and CB variants. In CB−/− neurons, an NL2-binding-deficient CB2SH3+/W52A mutant showed a significantly reduced capacity to rescue perisomatic gephyrin clustering, indicating that NL2-dependent activation is required for CB function in neurons. Accordingly, in comparison to CB−/− neurons, CB-induced gephyrin clustering was weaker in NL2−/− neurons, where overexpression of WT CB2SH3+ and its W52A mutant are equally ineffective in recruiting gephyrin to inhibitory postsynapses. In contrast, the constitutively active CB mutant (E262A or the W24A-E262A double mutant) outperformed CB2SH3+ in gephyrin clustering in every cell type tested (COS7 cells, CB−/− and NL2−/− neurons), indicating that its open conformation and increased capacity to bind PI(3)P endow this variant with a strong gephyrin-clustering activity. Gephyrin clusters induced by CB2SH3+/W24A-E262A were smaller in NL2−/− neurons than those induced by the same construct in CB−/− neurons, indicating that NL2 may exhibit further roles in augmenting the assembly of inhibitory postsynapses. This feature is most likely linked to the capacity of NL2 to form a ternary complex with gephyrin and CB, where it does not only act as a CB activator but also as a factor to consolidate the assembly of inhibitory postsynapses (Poulopoulos et al, 2009).
In contrast to their distinct postsynaptic effects, the different CB variants we tested had no or only very minor effects on presynaptic innervation as assessed by VIAAT cluster density. In CB−/− and NL2−/− neurons, VIAAT clusters were generally more dense (0.13–0.17 clusters per μm2) and larger (0.20–0.27 μm2) than gephyrin clusters (0.06–0.17 clusters per μm2, 0.13–0.27 μm2 in size). Correspondingly, new gephyrin clusters induced by CB2/ΔSH3 or CB2SH3+/W24A-E262A were still synaptic, as reflected by essentially unaltered proportions of synaptic gephyrin clusters, indicating that newly induced gephyrin clusters formed at sites with prior innervation. Beyond this, indications of transsynaptic signaling effects of the postsynaptic changes caused by the different CB variants were rare. We detected minor increases (˜10%) in the mean fluorescence intensity and average size of VIAAT clusters in CB−/− and NL2−/− neurons expressing GFP alone, as compared to neurons expressing CB2SH3+. These changes are likely of homeostatic nature and can be explained by a compensatory increase in the presynaptic input strength due to the corresponding postsynaptic perturbations. Overexpression of constitutively active CB2/ΔSH3 in CB−/− neurons also led to increased VIAAT cluster size and intensity, which is best compatible with the opposite scenario, where significantly larger gephyrin clusters may induce the expansion of the corresponding presynaptic input.
Our structural, biochemical, and cell biological data led us to formulate a model that describes how NL2 might regulate the number and size of gephyrin clusters (Fig8). According to this model, CB-mediated gephyrin clustering is dependent upon two differentially regulated processes that we termed CB on-rate and CB off-rate. The on-rate is directly correlated to the frequency of initiating a successful membrane-tethering event and becomes manifest in the number of gephyrin clusters. This depends on the accessibility of the PH domain of CB to bind plasma membrane phospholipids, a feature that is promoted by a conformational change in CB structure toward the open state and consequent PH domain exposure. The off-rate, on the other hand, represents membrane-dissociation events and depends on the stability of protein–protein interactions within existing CB/gephyrin/NL2 complexes. In NL2−/− neurons, CB has a low on-rate due to the SH3 domain-mediated autoinhibition, and a high off-rate due to the absence of NL2 as a factor that further stabilizes already formed CB/gephyrin clusters, resulting in gephyrin clusters of low density, small size, and low fluorescence intensity. Under the same circumstances, the open conformation mutant of CB, due to its increased on-rate, has an improved ability to initiate gephyrin clustering, although these clusters are not entirely stable and thus relatively small in size, due to the persisting high off-rate in the absence of NL2. In the presence of NL2, CB has an increased on-rate, since the conformational equilibrium is shifted toward the open state, and a decreased off-rate, due to the structural integrity of and binding avidities within the CB/gephyrin/NL2 holo-complex, resulting in a higher density of gephyrin clusters with a larger size. The NL2-binding-deficient CB mutant is still affected by its lower on-rate as compared to WT CB and thus has only a limited capacity to form stable gephyrin clusters, which becomes manifest in a lower density of gephyrin clusters with a smaller size.
Figure 8. A model for the role of NL2 and CB in the assembly of the gephyrin scaffold at postsynaptic membranes.

- CB-mediated gephyrin clustering is regulated by two mechanisms, the CB on-rate and CB off-rate. The on-rate is directly correlated with the frequency of a successful membrane-tethering event. This depends on the accessibility of the PH domain and its ability to bind to targets at the plasma membrane, which is more likely after a conformational change in the CB structure favoring the open state. The off-rate is a measure of membrane-dissociation events and depends on the protein–protein interactions within and the structural stability of the CB/gephyrin/NL2 complex. In NL2−/− neurons, CB has a low on-rate due to the SH3 domain-mediated autoinhibition, and a high off-rate due to the absence of NL2 as a factor that would further stabilize the clusters already formed (upper panel, left). The open conformation mutant of CB, due to its increased on-rate, has an improved ability to initiate gephyrin clustering, but these clusters are not entirely stable and thus relatively small in size and low in intensity, due to the high off-rate that persists in the absence of NL2 (upper panel, middle-right). In the presence of NL2, CB has an increased on-rate, since the conformational equilibrium is shifted toward a more open state, characterized by a decreased off-rate, due to the structural integrity of the CB/gephyrin/NL2 holo-complex that is consolidated by NL2 (lower panel, left). In contrast, compared to the WT protein, the NL2-binding-deficient CB mutant still exhibits a low on-rate and thus has a limited capacity of forming stable gephyrin clusters (lower panel, middle-left), and the autoactive CB mutant outperforms WT CB due to its high on-rate (lower panel, middle-right). The ΔSH3 mutant, due to its very high on-rate, leads to large-sized gephyrin clusters with higher density compared to WT CB. The mean size and the fluorescence intensity of these clusters are larger in CB−/− neurons (lower panel, right) compared to NL2−/− neurons (upper panel, right), once again reflecting the impact of NL2 in stabilizing membrane-tethered gephyrin clusters. The mean size (μm2), mean fluorescence intensity (arbitrary units), and density (number per μm2) of gephyrin clusters induced by each construct (as measured in Figs6 and 7) are indicated.
Our model poses that CB acts as a regulatory node in the formation of inhibitory synapses, upon which allosteric regulation by trans-synaptic neurexin–neuroligin complexes and intracellular lipid second messenger signaling converge to regulate the activity of CB in gephyrin recruitment and the formation of inhibitory synapses. These two regulatory pathways likely act in concert with other regulatory mechanisms that affect gephyrin synthesis, degradation, oligomerization, or function, such as alternative splicing (Herweg & Schwarz, 2012), phosphorylation (Tyagarajan et al, 2010, 2013; Kuhse et al, 2012), phosphorylation-dependent prolyl isomerization (Zita et al, 2007), or proteolytic processing (Tyagarajan et al, 2013) and may even play a key role beyond synaptogenesis during brain development, for example, in the maintenance and plasticity of inhibitory synapses.
Materials and Methods
Constructs
Epitope-tagged expression constructs of HA-NL2 in pcDNA3 and HA-NL3 in pCMV were provided by S. Jamain (Göttingen, Germany; now Paris, France). Myc-CB2SH3+, myc-CB2SH3− (Harvey et al, 2004), GFP-Gephyrin (Fuhrmann et al, 2002), NL2icd-Fc (icd = intracellular domain), and GST-CB2SH3+ (Hoon et al, 2011) constructs were described previously. The GST-tagged bacterial expression construct for the SH3 domain of CB was generated by subcloning the cDNA coding for residues 10–79 of rat CB2SH3+ into the pGEX-4T-1 vector. For large-scale protein purification, rat CBSH3+ cDNA coding for residues 1–456 was subcloned into the IMPACT system vector pTYB12 (New England Biolabs), and rat CB1SH3+ cDNA was subcloned into pTXB1 (New England Biolabs). For each construct, point mutations or domain deletions were introduced by site-directed mutagenesis using the QuikChange protocol (Stratagene). Sequencing of the full open reading frames was performed in each case to validate constructs.
Western blot analysis
Brain homogenates from WT and CB KO mice were prepared as described (Kneussel et al, 1999). Recombinant myc-CB2SH3+ and myc-CB2SH3− were expressed in HEK293FT cells and extracted with TNE buffer containing 1% SDS (50 mM Tris–HCl, 150 mM NaCl, 5 mM EDTA, 1 μM leupeptin, 1 μg/ml aprotinin, and 100 μM PMSF at pH 7.4). Lysates were separated on NuPAGE® Novex® 4–12% Bis-Tris gels (Invitrogen) (20 μg protein/lane), transferred to nitrocellulose membranes, and probed with rabbit anti-CB (1:500, Synaptic Systems) and mouse-anti-SH3(CB) (1:500, BD Biosciences) and secondary HRP-coupled goat-anti-rabbit and goat-anti-mouse antibodies (Bio-Rad, 1:10,000). Immunopositive bands were detected by enhanced chemiluminescence (Pierce).
Protein expression and purification
For crystallography, recombinant rat CBSH3+ and CB1SH3+ were purified from E. coli as described (Xiang et al, 2006). CB proteins were expressed in the E. coli strain BL21 (DE3). Cell lysates were subjected to affinity chromatography on chitin agarose beads (New England Biolabs) equilibrated in buffer A (50 mM HEPES pH 8, 250 mM NaCl, 5% glycerol, 5 mM EDTA). The beads were washed extensively with buffer B (buffer A plus 1 M NaCl), followed by equilibration with cleavage buffer (buffer A plus 50 mM DTT) and incubated for > 40 h at 4°C. After elution with buffer A containing 2 mM β-mercaptoethanol (BME), the protein was transferred to buffer M1 (50 mM HEPES pH 8, 50 mM NaCl, 5% (v/v) glycerol, 5 mM EDTA, 2 mM BME), applied to a MonoQ 10/100GL column (GE Healthcare), and eluted using a linear NaCl gradient from 50 mM to 1 M NaCl. Finally, CBSH3+ was subjected to size exclusion chromatography on a Superdex 200 column (GE Healthcare) and concentrated to 5–10 mg/ml by ultrafiltration with concentrators. To test phospholipid binding of CB, recombinant GST-tagged CB2SH3+ was produced. Glutathione-sepharose beads were incubated with cleared BL21 (DE3) lysates containing GST-fusion proteins for 2 h at 4°C, and the proteins were eluted in a buffer with 50 mM Tris–HCl, 150 mM NaCl, 1 mM DTT, 1 mM EDTA, 0.5% CHAPS, 50 mM reduced glutathione at pH 8.0, and adjusted to a final concentration of 0.2 mg/ml. Mutant variants of CBSH3+ and GST-CB2SH3+ were expressed and purified as described above.
Atomic force microscopy
All CB variants were diluted in AFM deposition buffer (25 mM HEPES pH 7.5, 25 mM sodium acetate, 10 mM magnesium acetate) to a final deposition concentration of 25 nM. Immediately after dilution, 20 μl volumes were deposited on freshly cleaved mica (Grade V; SPI Supplies), rinsed with ultra-pure deionized water, and dried in a gentle stream of nitrogen. The AFM images were captured with a molecular force probe 3D-Bio AFM (Asylum Research, Santa Barbara, CA) in tapping mode using OMCL-AC240TS (Olympus) non-contact/tapping mode silicon probes with spring constants of approximately 2 N/m and resonance frequencies of approximately 75 kHz. Images were collected at a scan speed of 2.5 μm/s at scan sizes of 1 × 1 and 2 × 2 μm, and at pixel resolutions of 512 × 512 and 1024 × 1024, respectively. AFM images were flattened and plane-fitted to 3rd order using the AFM software (Asylum Research on Igor Pro). AFM volumes of imaged particles were determined with the ImageSXM software (S. Barret, Liverpool, UK), and Gaussian fits to the resulting volume distributions provided the average volume of a protein as the center of the peak, using OriginPro 8.6G. Protein molecular weights can be calculated from these AFM volumes by experimentally derived calibration curves (Ratcliff & Erie, 2001). Utilizing proteins of known molecular weights, we determined a linear relationship between measured AFM volumes (V) and molecular weight (MW) of proteins for our AFM setup: V = 1.2 × (MW)−5.9 (Roth et al, 2012). Particles with volumes within ± 2 standard deviations from the center of the Gaussian peak corresponding to monomeric proteins were used for conformational analyses.
Small angle X-ray scattering
For SAXS measurements, proteins were extensively dialyzed against 50 mM HEPES pH 8, 250 mM NaCl, 10% glycerol, 5 mM EDTA, and 5 mM BME, and aggregates were removed by centrifugation (20 min at > 15,000 × g). Synchrotron X-ray scattering data of CB were collected at the EMBL X33 beamline (DESY, Hamburg) (Roessle et al, 2007; Blanchet & Svergun, 2013) and recorded at 10°C using a PILATUS 1M pixel detector (DECTRIS) at a sample-detector distance of 2.7 m and a wavelength of 1.5 Å. This setup covers a range of momentum transfer of 0.005 < s < 0.6/Å (s = 4π sin(θ)/λ, where 2θ is the scattering angle). A robotic sample changer (Round et al, 2008) was used, and the samples were measured in a concentration range from 6.6 to 0.8 mg/ml (6.6, 3.3, 1.6 and 0.8 mg/ml) for WT CBSH3+ and from 1 to 0.27 mg/ml (1.0, 0.5 and 0.27 mg/ml) for CBSH3+/E262A. Initial data preprocessing and reduction were performed using an automatic pipeline and theoretical extrapolation to infinite dilution for the analysis. For the calculation of the forward scattering I(0) and the radius of gyration (Rg), the Guinier approximation implemented in PRIMUS (Konarev et al, 2003) was used, assuming that at very small angles (s < 1.3/Rg), the intensity is represented as I(s) = I(0)*exp(−(sRg)2/3). The pair-distance distribution function P(r) was evaluated with GNOM (Svergun, 1992), and consecutively, the maximum particle dimension (Dmax) as well as Rg were estimated. Finally, the Porod volume was computed using the Porod invariant (Porod, 1952), and the molecular mass estimated as 0.6 times the Porod volume (Petoukhov et al, 2012). Ab initio models were computed with DAMMIF (Franke & Svergun, 2009), using low-resolution data in the range of 0.012/Å < s < 0.20/Å. The algorithm constructs bead models yielding a scattering profile with the lowest possible discrepancy (χ) to the experimental data while keeping beads interconnected and the model compact. Twenty independent ab initio reconstructions were performed and then averaged using DAMAVER (Volkov & Svergun, 2003), which also provides a value of normalized spatial discrepancy (NSD), representing a measure of similarity among different models. Model superimposition was computed using the program SUPCOMB (Kozin & Svergun, 2001). Flexibility was assessed with the Ensemble Optimization Method 2.0 (developed by G. Tria), an enhanced version of EOM (Bernadó et al, 2007), which assumes coexistence of a range of conformations in solution for which an average scattering intensity fits the experimental SAXS data. Using EOM 2.0, a pool of 10,000 independent models was initially generated. All models in the pool showed the individual DH/PH tandem fixed as in the crystal (CB1SH3− pdb code: 4MT7) and the SH3 domain (pdb code: 2YSQ) free to randomly move in order to cover the entire conformational space. The theoretical scattering curve was automatically computed for each model in the pool by CRYSOL (Svergun et al, 1995). Afterward, a genetic algorithm (GA) was employed to selected ensembles, randomly distributed in terms of size from 2 to 50 conformers, by calculating the average theoretical profiles and fitting them to the experimental SAXS data. The GA was repeated 100 times, and the ensemble with the lowest discrepancy was reported as the best solution out of 100 final ensembles.
Crystallization and structure solution
CBSH3+ crystals were obtained by hanging drop vapor diffusion in 20 mM Na-cacodylate, pH 6.5, 5 mM Co(III)hexaminechloride, and 2.4 M KCl. The crystals belonged to the cubic space group I213 with a = b = c = 217.4 Å and contained one molecule in the asymmetric unit. Crystals were transferred into mother liquor containing increasing concentrations of glycerol up to a final concentration of 25% (v/v) in 5% increments and were flash-cooled in liquid nitrogen. CB1SH3+ crystals were grown in 100 mM MES pH 6.5 and 15% PEG 20000. The protein setup for crystallization contained the SH3 domain; however, this domain was lost due to proteolytic degradation, and hence, we refer to the corresponding structure as CB1SH3−. The crystals belonged to the orthorhombic space group C2221 with a = 82.93 Å, b = 165.43 Å and c = 129.12 Å and contained one molecule in the asymmetric unit. Crystals were transferred into mother liquor containing 25% (v/v) glycerol and flash-cooled in liquid nitrogen. Diffraction data for both proteins were collected on beamline ID23-2 at the European Synchrotron Radiation Facility (ESRF) at a wavelength of 0.8726 Å on a MAR225 detector. Data were indexed, integrated, and scaled using Mosflm and Scala (Leslie & Powell, 2007; Winn, 2011). The individual SH3, DH, and PH domain (2YSQ for SH3, 2DFK for DH and PH) structures were subsequently used as search models for molecular replacement using Phaser (McCoy et al, 2007). The initial models were refined using Phenix (Afonine et al, 2012) by rigid body refinement, followed by TLS and grouped B-factor (one B-factor per residue) refinement (for CBSH3+) and individual B-factor refinement (for CB1SH3−), as well as positional refinement employing the individual DH and PH domains of Cdc42-CB2SH3− co-crystal structure (2DFK) and, when present, the NMR structure of the CB SH3 domain (2YSQ) as reference models. Structure illustrations and calculations were performed using Pymol (The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC).
Protein–lipid overlay assays
To assess phosphoinositide specificity of CB, PIP-Strips (Echelon) were used according to the manufacturer's instructions. To test relative PI(3)P binding strength of individual CB variants, custom-made PI(3)P strips were prepared by spotting 1, 0.75, 0.5, 0.25 and 0.1 μg of PI(3)P (Echelon) onto Hybond-C-extra membranes (GE Healthcare). Membranes were blocked with 5% milk in TBS and incubated with 0.5 μg of GST-tagged CB proteins for 1 h at RT. After 3 washes with 5% milk in TBST (0.1% Tween-20 in TBS), bound proteins were detected with standard dot-blot techniques using a mouse-anti-GST antibody (Sigma, 1:4,000), followed by an HRP-coupled goat-anti-mouse secondary antibody (Bio-Rad, 1:10,000).
Microscale thermophoresis
MST binding studies were performed using standard protocols on a Monolith NT.115 (Nanotemper Technologies). Briefly, recombinant CB2SH3− or CB2SH3−/E202A were labeled with the RED-NHS (amine reactive) protein labeling kit (Nanotemper Technologies). CB2SH3− or CB2SH3−/E202A were mixed with purified SH3, SH3/W24A, or SH3/R70A in 20 mM Tris pH 8.0, 150 mM NaCl, 1 mM BME, 0.05% Tween-20 and 0.1% BSA, incubated for 10 min at room temperature, and loaded into hydrophilic capillaries. The concentration of the labeled proteins was kept constant at 100 nM, while the concentration of the SH3 domain variants varied from 1.5 to 1500 μM. Measurements were repeated at least three times at 20 and 30% laser power, respectively, and 30% LED power. KD values were obtained by plotting the normalized fluorescence Fnorm against the logarithm of the different concentrations of the dilution series, and by fitting the data using OriginPro 9.1.
Transfection, immunostaining, and imaging of cell lines
COS7 cells were plated directly onto glass coverslips and cultured in DMEM with 10% fetal calf serum, and transfected using FuGENE6 (Roche) or Lipofectamine 2000 (Invitrogen) according to standard protocols. Cells were fixed 10 h post-transfection in 4% paraformaldehyde in 100 mM phosphate buffer pH 7.4 (PB) for 10 min at RT. Samples were blocked with 5% normal goat serum and 0.1% gelatin in PB. Prior to permeabilization, cells were stained with polyclonal rabbit anti-HA antibody (1:2,000, Zymed Laboratories, Invitrogen) in the same blocking solution for 2 h at RT to detect the surface pool of HA-tagged NLs. After 3 washes with PB, cells were permeabilized with 0.1% Triton X-100, 5% normal goat serum, and 0.1% gelatin in PB and stained with monoclonal mouse-anti-myc antibodies (clone 9E10, 1:1,000, Sigma-Aldrich) in the same buffer for 2 h at RT. Secondary antibody staining was performed for 1 h at RT with anti-isotypic fluorophore-conjugated antibodies Alexa-555 and Cy5 at dilutions of 1:2,000 (Molecular Probes). Samples were imaged using an inverse Leica DMIRE2 microscope equipped with a 63× oil-immersion objective connected to a Leica TCS SP2 AOBS confocal laser scanning setup. Intensity correlation analysis was performed on multichannel images using ImageJ. Briefly, a Gaussian blur was applied, and GFP and HA channels were thresholded. A standard Pearson's correlation coefficient was evaluated between the HA and GFP channels in the thresholded fields using the Intensity Correlation Analysis plugin for ImageJ (http://rsb.info.nih.gov/ij/) from T. Collins and E. Stanley (Toronto, ON, Canada).
Culture, transfection, immunostaining, and image analysis of cultured neurons
Hippocampal neurons were prepared from newborn CB−/− and NL2−/− mice for transfection experiments. Hippocampi were treated with 20 units/ml papain (Worthington) in DMEM (Gibco) containing 0.2 mg/ml cysteine, 1 mM CaCl2, and 0.5 mM EDTA, for 1 h at 37°C, followed by a 15-min incubation with the stop solution containing 2.5 mg/ml albumin, 2.5 mg/ml trypsin inhibitor, and 10% FCS (Gibco) in DMEM (Gibco). After triturating mechanically, the cells were plated on poly-L-lysine coated glass coverslips at a density of 30,000 cells/cm2. Neurons were cultured in Neurobasal medium (Gibco) supplemented with B27 (Gibco), glutamax (Gibco), and penicillin/streptomycin (Roche). Neurons were transfected at DIV3-4 with the calcium phosphate method using a custom-made 2× HBS buffer (274 mM NaCl, 10 mM KCl, 1.4 mM Na2HPO4, 15 mM glucose, 42 mM HEPES, pH 7.15) and 2 M CaCl2 solution. An established transfection protocol was used for enhanced transfection efficiency and reduced cytotoxicity (Jiang & Chen, 2006). For immunostaining, cultured neurons at DIV14 were washed in PBS, fixed with 4% paraformaldehyde in PB for 7 min, and permeabilized with 0.1% Triton X-100 in PBS containing 5% normal goat serum and 0.1% gelatin. The samples were incubated with mAb7a mouse monoclonal anti-gephyrin (1:3,000, Connex), rabbit anti-myc (1:1,000, Sigma-Aldrich) and polyclonal guinea-pig anti-VIAAT (1:2,000, Synaptic Systems) antibodies overnight at 4°C. After further washes with PBS, the neurons were incubated with fluorophore-conjugated goat-anti-rabbit, goat-anti-guinea-pig and IgG1 isotype-specific goat-anti-mouse secondary antibodies (Molecular Probes). Specimens were analyzed with the microscope setup described above for cell lines. Images were acquired as z-stacks (10 optical sections, 0.3 μm step size). Maximum intensity projections were created from the z-stacks and analyzed using ImageJ (http://rsb.info.nih.gov/ij/). For the analysis, both gephyrin and VIAAT channels were modified using the Gaussian blur filter and the subtract background function (with a rolling ball radius of 10 pixels) and thresholded (threshold values of 30 and 40 were selected for gephyrin and VIAAT channels, respectively). Somata of the transfected neurons were outlined and gephyrin, and VIAAT clusters ranging between 0.04–1 μm2 were counted using the analyze particles function. The number of gephyrin and VIAAT clusters per μm2 area of somata, as well as the mean size (in μm2) and the mean fluorescence intensity (in arbitrary units ranging between 30 and 255) of gephyrin clusters, was recorded. Gephyrin clusters overlapping with VIAAT clusters were regarded as synaptic.
Statistics
Statistical comparisons in this study were made using the unpaired, two-tailed Student's t-test unless mentioned otherwise. P-values < 0.05 were considered significant. For cell culture experiments and MST measurements, all values represent the mean ± standard error of the mean (SEM), unless mentioned otherwise. Asterisks indicate significant differences (*P < 0.05, **P < 0.01 and ***P < 0.001); n.s. indicates no significant difference. For AFM data, all values represent the mean ± one standard deviation (SD) from repeated experiments. For custom-made PI(3)P strip assays, a paired Student's t-test was performed. For MST measurements, an unpaired, one-tailed Student's t-test was performed.
Acknowledgments
We thank F. Benseler, I. Thanhäuser, and D. Schwerdtfeger for excellent technical support, and the staff of the MPIEM animal facility for the management of mouse colonies. This work was supported by the Deutsche Forschungsgemeinschaft (Rudolf Virchow Center for Experimental Biomedicine, FZ 82, H.S. and I.T.; Schi 425/8-1 and SFB487 C7, H.S.), by the Max Planck Society (N.Br.), by the European Commission EUROSPIN and SynSys Consortia (FP7-HEALTH-F2-2009-241498, FP7-HEALTH-F2-2009-242167, N.Br.), by the European Commission Innovative Medicines Initiative (EU-AIMS FP7-115300, N.Br.), by the Bundesministerium für Bildung und Forschung (BIOSCAT, 05K12YE1, D.Sv.), and by a European Union FP7 Infrastructure grant (BioStruct-X, 283570, D.Sv.).
Author contributions
The author(s) have made the following declarations about their contributions: TS, DSc, GT, DSv, AP, FV, HS, and NBr conceived and designed the experiments. TS, DSc, CB, NBa, GT, DSv, IT, and AP performed the experiments. TS, DSc, GT, CB, NBa, DSv, IT, AP, and HS analyzed the data. TP contributed reagents/materials/analysis tools. TS, DSc, HS, and NBr wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernadó P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- Blanchet CE, Svergun DI. Small-angle X-ray scattering on biological macromolecules and nanocomposites in solution. Annu Rev Phys Chem. 2013;64:37–54. doi: 10.1146/annurev-physchem-040412-110132. [DOI] [PubMed] [Google Scholar]
- Chiou T-T, Bonhomme B, Jin H, Miralles CP, Xiao H, Fu Z, Harvey RJ, Harvey K, Vicini S, De Blas AL. Differential regulation of the postsynaptic clustering of γ-aminobutyric acid type A (GABAA) receptors by collybistin isoforms. J Biol Chem. 2011;286:22456–22468. doi: 10.1074/jbc.M111.236190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro E, Peluso D, Via A, Ausiello G, Helmer-Citterich M. SH3-hunter: discovery of SH3 domain interaction sites in proteins. Nucleic Acids Res. 2007;35:451–454. doi: 10.1093/nar/gkm296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Crystallogr. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann JC, Kins S, Rostaing P, Far El O, Kirsch J, Sheng M, Triller A, Betz H, Kneussel M. Gephyrin interacts with Dynein light chains 1 and 2, components of motor protein complexes. J Neurosci. 2002;22:5393–5402. doi: 10.1523/JNEUROSCI.22-13-05393.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey K, Duguid IC, Alldred MJ, Beatty SE, Ward H, Keep NH, Lingenfelter SE, Pearce BR, Lundgren J, Owen MJ, Smart TG, Lüscher B, Rees MI, Harvey RJ. The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J Neurosci. 2004;24:5816–5826. doi: 10.1523/JNEUROSCI.1184-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herweg J, Schwarz G. Splice-specific glycine receptor binding, folding, and phosphorylation of the scaffolding protein gephyrin. J Biol Chem. 2012;287:12645–12656. doi: 10.1074/jbc.M112.341826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoon M, Soykan T, Falkenburger B, Hammer M, Patrizi A, Schmidt K-F, Sassoè-Pognetto M, Löwel S, Moser T, Taschenberger H, Brose N, Varoqueaux F. Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina. Proc Natl Acad Sci USA. 2011;108:3053–3058. doi: 10.1073/pnas.1006946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob TC, Bogdanov YD, Magnus C, Saliba RS, Kittler JT, Haydon PG, Moss SJ. Gephyrin regulates the cell surface dynamics of synaptic GABAA receptors. J Neurosci. 2005;25:10469–10478. doi: 10.1523/JNEUROSCI.2267-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal M, Dvorsky R, Ahmadian MR. Deciphering the molecular and functional basis of Dbl family proteins: a novel systematic approach toward classification of selective activation of the Rho family proteins. J Biol Chem. 2013;288:4486–4500. doi: 10.1074/jbc.M112.429746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedlicka P, Papadopoulos T, Deller T, Betz H, Schwarzacher SW. Increased network excitability and impaired induction of long-term potentiation in the dentate gyrus of collybistin-deficient mice in vivo. Mol Cell Neurosci. 2009;41:94–100. doi: 10.1016/j.mcn.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Jedlicka P, Hoon M, Papadopoulos T, Vlachos A, Winkels R, Poulopoulos A, Betz H, Deller T, Brose N, Varoqueaux F, Schwarzacher SW. Increased dentate gyrus excitability in neuroligin-2-deficient mice in vivo. Cereb Cortex. 2011;21:357–367. doi: 10.1093/cercor/bhq100. [DOI] [PubMed] [Google Scholar]
- Jiang M, Chen G. High Ca2+-phosphate transfection efficiency in low-density neuronal cultures. Nat Protoc. 2006;1:695–700. doi: 10.1038/nprot.2006.86. [DOI] [PubMed] [Google Scholar]
- Kalscheuer VM, Musante L, Fang C, Hoffmann K, Fuchs C, Carta E, Deas E, Venkateswarlu K, Menzel C, Ullmann R, Tommerup N, Dalprà L, Tzschach A, Selicorni A, Lüscher B, Ropers H-H, Harvey K, Harvey RJ. A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum Mutat. 2009;30:61–68. doi: 10.1002/humu.20814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Senda T, Ishidate T, Koyama R, Morishita T, Iwayama Y, Higuchi O, Akiyama T. Asef, a link between the tumor suppressor APC and G-protein signaling. Science. 2000;289:1194–1197. doi: 10.1126/science.289.5482.1194. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Sato R, Akiyama T. Mutated APC and Asef are involved in the migration of colorectal tumour cells. Nat Cell Biol. 2003;5:211–215. doi: 10.1038/ncb937. [DOI] [PubMed] [Google Scholar]
- Kins S, Betz H, Kirsch J. Collybistin, a newly identified brain-specific GEF, induces submembrane clustering of gephyrin. Nat Neurosci. 2000;3:22–29. doi: 10.1038/71096. [DOI] [PubMed] [Google Scholar]
- Kneussel M, Brandstätter JH, Laube B, Stahl S, Müller U, Betz H. Loss of postsynaptic GABA(A) receptor clustering in gephyrin-deficient mice. J Neurosci. 1999;19:9289–9297. doi: 10.1523/JNEUROSCI.19-21-09289.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneussel M, Betz H. Receptors, gephyrin and gephyrin-associated proteins: novel insights into the assembly of inhibitory postsynaptic membrane specializations. J Physiol. 2000;525:1–9. doi: 10.1111/j.1469-7793.2000.t01-4-00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr. 2003;36:1277–1282. [Google Scholar]
- Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Crystallogr. 2001;34:33–41. [Google Scholar]
- Kuhse J, Kalbouneh H, Schlicksupp A, Mükusch S, Nawrotzki R, Kirsch J. Phosphorylation of gephyrin in hippocampal neurons by cyclin-dependent kinase CDK5 at ser-270 is dependent on collybistin. J Biol Chem. 2012;287:30952–30966. doi: 10.1074/jbc.M112.349597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie AW, Powell H. Processing diffraction data with mosflm. In: Read R, Sussman J, editors. Evolving Methods for Macromolecular Crystallography. Netherlands: Springer; 2007. pp. 41–51. [Google Scholar]
- Maric H-M, Mukherjee J, Tretter V, Moss SJ, Schindelin H. Gephyrin-mediated γ-aminobutyric acid type A and glycine receptor clustering relies on a common binding site. J Biol Chem. 2011;286:42105–42114. doi: 10.1074/jbc.M111.303412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer S, Kumar R, Jaiswal M, Soykan T, Ahmadian MR, Brose N, Betz H, Rhee J-S, Papadopoulos T. Collybistin activation by GTP-TC10 enhances postsynaptic gephyrin clustering and hippocampal GABAergic neurotransmission. Proc Natl Acad Sci USA. 2013;110:20795–20800. doi: 10.1073/pnas.1309078110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier J, Vannier C, Sergé A, Triller A, Choquet D. Fast and reversible trapping of surface glycine receptors by gephyrin. Nat Neurosci. 2001;4:253–260. doi: 10.1038/85099. [DOI] [PubMed] [Google Scholar]
- Meyer G, Kirsch J, Betz H, Langosch D. Identification of a gephyrin binding motif on the glycine receptor beta subunit. Neuron. 1995;15:563–572. doi: 10.1016/0896-6273(95)90145-0. [DOI] [PubMed] [Google Scholar]
- Mitin N, Betts L, Yohe ME, Der CJ, Sondek J, Rossman KL. Release of autoinhibition of ASEF by APC leads to CDC42 activation and tumor suppression. Nat Struct Mol Biol. 2007;14:814–823. doi: 10.1038/nsmb1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss SJ, Smart TG. Constructing inhibitory synapses. Nat Rev Neurosci. 2001;2:240–250. doi: 10.1038/35067500. [DOI] [PubMed] [Google Scholar]
- Mukherjee J, Kretschmannova K, Gouzer G, Maric H-M, Ramsden S, Tretter V, Harvey K, Davies PA, Triller A, Schindelin H, Moss SJ. The residence time of GABA(A)Rs at inhibitory synapses is determined by direct binding of the receptor α1 subunit to gephyrin. J Neurosci. 2011;31:14677–14687. doi: 10.1523/JNEUROSCI.2001-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama K, Shirouzu M, Kawasaki Y, Kato-Murayama M, Hanawa-Suetsugu K, Sakamoto A, Katsura Y, Suenaga A, Toyama M, Terada T, Taiji M, Akiyama T, Yokoyama S. Crystal structure of the rac activator, Asef, reveals its autoinhibitory mechanism. J Biol Chem. 2007;282:4238–4242. doi: 10.1074/jbc.C600234200. [DOI] [PubMed] [Google Scholar]
- Panzanelli P, Gunn BG, Schlatter MC, Benke D, Tyagarajan SK, Scheiffele P, Belelli D, Lambert JJ, Rudolph U, Fritschy J-M. Distinct mechanisms regulate GABAA receptor and gephyrin clustering at perisomatic and axo-axonic synapses on CA1 pyramidal cells. J Physiol. 2011;589:4959–4980. doi: 10.1113/jphysiol.2011.216028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos T, Korte M, Eulenburg V, Kubota H, Retiounskaia M, Harvey RJ, Harvey K, O'Sullivan GA, Laube B, Hülsmann S, Geiger JRP, Betz H. Impaired GABAergic transmission and altered hippocampal synaptic plasticity in collybistin-deficient mice. EMBO J. 2007;26:3888–3899. doi: 10.1038/sj.emboj.7601819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos T, Eulenburg V, Reddy-Alla S, Mansuy IM, Li Y, Betz H. Collybistin is required for both the formation and maintenance of GABAergic postsynapses in the hippocampus. Mol Cell Neurosci. 2008;39:161–169. doi: 10.1016/j.mcn.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Papadopoulos T, Soykan T. The role of collybistin in gephyrin clustering at inhibitory synapses: facts and open questions. Front Cell Neurosci. 2011;5:11. doi: 10.3389/fncel.2011.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porod G. Die Röntgenkleinwinkelstreuung von dichtgepackten kolloiden Systemen. Kolloid-Zeitschrift. 1952;125:51–57. [Google Scholar]
- Poulopoulos A, Aramuni G, Meyer G, Soykan T, Hoon M, Papadopoulos T, Zhang M, Paarmann I, Fuchs C, Harvey K, Jedlicka P, Schwarzacher SW, Betz H, Harvey RJ, Brose N, Zhang W, Varoqueaux F. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron. 2009;63:628–642. doi: 10.1016/j.neuron.2009.08.023. [DOI] [PubMed] [Google Scholar]
- Ratcliff GC, Erie DA. A novel single-molecule study to determine protein–protein association constants. J Am Chem Soc. 2001;123:5632–5635. doi: 10.1021/ja005750n. [DOI] [PubMed] [Google Scholar]
- Reddy-Alla S, Schmitt B, Birkenfeld J, Eulenburg V, Dutertre S, Böhringer C, Götz M, Betz H, Papadopoulos T. PH-domain-driven targeting of collybistin but not Cdc42 activation is required for synaptic gephyrin clustering. Eur J Neurosci. 2010;31:1173–1184. doi: 10.1111/j.1460-9568.2010.07149.x. [DOI] [PubMed] [Google Scholar]
- Roessle MW, Klaering R, Ristau U, Robrahn B, Jahn D, Gehrmann T, Konarev P, Round A, Fiedler S, Hermes C, Svergun D. Upgrade of the small-angle X-ray scattering beamline X33 at the European Molecular Biology Laboratory, Hamburg. J Appl Crystallogr. 2007;40:190–194. [Google Scholar]
- Roth HM, Römer J, Grundler V, Van Houten B, Kisker C, Tessmer I. XPB helicase regulates DNA incision by the thermoplasma acidophilum endonuclease Bax1. DNA Repair. 2012;11:286–293. doi: 10.1016/j.dnarep.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Round AR, Franke D, Moritz S, Huchler R, Fritsche M, Malthan D, Klaering R, Svergun DI, Roessle M. Automated sample-changing robot for solution scattering experiments at the EMBL Hamburg SAXS station X33. J Appl Crystallogr. 2008;41:913–917. doi: 10.1107/S0021889808021018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiepour L, Fuchs C, Patrizi A, Sassoè-Pognetto M, Harvey RJ, Harvey K. Complex role of collybistin and gephyrin in GABAA receptor clustering. J Biol Chem. 2010;285:29623–29631. doi: 10.1074/jbc.M110.121368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander B, Tria G, Shkumatov AV, Kim EY, Grossmann JG, Tessmer I, Svergun DI, Schindelin H. Structural characterization of gephyrin by AFM and SAXS reveals a mixture of compact and extended states. Acta Crystallogr D Biol Crystallogr. 2013;69:2050–2060. doi: 10.1107/S0907444913018714. [DOI] [PubMed] [Google Scholar]
- Shimojima K, Sugawara M, Shichiji M, Mukaida S, Takayama R, Imai K, Yamamoto T. Loss-of-function mutation of collybistin is responsible for X-linked mental retardation associated with epilepsy. J Hum Genet. 2011;56:561–565. doi: 10.1038/jhg.2011.58. [DOI] [PubMed] [Google Scholar]
- Specht CG, Izeddin I, Rodriguez PC, Beheiry El M, Rostaing P, Darzacq X, Dahan M, Triller A. Quantitative nanoscopy of inhibitory synapses: counting gephyrin molecules and receptor binding sites. Neuron. 2013;79:308–321. doi: 10.1016/j.neuron.2013.05.013. [DOI] [PubMed] [Google Scholar]
- Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr. 1992;25:495–503. [Google Scholar]
- Svergun D, Barberato C, Koch MHJ. CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- Tretter V, Jacob TC, Mukherjee J, Fritschy J-M, Pangalos MN, Moss SJ. The clustering of GABA(A) receptor subtypes at inhibitory synapses is facilitated via the direct binding of receptor alpha 2 subunits to gephyrin. J Neurosci. 2008;28:1356–1365. doi: 10.1523/JNEUROSCI.5050-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V, Kerschner B, Milenkovic I, Ramsden SL, Ramerstorfer J, Saiepour L, Maric HM, Moss SJ, Schindelin H, Harvey RJ, Sieghart W, Harvey K. Molecular basis of the γ-aminobutyric acid a receptor 3 subunit interaction with the clustering protein gephyrin. J Biol Chem. 2011;286:37702–37711. doi: 10.1074/jbc.M111.291336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V, Mukherjee J, Maric H-M, Schindelin H, Sieghart W, Moss SJ. Gephyrin, the enigmatic organizer at GABAergic synapses. Front Cell Neurosci. 2012;6:23. doi: 10.3389/fncel.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagarajan SK, Ghosh H, Yévenes GE, Nikonenko I, Ebeling C, Schwerdel C, Sidler C, Zeilhofer HU, Gerrits B, Muller D, Fritschy J-M. Regulation of GABAergic synapse formation and plasticity by GSK3{beta}-dependent phosphorylation of gephyrin. Proc Natl Acad Sci USA. 2010;108:379–384. doi: 10.1073/pnas.1011824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagarajan SK, Ghosh H, Harvey K, Fritschy J-M. Collybistin splice variants differentially interact with gephyrin and Cdc42 to regulate gephyrin clustering at GABAergic synapses. J Cell Sci. 2011;124:2786–2796. doi: 10.1242/jcs.086199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagarajan SK, Ghosh H, Yévenes GE, Imanishi SY, Zeilhofer HU, Gerrits B, Fritschy J-M. ERK and GSK3β regulate gephyrin postsynaptic aggregation and GABAergic synaptic function in a calpain-dependent mechanism. J Biol Chem. 2013;288:9634–9647. doi: 10.1074/jbc.M112.442616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD. Overview of the CCP4 suite and current developments. Acta Cryst. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang S, Kim EY, Connelly JJ, Nassar N, Kirsch J, Winking J, Schwarz G, Schindelin H. The crystal structure of Cdc42 in complex with collybistin II, a gephyrin-interacting guanine nucleotide exchange factor. J Mol Biol. 2006;359:35–46. doi: 10.1016/j.jmb.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chen L, Gao L, Lin K, Zhu L, Lu Y, Shi X, Gao Y, Zhou J, Xu P, Zhang J, Wu G. Structural basis for the recognition of Asef by adenomatous polyposis coli. Cell Res. 2012;22:372–386. doi: 10.1038/cr.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zita MM, Marchionni I, Bottos E, Righi M, Del Sal G, Cherubini E, Zacchi P. Post-phosphorylation prolyl isomerisation of gephyrin represents a mechanism to modulate glycine receptors function. EMBO J. 2007;26:1761–1771. doi: 10.1038/sj.emboj.7601625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.