

Abstract
According to current belief, the molecular networks orchestrating cell death or exit from mitosis upon extended mitotic arrest do not interact, stubbornly executing two parallel biological programs and competing to define a stochastic decision between death and a chance for survival with uncertain destiny. However, recent findings by Diaz-Martinez et al (2014) in this issue of The EMBO Journal now call for a reassessment of the “competing network” hypothesis.
See also: LA Diaz-Martinez et al (September 2014)
Anti-mitotic drugs are essential ingredients of current anti-cancer therapy. While the molecular basis of the clinical benefit elicited by these drugs is still debated (Mitchison, 2012), cells exposed to taxanes or vinca-alkaloids in experimental settings usually undergo one of two fates after prolonged mitotic arrest: cell death, usually by apoptosis, or adaptation, that is exit from mitosis without cellular division, a process also known as mitotic slippage and a possible cause for long-term treatment failure.
The “competing network” hypothesis developed by Gascoigne and Taylor suggests that two independent molecular circuits control cell death or slippage upon extended mitotic arrest (Gascoigne & Taylor, 2008). In this model, cell fate solely depends on the time needed by either program to reach a critical threshold. Gradual decline in cyclin B1 levels defines the time period to mitotic exit, since even in arrested cells, the spindle assembly checkpoint (SAC) is unable to fully restrain the activity of the APC/CCdc20 ubiquitin ligase toward cyclin B1 (Brito & Rieder, 2006). In parallel, apoptotic cell death is initiated through the integration of largely undefined signals leading to activation of the two key pro-apoptotic effectors within the Bcl-2 family, Bax and/or Bak, required for mitochondrial outer membrane permeabilization (MOMP) and subsequent caspase activation (reviewed in Czabotar et al, 2013). Different cancer cells and cell lines vary considerably in their responsiveness to anti-mitotic drug treatment, with, for example, HeLa and RKO cells being highly apoptosis-prone, while U2OS or DLD1 cells tend to adapt. This has been in part explained by differences in individual apoptosis and adaptation thresholds, which may in turn be based on different expression levels and activities of key components of the respective pathways (Gascoigne & Taylor, 2008).
Several studies have highlighted possible molecular crosstalk between the cell death and cell cycle machineries: on one hand, pro-survival Bcl-2 family members and initiating cell death caspases are targets of Cdk1-dependent phosphorylation (reviewed in Topham & Taylor, 2013); on the other hand, the SAC protein BubR1 is cleaved by caspases during apoptosis (Kim et al, 2005). Despite such “opportunities to communicate”, negatively interfering with adaptation (e.g. by overexpressing non-degradable cyclin B1 or by depleting the APC/C activator Cdc20) leaves mitotic cell death unaffected, and similarly, inhibiting caspases does not affect the kinetics of checkpoint adaptation (Huang et al, 2010). The further fate of such post-slippage cells can be highly variable, but this will not be discussed here in more detail (for review, please refer to Vitale et al, 2011). Although well-supported by the above observations, the “competing network” model becomes less coherent upon interference with cell death upstream of mitochondria, as Diaz-Martinez et al now clearly show that inhibition of MOMP impacts on the timing by which cells adapt, in direct contradiction to the model.
Performing a genome-wide RNAi screen and monitoring survival of Taxol-treated HeLa cells, Hongtao Yu's laboratory identified a number of known and novel candidate genes involved in mitotic cell death and adaptation (Diaz-Martinez et al, 2014). Knockdown of regulators of mitochondrial apoptosis (Bad, Noxa, and Bax) and SAC fidelity (Mps1, Mad2, and BubR1) increased HeLa cell viability, as did depletion of factors that delayed mitotic entry. In contrast, knockdown of APC/C components (ANAPC1/5/13, CDC23, and CDC26), which participate in the adaptation network, reduced survival, as did knockdown of the mitotic regulator Plk1 or the MAD2-inhibitor p31comet (Diaz-Martinez et al, 2014).
p31comet prevents conformational activation of MAD2, a key SAC protein and component of the mitotic checkpoint complex (MCC), and by promoting MCC disassembly both during and after checkpoint arrest controls the amount of assembled MCC, favouring APC/CCdc20 activation and mitotic exit (Varetti et al, 2011). Consistently, p31comet silencing was reported to sensitize cancer cells to anti-mitotic drugs, suggesting critical roles in adaptation. In slippage-prone U2OS cells, p31comet knockdown readily reduced adaptation and increased rates of mitotic cell death. As predicted, enforced arrest upon p31comet knockdown was associated with prolonged APC/CCdc20 inhibition reflected by reduced cyclin B1 degradation. Surprisingly however, p31comet knockdown also significantly shortened the time to cell death, and in cell death-prone HeLa cells accelerated caspase activation, with neither effect being phenocopied when APC/C activation was prevented by Cdc20 knockdown (Diaz-Martinez et al, 2014). Taken together, these findings thus demonstrate that p31comet exerts a previously unappreciated anti-apoptotic function in mitotically arrested cells, independent of its role in promoting mitotic slippage by SAC inhibition and APC/C activation.
The comparable death-prone phenotype seen when depleting only p31comet or co-depleting p31comet and CDC20 may suggest that p31comet affects apoptosis independently of APC/CCdc20; however, since Bcl-2 family members such as the pro-survival molecule Mcl-1 (Harley et al, 2010) and the pro-apoptotic BH3-only protein Bim (Wan et al, 2014) have also been reported to be APC/C degradation targets, it can at this point not yet be excluded that p31comet acts by indirectly controlling the protein abundance of cell death regulators. As such, it would have been interesting to see whether depletion of p31comet, Cdc20, or both, in Taxol-arrested cells would differentially affect Bim and Mcl-1 levels, something that was, however, not pursued in more detail. In the case of Bim, the authors observed insignificant cell death protection upon knockdown to begin with, and they also failed to reproduce Mcl-1 stabilization upon Cdc20 depletion in their experimental conditions, consistent with the existence of multiple redundant mechanisms acting to degrade pro-survival Mcl-1 during mitotic arrest (Topham & Taylor, 2013). Hence, it remains possible that p31comet controls additional pro-apoptotic targets that act in concert with Bim to kill mitotically arrested cells, given that even the most potent BH3-only proteins usually display significant redundancy with other members of this group.
Amongst all pro-survival Bcl-2 family members, Mcl-1 is the one that displays the highest affinity for the BH3-only protein Noxa, while it does not interact with Bad, another pro-apoptotic homolog picked up by the authors' initial screen. Although reported to be a critical effector of p53-mediated cell death upon DNA damage, recent studies suggest Noxa roles also in cell death upon glucose-deprivation or proteasome inhibition, where it neutralizes Mcl-1 (reviewed in Ploner et al, 2008). Strikingly, Noxa knockdown in HeLa cells proved as efficient in preventing Taxol-mediated apoptosis and increasing adaptation rate as did combined Bax/Bak knockdown or Mcl-1 overexpression. Moreover, inhibition of the Noxa/Mcl-1/Bax/Bak axis in slippage-prone U2OS cells resulted not only in suppression of apoptosis but also in a reduction of mitotic adaptation, demonstrating once more how key players of one pathway impact on the other (Diaz-Martinez et al, 2014). Although the lack of suitable reagents (in particular anti-Noxa antibodies of sufficient quality) prevented the authors from showing Noxa accumulation or a direct interaction between p31comet and Noxa in mitotically arrested cells, it remains of interest to test whether and how components of the p31comet-targeted MCC or mitotic kinases might control Noxa abundance and thereby regulate Mcl-1 levels upon enforced mitotic arrest.
Of note, Mcl-1 inhibition by Noxa may not suffice to prime all cell types stalled in mitosis to apoptosis nor may Bim accumulation be rate-limiting in all settings. Accordingly, RKO cell sensitisation to Taxol-induced death was recently reported to involve a Cdk1-dependent mitotic priming phosphorylation on Bid (Wang et al, 2014). Similarly, direct inhibitory phosphorylation of Bcl-2 and Bcl-x by Cdk1 (Topham & Taylor, 2013) may be more or less critical dependent on a given cell type or stimulus. Together, this suggests that cells during mitosis become highly primed to apoptosis and ready to self-destruct any time in the face of trouble, with timely exit from mitosis seemingly the only way to survive.
While these findings together with the new results presented here provide a plausible model for mitotic cell death (Fig1), it remains enigmatic how upstream elements of the apoptotic machinery may directly interfere with mitotic adaptation. Having excluded several proteins acting downstream of MOMP, Diaz-Martinez and colleagues propose non-apoptotic Bax/Bak functions in mitochondrial dynamics as a possible cause for this phenomenon. Bax/Bak impinge on mitochondrial fission by facilitating activation of dynamin-related protein 1 (Drp1), and concerted fission is a prerequisite for the successful completion of mitosis (Kashatus et al, 2011; and references therein). Consistent with this hypothesis, Drp1 knockdown caused mitotic phenotypes comparable to those caused by Bax/Bak depletion but did not affect cell death (Diaz-Martinez et al, 2014). In the absence of Noxa, increased availability of Mcl-1 may keep Bax/Bak from interacting with the fission machinery and hence phenocopy the effect of Bax/Bak depletion. Whether mitochondrial fission, as speculated by the authors, weakens the spindle assembly checkpoint through a drop in ATP levels and subsequent global reduction of protein synthesis affecting cyclin B1 levels remains to be experimentally established.
Figure 1. The molecular circuitry determining cell fate upon extended mitotic arrest.
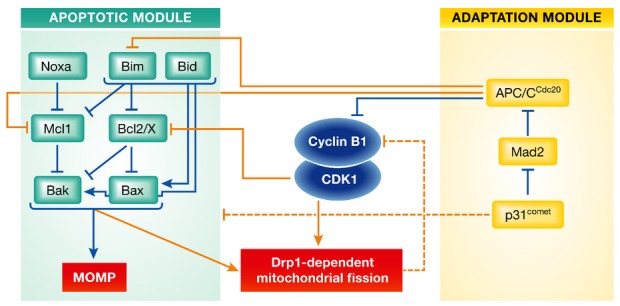
Two molecular modules contribute to the decision whether a mitotically arrested cell undergoes apoptosis after mitochondrial outer membrane permeabilization (MOMP), or exhibits mitotic slippage following cyclin B1 degradation below a critical threshold. Inhibitory and activating interactions are displayed by blue lines whenever taking place within a module and by orange lines whenever coupling both modules. Dashed lines represent functionally identified interactions of indirect or yet to be defined molecular nature.
Collectively, these observations argue against a strict separation of two competing molecular networks that would act independently of each other to define the fate of mitotically arrested cells. Instead, Diaz-Martinez et al demonstrate how individual proteins thought to belong to one given network actually act in both, and ultimately that components of both circuitries are hard-wired together, even if they may not exclusively execute their presumed bona fide biological functions.
Acknowledgments
The work in our laboratory is supported by grants from the Austrian Science Fund (FWF), Tiroler Wissenschaftsfond (TWF), and Krebshilfe Tirol. LLF was supported by the EMBO-LTF program. We apologize to all scientists in the field whose work could not be cited due to space constraints.
References
- Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16:1194–1200. doi: 10.1016/j.cub.2006.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2013;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- Diaz-Martinez LA, Karamysheva Z, Warrington R, Li B, Wei S, Xie XJ, Roth MG, Yu H. Genome-wide siRNA screen reveals coupling between mitotic apoptosis and adaptation. EMBO J. 2014;33:1960–1976. doi: 10.15252/embj.201487826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–122. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29:2407–2420. doi: 10.1038/emboj.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Mitchison TJ, Shi J. Stochastic competition between mechanistically independent slippage and death pathways determines cell fate during mitotic arrest. PLoS ONE. 2010;5:e15724. doi: 10.1371/journal.pone.0015724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, Counter CM. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol. 2011;13:1108–1115. doi: 10.1038/ncb2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Murphy K, Liu F, Parker SE, Dowling ML, Baff W, Kao GD. Caspase-mediated specific cleavage of BubR1 is a determinant of mitotic progression. Mol Cell Biol. 2005;25:9232–9248. doi: 10.1128/MCB.25.21.9232-9248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell. 2012;23:1–6. doi: 10.1091/mbc.E10-04-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploner C, Kofler R, Villunger A. Noxa: at the tip of the balance between life and death. Oncogene. 2008;27(Suppl. 1):S84–S92. doi: 10.1038/onc.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham CH, Taylor SS. Mitosis and apoptosis: how is the balance set? Curr Opin Cell Biol. 2013;25:780–785. doi: 10.1016/j.ceb.2013.07.003. [DOI] [PubMed] [Google Scholar]
- Varetti G, Guida C, Santaguida S, Chiroli E, Musacchio A. Homeostatic control of mitotic arrest. Mol Cell. 2011;44:710–720. doi: 10.1016/j.molcel.2011.11.014. [DOI] [PubMed] [Google Scholar]
- Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–392. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- Wan L, Tan M, Yang J, Inuzuka H, Dai X, Wu T, Liu J, Shaik S, Chen G, Deng J, Malumbres M, Letai A, Kirschner MW, Sun Y, Wei W. APC(Cdc20) suppresses apoptosis THROUGH targeting bim for ubiquitination and destruction. Dev Cell. 2014;29:377–391. doi: 10.1016/j.devcel.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Lindsay J, Owens TW, Mularczyk EJ, Warwood S, Foster F, Streuli CH, Brennan K, Gilmore AP. Phosphorylation of the proapoptotic BH3-only protein bid primes mitochondria for apoptosis during mitotic arrest. Cell Rep. 2014;7:661–671. doi: 10.1016/j.celrep.2014.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]