

Abstract
Natural killer (NK) cells are involved in immune responses against tumors and microbes. NK-cell activation is regulated by intrinsic and extrinsic mechanisms that ensure NK tolerance and efficacy. Here, we show that the cytoplasmic signaling molecules Dok1 and Dok2 are tyrosine phosphorylated upon NK-cell activation. Overexpression of Dok proteins in human NK cells reduces cell activation induced by NK-cell-activating receptors. Dok1 and Dok2 gene ablation in mice induces an NK-cell maturation defect and leads to increased IFN-γ production induced by activating receptors. Taken together, these results reveal that Dok1 and Dok2 proteins are involved in an intrinsic negative feedback loop downstream of NK-cell-activating receptors in mouse and human.
Keywords: adaptor proteins, cell signaling, cytokine production, Dok molecules, natural killer cells
Introduction
Natural killer (NK) cells are innate lymphocytes that participate in the first line of defense against tumors or microbe-infected cells. Targeting NK-cell receptors represents an attractive therapeutic strategy for the treatment of cancers (Ljunggren & Malmberg, 2007; Vey et al, 2012; Zitvogel et al, 2008). Understanding the mechanisms underlying NK-cell maturation and function may improve the design of clinical trials (Vivier et al, 2012).
Engagement of activating and inhibitory NK-cell receptors triggers intracellular signals that regulate NK-cell cytokine production and cytotoxicity (Long et al, 2013; Vivier et al, 2004). A first level of regulation of these signals occurs via the engagement of protein tyrosine kinases (PTK) downstream of activating NK receptors and protein tyrosine phosphatases (PTP) downstream of inhibitory receptors. This control of cytoplasmic protein tyrosine phosphorylation of key signaling elements such as Vav-1 is critical to transduce functional signals in NK cells. More recently, non-enzymatic adaptor proteins that are PTK substrates, such as Crk, have also been involved in the regulation of both activation and inhibition of NK cells (Liu et al, 2012). Crk tyrosine phosphorylation induced by inhibitory receptors shuts down the activation signals. Among the negative adaptor proteins known to be expressed in leukocytes, the group of Dok (downstream of tyrosine kinase) proteins, Dok1, Dok2, and Dok3, is involved in T- and B-cell signaling (Mashima et al, 2009). Here, we report that DOK1 and DOK2 genes are expressed in both human and mouse NK cells. During T-cell activation, Dok1 and Dok2 proteins are tyrosine phosphorylated (Dong et al, 2006; Nemorin et al, 2001; Nunes et al, 1996). Dok1 and Dok2 overexpression in T cells decreases IL-2 production in activated cells (Gerard et al, 2004; Guittard et al, 2009; Nemorin et al, 2001), and loss or reduction of Dok1 and Dok2 expression in primary T cells enhances TCR-mediated functions and signaling (Dong et al, 2006; Yasuda et al, 2007). While most studies show that Dok1 and Dok2 act as negative regulators in leukocytes (Mashima et al, 2009), there is also evidence that these signaling molecules are involved in positive regulation of leukocyte signaling (Inoue et al, 2007; Lee et al, 2012).
Here, we provide evidence that Dok1 and Dok2 are tyrosine phosphorylated upon engagement of activating receptors in NK cells. Dok1 and Dok2 act as negative feedback regulators of NK activation signals in human NK cells. In addition, NK-cell development is impaired in Dok1-/Dok2-deficient (DKO) mice. Remarkably, experiments in these mice reveal that Dok1 and Dok2 downregulate NK-cell activation induced by NK-cell-activating receptors, but upregulate cell activation induced by IL-12 and IL-18. This differential impact on NK-cell-activating pathways provides new insights into regulation of NK-cell effector function.
Results
Dok1 and Dok2 proteins are substrates of protein tyrosine kinases in NK cells
Dok family members are involved in immune cell signaling; however, their expression in NK cells has not been investigated (Mashima et al, 2009). DOK1 and especially DOK2 transcripts are expressed in both human and mouse NK cells. Other DOK genes appear not to be expressed in NK cells (Supplementary Fig S1). Dok1 and Dok2 can be detected in primary NK cells and human NK-cell lines by immunoblot (Fig1A). To test whether Dok1/2 are PTK substrates in NK cells, the human NK-cell lines KHYG-1 and NKL were stimulated with antibodies against the NKp30, NKG2D, or 2B4 activating NK-cell-surface receptors (Fig1B, Supplementary Fig S2), then Dok immunoprecipitates were revealed by anti-phosphotyrosine immunoblots. Dok1 was tyrosine phosphorylated upon NKp30, NKG2D, or 2B4 triggering, but not following cross-linking of the CD94/NKG2A inhibitory receptor (Fig1B, Supplementary Fig S2). As for Dok1, Dok2 tyrosine phosphorylation was also detected in KHYG-1 cells (data not shown). The level of NKp30-induced Dok1 tyrosine phosphorylation decreased upon co-engagement of NKp30 and CD94/NKG2A (Fig1B), suggesting that DOK1/2 are substrates of the SHP-1/2 protein tyrosine phosphatases reported to be associated with the CD94/NKG2A inhibitory receptor signaling (Le Drean et al, 1998).
Figure 1. Dok1 and Dok2 are substrates of protein tyrosine kinases upon ITAM receptors triggering in NK cells.
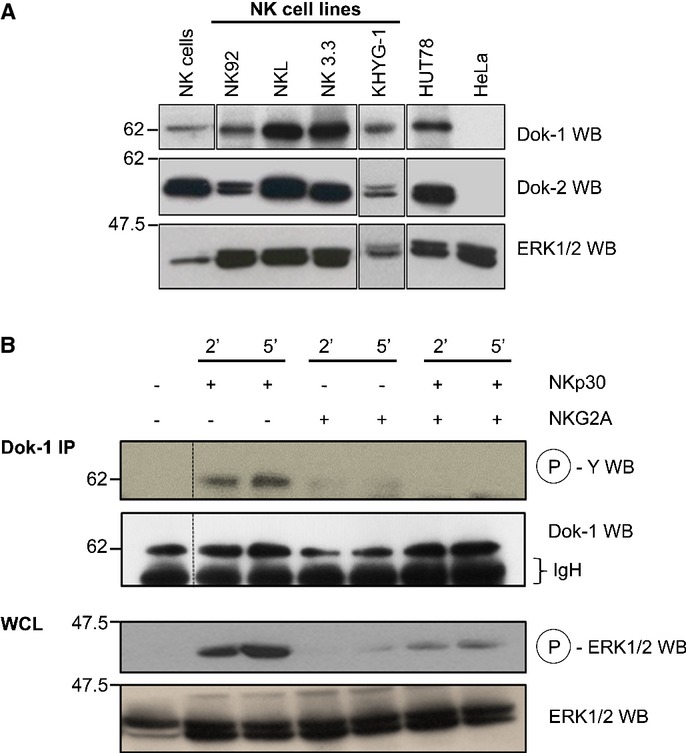
- Whole-cell lysates (WCL) from primary human NK cells and NK lines were separated by SDS–PAGE and immunoblotted for Dok1 and Dok2 proteins. Protein bands identified at molecular weight of 62 and 56 kDa correspond to Dok1 and Dok2 proteins, respectively. WCL from primary NK cells and KHYG-1 cell line were treated separately of the other WCLs. Human tumor cell lines, HUT-78 (T-cell lymphoma) and HeLa (cervical cancer), known to express or not Dok1/Dok2 proteins were included as positive and negative controls, respectively. This experiment is representative of three independent experiments.
- KHYG-1 cells, IL-2-starved for at least 18 h, were incubated with either anti-NKp30 or anti-NKG2A Abs (alone or in combination) and then stimulated by cross-linking with goat anti-mouse F(ab')2 at 37°C for indicated times. After stimulation, NKL cells were lysed and lysates were immunoprecipitated with an anti-Dok1 Ab, resolved on SDS–PAGE and immunoblotted for anti-phosphotyrosine mAb (clone 4G10). A fraction from WCL was immunoblotted for phospho-ERK1/2 and then reprobed for total ERK1/2. This experiment is representative of two independent experiments.
Dok proteins act as negative feedback regulators of activating NK-cell receptor signaling
In T cells, Dok1 and Dok2 proteins are mostly involved in negative regulation of TCR-induced T-cell activation (Acuto et al, 2008; Gerard et al, 2004). By recruiting enzymes transducing negative signals such as Ras GTPase-activating protein (RasGAP) and SH2 (Src homology 2)-containing inositol phosphatase-1 (SHIP1), Dok1 and Dok2 inhibit Ras/ERK and PI3K/Akt signaling pathways (Acuto et al, 2008; Mashima et al, 2009). To investigate the role of these signaling molecules in NK-cell signaling, we transfected the human NK-cell line KHYG-1 with GFP-tagged Dok2 either full-length or deleted of its pleckstrin homology (PH) domain (ΔPH-Dok2, a loss-of-function mutant of Dok2) (Guittard et al, 2009). Following NKp30 triggering, tyrosine phosphorylation of full-length Dok2 but not of ΔPH-Dok2 was detected (Fig2A). The level of NKp30-induced ERK or AKT phosphorylation was reduced when full-length Dok2 was overexpressed. This inhibitory effect was not detected with the Dok2 loss-of-function version ΔPH-Dok2. The results show that Dok2 overexpression in NK cells inhibits cell signaling induced by NKp30.
Figure 2. Dok1 and Dok2 negatively regulate the NK-cell signaling and function triggered by ITAM receptors.
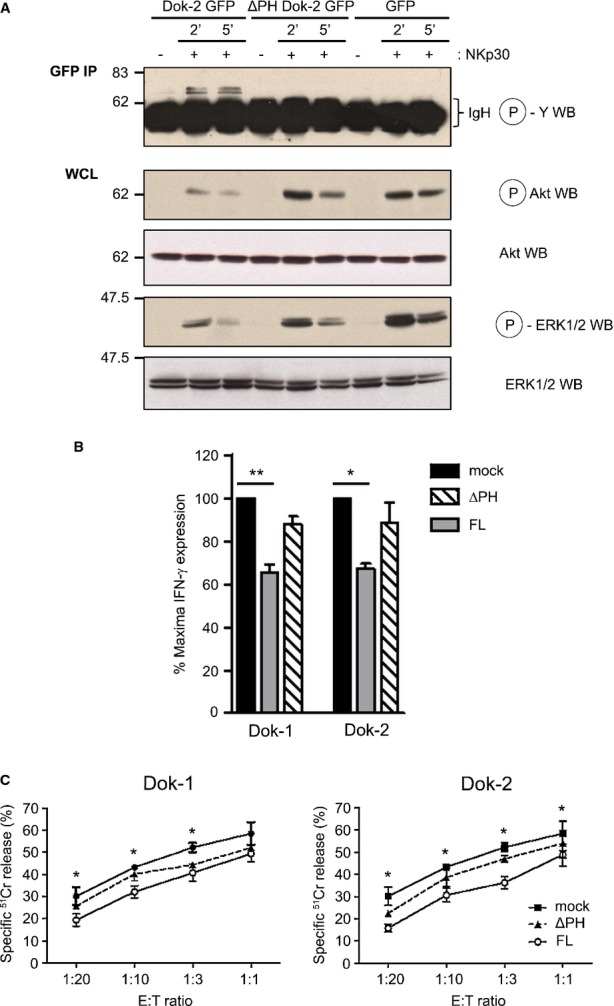
- A KHYG-1 cells were transfected by nucleofection with plasmids encoding for GFP-tagged full-length (FL) Dok2, its mutant lacking the PH domain, ΔPHDok2-GFP, or GFP alone. KHYG-1 cells were then stimulated by Ab-mediated cross-linking using a mouse anti-NKp30 Ab and a goat anti-mouse F(ab')2 Ab at 37°C for indicated times. Finally, KHYG-1 cells were lysed and lysates were immunoprecipitated using an anti-GFP Ab followed by SDS–PAGE and anti-phosphotyrosine immunoblot allowing the identification of phospho-Dok2. In parallel, WCL were separated by SDS–PAGE and subsequently immunoblotted for phosphoSer473-Akt and phospho-ERK1/2. The blots were reprobed for total Akt and ERK1/2. This panel shows representative blots of at least three independent experiments.
- B, C KHYG-1 cells expressing GFP-tagged Dok1 or Dok2 full-length (FL) proteins or lacking the PH domain (ΔPH) were stimulated with plate-bound anti-NKp30 mAb during 4-5 h and then stained for IFN-γ production (B) or were incubated with K562 target cells at different E:T ratios to analyze NK-cell cytotoxicity activity in a 4-h 51Cr release assay (C). Significant differences are detected between cells transfected with the empty vector (mock) and plasmid encoding for Dok proteins (FL); **P < 0.001; *P < 0.05.
We then investigated the role of Dok1 and Dok2 in NK-cell effector function. KHYG-1 cells transfected with full-length or PH-deleted Dok1/Dok2 were stimulated with anti-NKp30 Ab for 4–5 h, and their production of IFN-γ was measured by flow cytometry (Fig2B). Dok1 or Dok2 overexpression decreased NKp30-induced IFN-γ production. As a control, both ΔPH-Dok1 and ΔPH-Dok2 loss-of-function mutants did not alter IFN-γ production. Next, we measured the cytolytic activity of KHYG-1 transfected cells against K562 human tumor cells using a standard 4 h 51Cr release assay (Fig2C). Both for Dok1 (left panel) and Dok2 (right panel), the full-length form was more efficient than the ΔPH-Dok loss-of-function mutants to reduce NK-cell cytotoxicity. Altogether, these results demonstrate that Dok1 and Dok2 signaling molecules are negative regulators of NK-cell function in human cells.
Dok1 and Dok2 function in NK-cell maturation
Dok1 and Dok2 genes are expressed in mouse NK cells (Supplementary Fig S1B). As Dok1 and Dok2 have overlapping functions and single Dok1- or Dok2-deficient mice did not show obvious phenotypes (Mashima et al, 2009), we thus used mice deficient for both Dok1 and Dok2 (DKO) mice, to investigate the role of Dok1 and Dok2 in NK cells. The absolute and relative number of NK cells in several organs such as spleen, lymph nodes, blood, and liver was decreased in DKO mice as compared to WT mice (Fig3A–C). The percentage of NK cells was however normal in the BM of DKO mice (Fig3C). Heterozygous Dok1+/−Dok2+/− mice showed an intermediate phenotype, suggesting a dosage-dependent effect of DOK proteins. These results show that Dok1-/Dok2-deficient mice display reduced numbers of peripheral NK cells.
Figure 3. Reduced numbers of peripheral NK cells in Dok1-/Dok2-deficient mice.
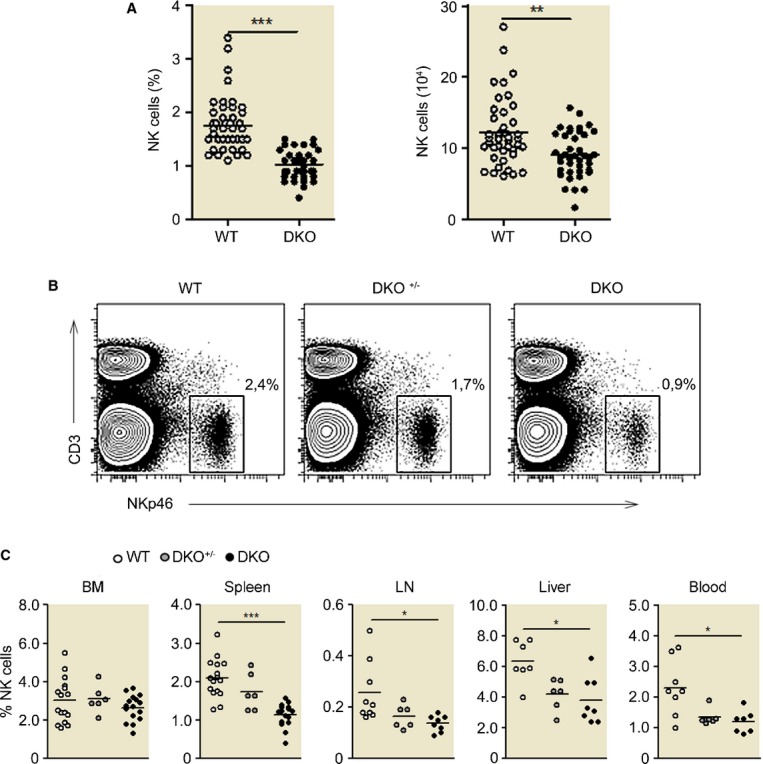
- A Percentage and absolute numbers of CD3− NKp46+ NK cells in spleen from WT and DKO 129/Sv mice. Each plot represents the data obtained from 1 mouse. ***P < 0.0001; **P < 0.01.
- B, C The percentage of CD3− NKp46+ NK cells resident in different organs has been analyzed from the three types of mice (WT, DKO+/−,and DKO). (B) Gating is shown for the spleen. (C) Histograms corresponding to the NK-cell frequency for the indicated organs ± SD (n = 9–20 mice, depending on the organ).
NK-cell development mainly occurs in the BM (Di Santo & Vosshenrich, 2006). Precursors committed to the NK-cell lineage express the γ-subunit of IL-2/IL-15 receptor, CD122, and lack other lineage markers. Subsequently, these precursors reach an immature NK-cell phenotype, characterized by the sequential acquisition of NK receptor expression at the cell surface, such as NK1.1, NKp46, CD94-NKG2, and Ly49. NK cells then upregulate the CD11b β2 integrin, as well as CD43 and KLRG-1, cell surface molecules that define the mature NK-cell phenotype (Kim et al, 2002; Narni-Mancinelli et al, 2011). Monitoring CD11b and CD27 expression on NK-cell surface allows the classification of NK-cell maturation in three sequential stages, namely CD27highCD11blow (immature), CD27highCD11bhigh (semi-mature), and CD27lowCD11bhigh (mature) (Chiossone et al, 2009; Hayakawa & Smyth, 2006). We studied the impact of the absence of Dok1 and Dok2 on the maturation status of NK cells in various lymphoid compartments (Fig4A and B). A significant accumulation of immature CD27highCD11blow NK cells and a corresponding reduction of mature CD27lowCD11bhigh cells were observed in the lymphoid organs of DKO mice as compared with WT mice. Moreover, we measured the expression of the terminal differentiation markers CD43 and KLRG-1 on CD27lowCD11bhigh NK cells (Fig4C). A block in the NK transition from CD27highCD11bhighKLRG-1low toward the CD27lowCD11bhighKLRG-1high stage was observed in the BM or spleen from DKO versus WT mice (Fig4C, left panel). Similar results were obtained using CD43 as a marker of terminal NK-cell differentiation (Fig4C, right panel). Again, heterozygous mice displayed an intermediate phenotype. Altogether, these results demonstrate a role of Dok1/Dok2 in the maturation of NK cells.
Figure 4. Dok1-/Dok2-deficient NK cells show a developmental blockade in the last stages.
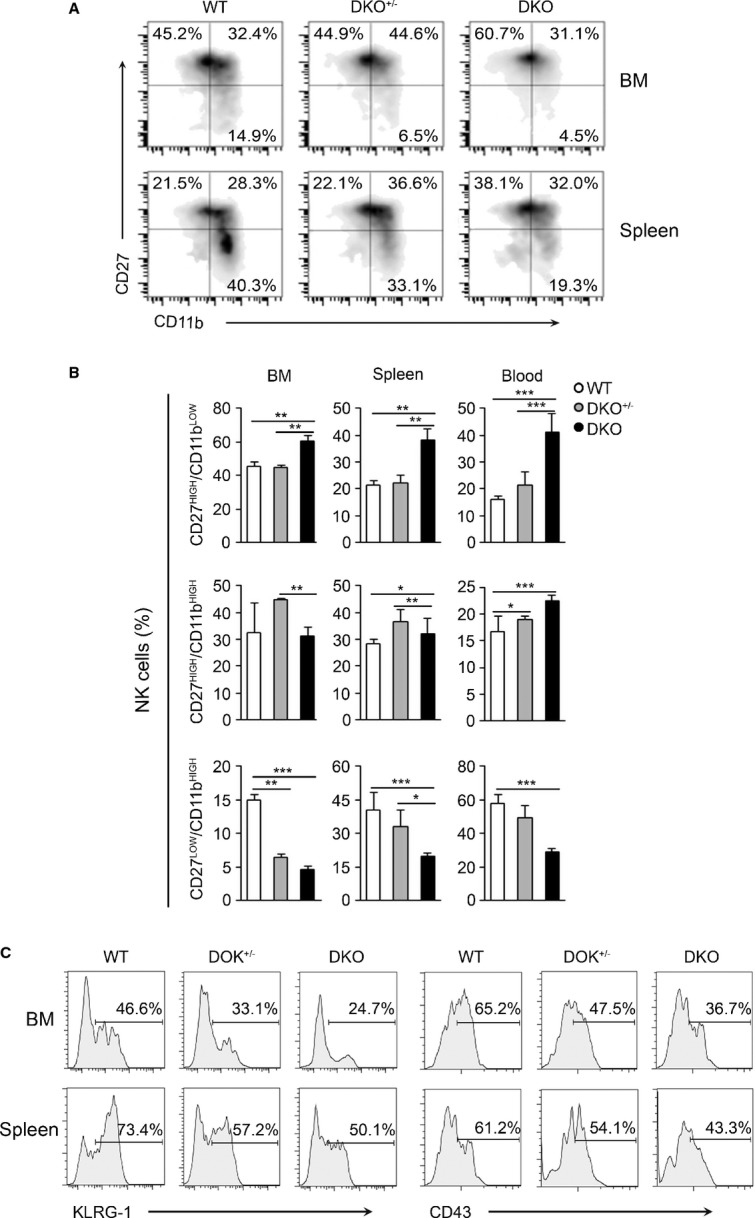
- Flow cytometry analysis of bone marrow (BM)-resident and splenic NK-cell subsets according to CD27 and CD11b staining as: CD27High/CD11bLow (immature); CD27High/CD11bHigh (semi-immature); and CD27Low/CD11bHigh (mature) in WT, DKO+/−. and DKO mice.
- The numbers in the panels represent the frequency ± SD of each NK subset of the indicated organs from WT, DKO+/−, and DKO mice (n = 9–20). ***P < 0.0001; **P < 0.01, and *P < 0.05 (one-way ANOVA test).
- The histograms shown correspond to the expression of CD27 and CD43 markers in gated CD11bHigh NK-cell population from BM and spleen of WT, DKO+/−, and DKO mice. This experiment is representative of four independent experiments.
Dok1-/Dok2-deficient NK cells present an intrinsic developmental defect
The impaired NK-cell development observed in Dok1-/Dok2-deficient mice could be due to a role of Dok proteins in NK cells or in their environment. To discriminate between these possibilities, we generated mixed BM chimeric mice by reconstituting lethally irradiated C57BL/6 (CD45.1+) mice with a 1:1 mix of BM from C57BL/6 CD45.1+ and 129/Sv CD45.2+ mice or C57BL/6 CD45.1+ and DKO (CD45.2+) mice. Eight to ten weeks after reconstitution, we measured the percentages of NK cells within CD45.2+ cells and their maturation status in each BM chimera type. The percentage of NK cells within CD45.2 cells was significantly reduced in C57BL/6:DKO chimera mice as compared to C57BL/6:129/Sv chimera mice (Fig5A). This defect was accompanied by a significant reduction of the mature CD27low CD11bhigh NK-cell subset and thus an increased frequency of the CD27high CD11blow cell subset through all examined organs (Fig5B). These observations indicate that the impairment of NK development in Dok1-/Dok2-deficient mice is due to an intrinsic NK-cell defect.
Figure 5. The maturation defect of Dok1-/Dok2-deficient NK cells is a cell-intrinsic defect and is associated with increased apoptosis.
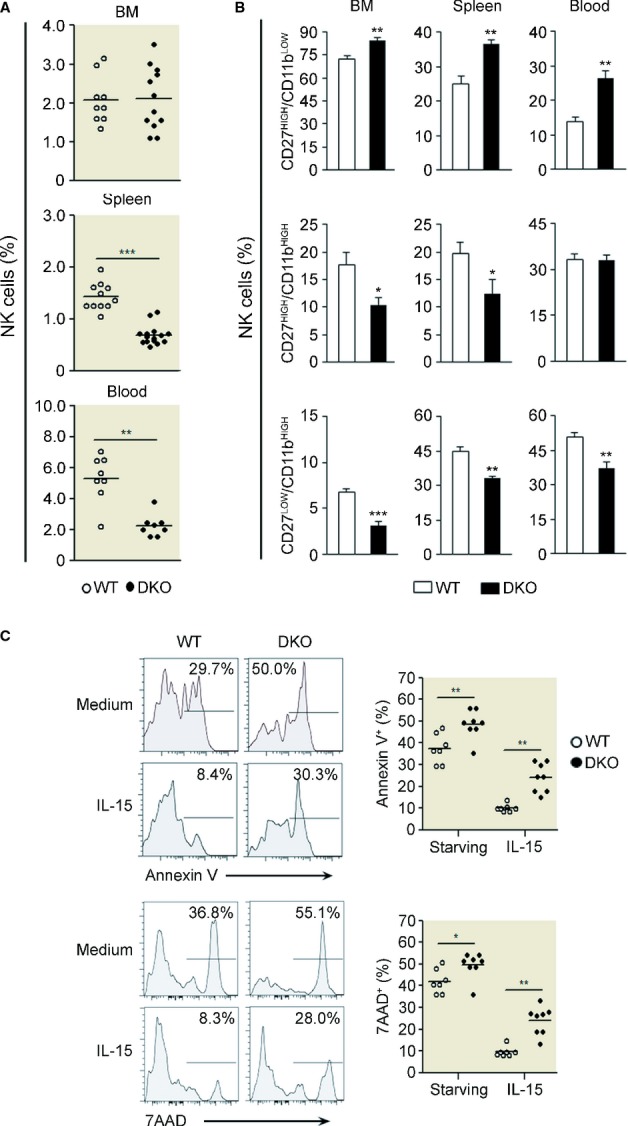
- A, B WT (white circles/bars) or DKO (black circles/bars) 129/Sv (CD45.2+) cells from the bone marrow were isolated and mixed at an equal ratio with B6 (CD45.1+) cells from the bone marrow before transfer into irradiated B6 (CD45.1+) recipients. Spleen cells from chimeric mice were isolated and analyzed 8–10 weeks after reconstitution. NK-cell frequency within CD45.2 cells (A) and NK-cell subset distribution according to CD27 and CD11b staining in the indicated organs (B) are shown. The data are the results of a pool of 3–4 independent experiments and are presented as mean ± SD (n = 4–12, depending on the organ). ***P < 0.0001; **P < 0.01, and *P < 0.05 (Mann–Whitney test).
- C Splenocytes from WT or DKO mice are cultured in the presence or in absence of IL-15 (10 ng/ml) for at least 18 h, and cells were stained for 7-AAD and Annexin V. Histograms corresponding to this staining in gated CD11bHigh NK-cell population from splenocytes of WT and DKO mice are shown. This experiment is representative of three independent experiments.
The reduced frequency of NK cells and mostly of mature NK cells in DKO mice could be accounted for by different cellular mechanisms, such as increased apoptosis. To test this hypothesis, we analyzed NK cell death ex vivo in overnight culture of splenocytes from WT and DKO mice gating on CD11bhigh NK-cell subset (Fig5C). DKO mice displayed higher levels of apoptotic and dead CD11bhigh NK cells as compared to WT mice according to the Annexin V and 7-AAD stainings. Moreover, overnight culture with anti-apoptotic IL-15 cytokine weakly rescued DKO CD11bhigh NK cells from cell death (Fig5C). Altogether, these results suggest that the reduced frequency of mature NK cells could be due to a high rate of cell apoptosis in this subset.
Loss of Dok1 and Dok2 induces the upregulation of IFN-γ production downstream of NK receptor stimulation
We then tested the role of Dok1/2 in mouse NK-cell effector function. Resting or in vivo poly(I:C)-primed NK cells were stimulated in vitro using mAb-mediated cross-linking of activation receptors or using IL-12 alone or in combination with IL-2 or IL-18. A higher proportion of DKO CD11bhigh NK cells produced IFN-γ upon Ly49D receptor cross-linking as compared to WT CD11bhigh NK cells. Similarly, incubation with YAC-1 tumor cells and cytokine stimulation (IL-12 and IL-12/IL-2) induced a higher IFN-γ response in DKO NK cells versus WT NK cells. In contrast, upon stimulation with IL-12 plus IL-18, a strong synergistic stimulus for IFN-γ production, DKO CD11bhigh NK cells produced less IFN-γ as compared to WT NK cells (Fig6B, right panel; Supplementary Fig S3). In vivo poly(I:C) priming significantly increased NK responsiveness in both groups, but did not change the differences detected between DKO and WT CD11bhigh NK cells (Fig6B). These data indicate that Dok1/Dok2 proteins inhibit IFN-γ production induced by NK-cell-activating receptors, but increase IFN-γ production induced by IL-12 and IL-18 receptors.
Figure 6. IFN-γ production via activating receptor stimulation is increased in Dok1-/Dok2-deficient NK cells.

- A, B Splenocytes from DKO or WT mice pretreated with poly(I:C) or with a PBS control were incubated on antibody-coated plates using anti-NKp46 (10 μg/ml) and anti-Ly49D (5 μg/ml) antibodies or with IL-12 (20 ng/ml) or a mixture of IL-12 + IL-2 (3,000 U/ml) or IL-12 + IL-18 (5 ng/ml) or YAC-1 tumor cells (E:T ratio of 2:1) during 4–5 h. At the end of incubation, the splenocytes were stained for CD3, NKp46 or CD49b (DX5), and CD11b and then analyzed by flow cytometry. IFN-γ expression was determined by intracellular staining. (A) Representative flow cytometry dot plots in WT or DKO CD11b+ NK cells incubated with anti-activating receptors antibodies are shown. (B) Histograms represent the WT or DKO NK-cell frequency ± SEM corresponding to different stimulation conditions. The data are the results of a pool of 4–5 independent experiments (n = 10–26). ***P < 0.0001; **P < 0.01, and *P < 0.05.
- C Splenocytes were stimulated with plate-bound anti-NKp46 mAb at the indicated concentrations during 4–5 h. IFN-γ NK production was tested by flow cytometry gating on CD11b+ NK cells (CD3− NKp46+) from WT (white squares, dotted line) and DKO (black squares) mice. Representative data from three independent experiments (n = 7–8 mice each genotype/ experiment; mean ± SEM).
- D Flow cytometry of ERK and Akt phosphorylation (Phosphoflow analysis). Splenocytes from WT (white squares, dotted line) or DKO (black squares) mice were incubated with anti-NKp46 mAb at the indicated concentrations and stimulated with cross-linking Abs (goat anti-mouse 15 μg/ml) at 37°C for 3 min. Splenocytes were cell surface-stained for CD3, NKp46, CD49b (DX5), and CD11b and then intracellularly stained with phosphoSer473-Akt and phospho-ERK mAbs. The results shown represent mean Mean Fluorescence Intensity (MFI) ± SEM gating on the CD11b+ NK-cell population (Supplementary Fig S3). Representative data from two independent experiments (n = 6–7 mice each genotype/experiment). **P < 0.01 and *P < 0.05 (Mann–Whitney test).
Upon NKp46 triggering with saturating mAb concentrations, a similar IFN-γ expression was detected in WT and DKO NK cells (Fig6A and B, right panel). But, with lower concentrations of the NKp46 agonistic mAb (2.5 μg/ml instead of 10 μg/ml), a difference in IFN-γ production in CD11bhigh NK cells was observed between WT and DKO mice (Fig6C). At low doses of stimuli, IFN-γ production was higher in CD11bhigh DKO NK cells than in CD11bhigh WT NK cells. Similar observations were made with a Ly49D mAb titration (Supplementary Fig S5A). Thus, Dok1 and Dok2 proteins are negative regulators of NK-cell response downstream of NK-cell-activating receptors.
Dok1 and Dok2 proteins are involved in the negative regulation of the Ras/ERK-1/2 and PI3K/AKT pathways (Mashima et al, 2009, Fig2A). To assess whether Dok1 and Dok2 fulfill the same function in mouse NK cells, we performed phosphoflow analysis for two phosphoproteins, AKT and ERK-1/2 on CD11bhigh NK cells from WT and DKO mice following engagement of NKp46 (Fig6D, Supplementary Fig S4). The engagement of NKp46 at low doses of cross-linking mAb triggered higher AKT and ERK phosphorylation levels in DKO than in WT NK cells (Fig6D). However, when doses of cross-linking mAbs reached saturation levels, the AKT and ERK phosphorylation levels were similar to those detected in WT CD11bhigh NK cells. Similar results were obtained with anti-Ly49D mAb stimulation (Supplementary Fig S5B). Hence, Dok1/Dok2 adaptors are able to negatively regulate NK-cell-activating signaling pathways, leading in turn to a decrease in IFN-γ secretion.
Control of MCMV infection in Dok1-/Dok2-deficient mice
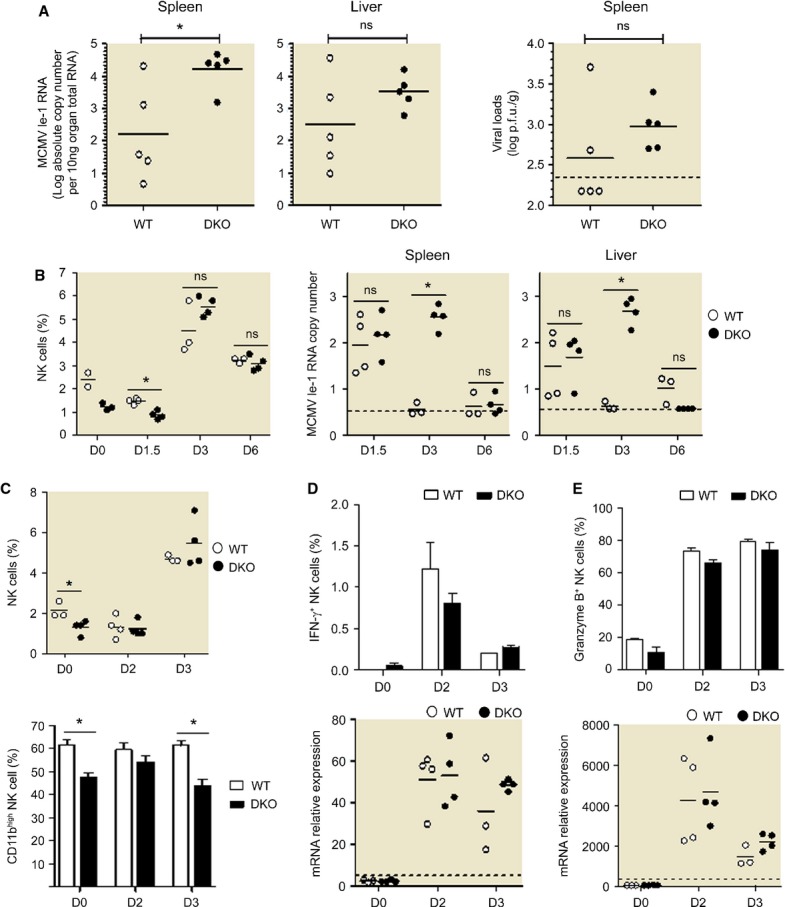
NK cells are involved in the early control of viral infections induced by the mouse cytomegalovirus (MCMV), both through IFN-γ production and through killing of infected cells (Loh et al, 2005), provided that the combination of the mouse and virus strains used allows NK-cell recognition of, activation by, and killing of infected cells (Miletic et al, 2013). In C57BL/6 mice, NK cell-mediated resistance against MCMV infection is naturally allowed by the interaction between the activating NK receptor Ly49H and its virally encoded ligand m157 (Arase et al, 2002; Smith et al, 2002). In 129 strains infected by WT viruses, NK cells are activated but fail to control viral replication due to the absence on infected cells of a proper ligand able to trigger NK-cell-activating receptors. However, this situation can be overcome by using a virus strain (Δm152Rae1γ MCMV) engineered to promote a strong expression of NKG2D ligands on the surface of infected cells (Slavuljica et al, 2010), enabling the study of in vivo NK-cell reactivity during a viral infection in our 129/Sv background DKO mice. We inoculated DKO and WT 129/Sv mice with Δm152Rae1γ MCMV and measured viral titers in the spleen and liver, and NK-cell activation in the spleen at different time points after infection. MCMV replication was identical at d1.5 and d2 post-infection in both mouse strains (partially shown in Fig7B). However, at d3 post-infection, MCMV titers were still high in DKO mice, while WT animals had already controlled the infection (Fig7A and B). The plaque assay also shows a trend toward lower viral loads in the spleen of DKO mice at day 3 post-infection although it did not reach significance. This enhanced susceptibility of DKO mice to MCMV infection was only transient, since the mice had achieved complete control of MCMV replication by day 6 under our experimental conditions (Fig7B). The proportion of mature, CD11b+, NK cells was lower in the spleen of DKO mice at all time points examined (Fig7C and not shown). However, no other difference in NK-cell activation could be detected between DKO and WT animals at any of the time points tested, in terms of IFN-γ (Fig7D) and granzyme B expression (Fig7E), proliferation as measured by Ki-67 intracellular staining, apoptosis as measured by 7-AAD staining, and NKG2D expression (data not shown). Of note, no deficit in the production of IFN-β and in the induction of its downstream target gene Mx1 was observed in MCMV-infected DKO mice, ruling out a deficit of myeloid or infected cells in the induction of the antiviral type I interferons responses in these animals (Supplementary Fig S6). Although the NK-cell numbers are strongly decreased in the DKO mice under steady-state conditions, NK cells accumulate in the spleen upon MCMV infection in both WT and DKO animals to reach high levels similar between the two mouse strains between days 3 and 6 which may correlate with the delayed but ultimately similar ability of DKO mice to control viral replication. Thus, these results show that Dok1/Dok2 proteins are necessary to promote early control of MCMV replication, potentially in part by promoting the constitution of a pool of mature CD11b+ NK cells able to more rapidly control viral replication.
Figure 7. Dok1/Dok2 proteins are required to early control of MCMV infection#.
- Viral loads were tested in spleen and liver of WT (white circle) or DKO (black circle) 129/Sv mice 3 days after infection with 2 × 105 plaque-forming units (p.f.u.) of Δm152-Rae1γ MCMV inoculated intraperitoneally. Data are representative of four independent experiments (n = 5 mice per group). Horizontal line indicates the mean of each group. *P < 0.05 (Mann–Whitney test). Viral loads were assessed by qRT-PCR (left panels) or plaque-forming assays (right panel).
- Percentage of NK cells in the lymphocyte population were measured in spleen at days 0, 1.5, 3, and 6 post-infection (left panel). Viral loads were assessed by qRT-PCR from spleen and liver as log absolute MCMV Ie-1 RNA copy number/10 ng organ total RNA (right panels). *P < 0.05 (Mann–Whitney test).
- Percentage of NK cells in the lymphocyte population were measured in spleen at days 0, 2 and 3 post-infection (top panel), and the proportion of CD11b+ cells were calculated from the NK subsets in spleen before (D0) and 2 or 3 days (D2, D3) post-infection (bottom panel).
- From the experiment shown in (C) percentage of IFN-γ-producing NK cells were measured from spleen of WT (white bars) or DKO (black bars) mice (top panel), and IFN-γ production was evaluated by qRT-PCR from spleen of WT (white circles) or DKO (black circles) mice (bottom panel) before (D0) and 2 or 3 days (D2, D3) post-infection.
- From the experiment shown in (C), percentage of granzyme B-expressing NK cells were measured from spleen of WT (white bars) or DKO (black bars) mice (top panel), and granzyme B production was evaluated by qRT-PCR from spleen of WT (white circles) or DKO (black circles) mice (bottom panel) before (D0) and 2 or 3 days (D2, D3) post-infection.
Correction added on 27 June 2014, after first online publication. The correct data for liver in panel B are now shown replacing the duplicated spleen data that were inadvertently previously shown. In the legend, all indications [(left panel)] and [(right panel)] have been corrected to reflect the Figure arrangement.
Discussion
In the present study, we identified two members of the Dok adaptor proteins, Dok1 and Dok2, as substrates of protein tyrosine kinases upon engagement of NK-cell-activating receptors. To determine the role of these proteins in NK cells, experiments were performed both in human NK-cell lines by overexpressing wild-type or transdominant negative forms of Dok1 or Dok2, and in Dok1/Dok2 DKO mice. Dok1 and Dok2 negatively regulate NK-cell activation induced by the engagement of NK-cell-activating receptors in both mice and humans. These results are consistent with those described in other immune cell types, such as T cells (Acuto et al, 2008; Mashima et al, 2009). Dok proteins thus act as negative feedback loops downstream of NK-cell-activating receptors. Therefore, in addition to the regulation of activating NK-cell receptors by their co-engagement with inhibitory ITIM-bearing receptors (Long et al, 2013; Vivier et al, 2004), we described here another negative checkpoint that controls NK-cell-activating receptor signaling as an auto-regulatory loop.
Combined deficiencies in both Dok1 and Dok2 impair NK-cell development in vivo. There are less mature NK cells in the lymphoid organs of DKO mice than in WT mice, and as demonstrated by data obtained in mixed BM chimeras experiments, this defect is intrinsic to the NK-cell compartment. Furthermore, DKO mice display higher counts of dying mature NK cells as compared to WT mice, suggesting that the Dok proteins regulate apoptosis during NK-cell maturation.
Besides its negative regulatory function, Dok1 has been previously reported to act as a positive regulator of IL-4 signaling in T cells via unknown molecular mechanisms (Inoue et al, 2007). Consistent with these data, we report here that the loss of Dok1 and Dok2 leads to decreased IFN-γ production induced by IL-12 and IL-18 in CD11bhigh NK cells. Similarly, impairment of cytokine-mediated NK-cell stimulation has been also reported in SHIP1-deficient mice (Banh et al, 2012). Dok1 and Dok2 proteins bind to SHIP1 (Mashima et al, 2009); this signaling complex has been involved in the regulation of B- and T-cell functions (Dong et al, 2006; Yarkoni et al, 2010). In light of these observations, it will be of interest to investigate the potential role of this SHIP/Dok regulatory pathway in NK-cell signaling.
Therefore, Dok1 and Dok2 differentially regulate NK-cell activation induced by activating cell surface receptors or cytokine receptors. These differences could be due to different signaling pathways involved downstream of activating cell surface receptors and cytokine receptors (Vivier et al, 2013). To dissect the impact of Dok1 and Dok2 on NK cell-dependent immune responses in vivo, we challenged DKO mice using MCMV. The control of MCMV replication was delayed in DKO as compared to WT mice, despite the absence of overt alteration in classically studied markers of NK-cell antiviral responses in the spleen. However, the proportion of mature CD11b+ NK cells was lower in the spleen of DKO mice. Hence, it is possible that the lower number of fully mature NK cells in DKO mice contributed to the delay in the control of MCMV observed in these animals. CD11b− and CD11b+ NK cells differ in their expression of many genes and therefore in several biological processes (Chiossone et al, 2009), some of which may influence their ability to control MCMV replication. Further studies beyond the scope of the current manuscript will be required to identify the cause of the delay in the control of MCMV replication in DKO mice, including the analysis of the micro-anatomical location of NK cells in the spleen and liver of these animals as compared to WT controls.
In the absence of Dok1/2 proteins, NK-cell sensitivity increases when activating receptor triggering is suboptimal. This suggests that negative feedback loops induced by Dok proteins in NK cells are particularly important at low level of NK-cell activation. An attractive hypothesis would be that a weak stimulation of an activating NK receptor could be self-controlled, whereas a stronger stimulation would be controlled by the engagement of inhibitory NK receptors. Additionally, signals from activating receptors (such as BCR or TCR) in B and T cells are controlled by Dok proteins that could avoid response to self-antigen (Acuto et al, 2008; Yarkoni et al, 2010). Lymphocyte tolerance is also documented in NK-cell biology, where self-antigen recognition by several activating receptors could potentially participate to NK cell-mediated auto-reactivity (Orr & Lanier, 2010). It is tempting to speculate that Dok proteins could participate to these mechanisms of NK-cell tolerance by controlling the threshold of NK-cell activation.
Materials and Methods
Mice and cell lines
Dok1−/−/Dok2−/− double-knockout mice (DKO mice) were obtained by interbreeding Dok1−/− mice with Dok2−/− mice both in 129/Sv background as described previously (Niki et al, 2004). This DKO mouse strain has been backcrossed once with 129/Sv mouse strain to generate Dok1+/−/Dok2+/− heterozygous double-knockout mice (DKO+/− mice). 129/Sv and CD45.1+ C57BL/6 mouse strains were purchased from Charles River. All experiments were performed in agreement with the French Guidelines for animal handling and were approved by the Inserm ethical committee. KHYG-1 NK cells (Yagita et al, 2000) were grown in RPMI-1640 (Invitrogen) medium supplemented with 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 20% heat-inactivated FCS plus 400 U/mL IL-2 (Roche). The K562 and YAC-1 cell lines were cultured in RPMI-1640 containing 10% heat-inactivated FCS with 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Antibodies and reagents
The anti-mouse antibodies used for cytometry were purchased from BD Biosciences, eBioscience and Life Technologies, and consisted of the following: NKp46 (29A1.4), CD49b (DX5), CD11b (M1/70), CD27 (LG.3A10, LG7.F9), KLRG-1 (2F1), CD43 (S7), CD122 (TM-beta1), CD3 (145-2C11), TCRβ (H57-597), CD19 (1D3), CD107a (1D4B), IFN-γ (XMG1.2), granzyme B (GB11). For in vitro mouse NK-cell stimulation assays, we used purified NKp46 (2941.4), Ly49A/D (12A8), and Ly49D (4E5) mAbs; recombinant IL-12, IL-18, IL-15 (R&D Systems), and IL-2 (Proleukin). For human NK-cell stimulation and analysis, we used anti-NKp30 (210845) and anti-NKG2D (149810, R&D Systems); anti-2B4 (C1.7) and anti-NKG2A (Z199, Beckman Coulter); and anti-IFN-γ (42.15) mAbs. Western blots were performed using Abs from Cell Signaling Technology: ERK-1/2 (#9102), phospho-ERK-1/2 (Thr202/Thr204; #9101), AKT (#9272) and phospho-AKT (Ser473, #4058), β-tubulin (#2128) and to immunoprecipitation assays, anti-Dok1 (Ab8112, Abcam) and anti-Dok2 (H-12, Santa Cruz Technology). Poly(I:C) (InvivoGen) is used for in vivo mouse pre-activation.
Cell preparation and flow cytometry
Single-cell suspensions were prepared from spleen, inguinal lymph nodes (iLNs), liver, bone marrow (BM), and blood from DKO and wild-type mice. Cells were incubated with an anti-FcRγII/III mAb (clone 2.4G2) and normal rat serum on ice for 15 min to block the Fc receptors prior to extracellular and intracellular staining. As DKO mice are on a 129/Sv genetic background, we used the NKp46 marker to detect NK cells in the different tissues (CD3− NKp46+). Samples were analyzed on LSRII or FACSCalibur flow cytometers (BD Biosciences), and data were processed using FlowJo version 9.0 (TreeStar). Phosphoproteins in mouse NK cells were detected by phosphoflow analysis (Firaguay & Nunes, 2009). Briefly, 1.5 × 106 splenocytes were incubated with anti-NKp46 mAbs on ice for 30 min followed by incubation with goat anti-rat F(ab')2' (15 μg/ml) for 3 and 5 min at 37°C. The cells were then stained for surface markers CD3, CD122, CD49b, or NKp46 and CD11b, fixed/permeabilized with Cytofix/Cytoperm™ (BD Biosciences) and indirectly intracellular stained for specific phosphoproteins. Ex vivo cell death in mouse NK cells was analyzed by Annexin V and 7-AAD staining in accordance with manufacturer's instructions (BD Biosciences).
Plasmids and transfection
The constructs encoding GFP-tagged full-length Dok1 or Dok2 or the mutants lacking the PH domain, ΔPHDok1-GFP or ΔPHDok2-GFP were described previously (Guittard et al, 2009). For overexpression of Dok proteins, 5 × 106 KHYG-1 cells were transfected with 1.5–2 μg of plasmids by nucleofection using the Amaxa Technology (Solution T, program Y-001; Lonza Cologne AG) (Messal et al, 2011). Transfection efficacy was quantified by flow cytometry (> 65% GFP positive cells for all constructs).
Cell stimulation, immunoprecipitation, and immunoblotting
For receptor cross-linking experiments, KHYG-1 or NKL cells were starved in medium 5% FCS without IL-2 for at least 20 h. Then, NK cells were pre-incubated with NK-cell receptor agonist mAb (10 μg/ml) on ice for 30 min, washed to eliminate the Ab overage and incubated with goat anti-mouse F(ab')2 (15 μg/ml) at 37°C for the indicated times. For KHYG-1 cells expressing GFP-tagged Dok1/Dok2 proteins, the cells were starved in medium 5% FCS without IL-2 following the transfection for at least 12 h and then stimulated as above. After stimulation, cells were lysed and post-nuclear lysates were incubated either with anti-GFP mAbs coupled to protein G-Sepharose or with anti-Dok1 Abs coupled to protein A-Sepharose for 2 h at 4°C. Immunoblots were performed as previously described (Gerard et al, 2009).
In vitro human NK-cell function
KHYG-1 cells (5 × 105 cell), expressing GFP-tagged WT Dok1/Dok2 proteins, or mutants lacking the PH domains, were incubated on 96-well plates precoated with anti-NKp30 mAb (2.5 μg/ml) for 4 h at 37°C in the presence of brefeldin A (BD Biosciences). KHYG-1 cells were then harvested and stained for IFN-γ (mAb PE-Cy7 conjugate) after fixation/permeabilization (BD Biosciences). IFN-γ production was measured gating on GFP+ cells by flow cytometry.
For direct cytotoxicity assay, K562 target cells were labeled with 100 μCi of 51Cr (Amersham, Buckinghamshire, UK) for 60–90 min at 37°C, washed three times with RPMI medium and then plated at 2,000 cells per well. Effector KHYG-1 cells, overexpressing either Dok proteins or their mutants, were added at the indicated ratios to triplicate wells. After 4 h of incubation at 37°C, 50 μl supernatant of each sample was harvested and radioactivity was determined by a gamma counter. Percent-specific lysis was calculated using standard methods.
In vitro mouse NK-cell function
Splenocytes were stimulated in 96-well plates precoated with either anti-NKp46 or anti-Ly49D mAbs at indicated concentrations for 4–5 h at 37°C. Similar expression levels of NKp46 and Ly49D were detected at the surface of both WT and DKO NK cells (data not shown). For cytokine stimulation, splenocytes were incubated with IL-12 alone or in combination with IL-12 or IL-18 at indicated concentrations. Finally, the NK-cell reactivity was also assessed by incubation with YAC-1 tumor cells at ratio E:T 2:1. All stimulations were developed in the presence of monensin and brefeldin A (BD Biosciences). Following incubation, the cells were harvested and surface-stained with CD3, NKp46 or CD49b, CD19 and CD11b mAbs, and then intracellular stained for IFN-γ using Cytofix/Cytoperm™ kit (BD Biosciences). In some experiments, mice were pretreated with intraperitoneal (i.p.) injection of 150 μg poly(I:C) to induce in vivo pre-activation of NK cells at least 14 h before sacrifice.
Generation of mixed BM chimera
Femoral BM cells were collected from CD45.1+ C57BL/6 and CD45.2+ Dok1/Dok2 double-knockout (DKO) 129/Sv or wild-type (WT) 129/Sv mice and then depleted for T and B cells using rat anti-CD3, anti-CD4, anti-CD8, and anti-CD5 mAbs, followed by incubation with goat anti-rat IgG magnetic beads (QIAGEN). Chimeric mice were generated by reconstituting lethally irradiated CD45.1+ C57BL/6B6 recipient mice (500 rads × 2) with a 1:1 mixture of C57BL/6 (CD45.1+; 106 cells) and [DKO or WT 129/Sv (CD45.2+, 106 cells)] BM cells. Eight to 10 weeks after reconstitution, spleen, liver, BM, iLNs, and blood were harvested of the recipient mice for phenotyping (flow cytometry) and functional tests.
MCMV infection and viral load titration
The recombinant MCMV lacking expression of the viral m152 gene and expressing mouse NKG2D-ligand Rae-1γ was generated by BAC technology as described previously (Slavuljica et al, 2010). Mice were infected with 2 × 105 plaque-forming units (p.f.u.) per mouse by i.p. injection. Spleens and livers were collected 3 days after inoculation for viral load titration by quantitative real-time PCR as previously described (Baranek et al, 2012).
Real-time PCR experiments
RNA was reverse transcribed into cDNA with the QuantiTect reverse transcription kit (QIAGEN), and the cDNA was analyzed by real-time PCR using the SYBR Premix Ex Taq (Takara). Primers were as follows: mouse Ifnγ 5′-CAACAGCAAGGCGAAAAAGG-3′ and 5′-CCTGTGGGTTGTTGACCTCAA-3′, mouse Gzmb 5′-CCACTCTCGACCCTACATGG-3′ and 5′-GGCCCCCAAAGTGACATTTATT-3′, mouse Mx1 5′-AGACTTGCTCTTTCTGAAAAGC-3′ and 5′-GACCATAGGGGTCTTGACCAA-3′; mouse Ifnβ 5′-CAGTTTTGGAAGTTTCTGGTAA-3′ and 5′-GGTGGTCCGAGCAGAGATCTT-3′, and mouse Hprt 5′-GGCCCTCTGTGTGCTCAAG-3′ and 5′-CTGATAAAATCTACAGTCATAGGAATGGA-3′. Relative gene expression was calculated using the ΔΔCt method with Hprt as the endogenous control housekeeping gene.
Statistics
Data were analyzed by two-tailed non-parametric tests using Prism software (GraphPad Software, Inc.), unless otherwise indicated. For analysis of ≥ 3 groups, a one-way analysis of variance was used. A P-value < 0.05 was considered significant, and the degree of significance is indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Acknowledgments
The authors are thankful to the research facilities of the Centre de Recherche en Cancérologie de Marseille for technical support (animal facility, cytometry, and immunomonitoring); Patrick Gibier, Jean-Christophe Orsoni and Fabrice Gianardi for animal care; Baptiste Jaeger and Cyril Fauriat for helpful discussions; Stéphanie Morin and Emilie Coppin for their help to conduct the mouse experiments; Florence Orlanducci for cell cytotoxicity assays; Astrid Krmpotić for the preparation of Δm152Rae1γ MCMV batches. This work was supported by grants from Institut National de la Santé et de la Recherche Médicale (INSERM) and the Institut National du Cancer (INCa #PL-06026 and #2009-112 to J.A.N., #2011-155 to M.D.). D.O. and J.A.N. laboratory is supported by the Fondation pour la Recherche Médicale (Equipe FRM 2014). J.C.-G. was supported by a fellowship from the Fundayacucho-CNOUS joint program then by the Société Française d'Hématologie (SFH). E.V. laboratory is supported by the European Research Council (THINK Advanced Grant) and by institutional grants from INSERM, CNRS and Aix-Marseille University to CIML. E.V. is a scholar of the Institut Universitaire de France.
Author contributions
JC-G, TW, and MD designed the research, performed experiments, analyzed the data, and wrote the manuscript; MB performed experiments and analyzed the data; PPP and SJ provided vital reagents; DO assisted the research design; EV and JAN directed and supervised the studies, analyzed the data, and wrote the manuscript.
Conflict of interest
EV is a co-founder and shareholder in Innate Pharma. All remaining authors declare no conflicting financial interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Acuto O, Di Bartolo V, Michel F. Tailoring T-cell receptor signals by proximal negative feedback mechanisms. Nat Rev Immunol. 2008;8:699–712. doi: 10.1038/nri2397. [DOI] [PubMed] [Google Scholar]
- Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- Banh C, Miah SM, Kerr WG, Brossay L. Mouse natural killer cell development and maturation are differentially regulated by SHIP-1. Blood. 2012;120:4583–4590. doi: 10.1182/blood-2012-04-425009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranek T, Manh TP, Alexandre Y, Maqbool MA, Cabeza JZ, Tomasello E, Crozat K, Bessou G, Zucchini N, Robbins SH, Vivier E, Kalinke U, Ferrier P, Dalod M. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe. 2012;12:571–584. doi: 10.1016/j.chom.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood. 2009;113:5488–5496. doi: 10.1182/blood-2008-10-187179. [DOI] [PubMed] [Google Scholar]
- Di Santo JP, Vosshenrich CA. Bone marrow versus thymic pathways of natural killer cell development. Immunol Rev. 2006;214:35–46. doi: 10.1111/j.1600-065X.2006.00461.x. [DOI] [PubMed] [Google Scholar]
- Dong S, Corre B, Foulon E, Dufour E, Veillette A, Acuto O, Michel F. T cell receptor for antigen induces linker for activation of T cell-dependent activation of a negative signaling complex involving Dok2, SHIP-1, and Grb-2. J Exp Med. 2006;203:2509–2518. doi: 10.1084/jem.20060650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firaguay G, Nunes JA. Analysis of signaling events by dynamic phosphoflow cytometry. Sci Signal. 2009;2:pl3. doi: 10.1126/scisignal.286pl3. [DOI] [PubMed] [Google Scholar]
- Gerard A, Favre C, Garcon F, Nemorin JG, Duplay P, Pastor S, Collette Y, Olive D, Nunes JA. Functional interaction of RasGAP-binding proteins Dok1 and Dok2 with the Tec protein tyrosine kinase. Oncogene. 2004;23:1594–1598. doi: 10.1038/sj.onc.1207283. [DOI] [PubMed] [Google Scholar]
- Gerard A, Ghiotto M, Fos C, Guittard G, Compagno D, Galy A, Lemay S, Olive D, Nunes JA. Dok4 is a novel negative regulator of T cell activation. J Immunol. 2009;182:7681–7689. doi: 10.4049/jimmunol.0802203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guittard G, Gerard A, Dupuis-Coronas S, Tronchere H, Mortier E, Favre C, Olive D, Zimmermann P, Payrastre B, Nunes JA. Cutting edge: Dok1 and Dok2 adaptor molecules are regulated by phosphatidylinositol 5-phosphate production in T cells. J Immunol. 2009;182:3974–3978. doi: 10.4049/jimmunol.0804172. [DOI] [PubMed] [Google Scholar]
- Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol. 2006;176:1517–1524. doi: 10.4049/jimmunol.176.3.1517. [DOI] [PubMed] [Google Scholar]
- Inoue A, Yasuda T, Yamamoto T, Yamanashi Y. Dok1 is a positive regulator of IL-4 signalling and IgE response. J Biochem. 2007;142:257–263. doi: 10.1093/jb/mvm127. [DOI] [PubMed] [Google Scholar]
- Kim S, Iizuka K, Kang HS, Dokun A, French AR, Greco S, Yokoyama WM. In vivo developmental stages in murine natural killer cell maturation. Nat Immunol. 2002;3:523–528. doi: 10.1038/ni796. [DOI] [PubMed] [Google Scholar]
- Le Drean E, Vely F, Olcese L, Cambiaggi A, Guia S, Krystal G, Gervois N, Moretta A, Jotereau F, Vivier E. Inhibition of antigen-induced T cell response and antibody-induced NK cell cytotoxicity by NKG2A: association of NKG2A with SHP-1 and SHP-2 protein-tyrosine phosphatases. Eur J Immunol. 1998;28:264–276. doi: 10.1002/(SICI)1521-4141(199801)28:01<264::AID-IMMU264>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Lee CM, Jung ID, Noh KT, Lee JS, Park JW, Heo DR, Park JH, Chang JH, Choi IW, Kim JS, Shin YK, Park SJ, Han MK, Lee CG, Cho WK, Park YM. An essential regulatory role of downstream of kinase-1 in the ovalbumin-induced murine model of asthma. PLoS ONE. 2012;7:e34554. doi: 10.1371/journal.pone.0034554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Peterson ME, Long EO. The adaptor protein Crk controls activation and inhibition of natural killer cells. Immunity. 2012;36:600–611. doi: 10.1016/j.immuni.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljunggren HG, Malmberg KJ. Prospects for the use of NK cells in immunotherapy of human cancer. Nat Rev Immunol. 2007;7:329–339. doi: 10.1038/nri2073. [DOI] [PubMed] [Google Scholar]
- Loh J, Chu DT, O'Guin AK, Yokoyama WM, Virgin HWt. Natural killer cells utilize both perforin and gamma interferon to regulate murine cytomegalovirus infection in the spleen and liver. J Virol. 2005;79:661–667. doi: 10.1128/JVI.79.1.661-667.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashima R, Hishida Y, Tezuka T, Yamanashi Y. The roles of Dok family adapters in immunoreceptor signaling. Immunol Rev. 2009;232:273–285. doi: 10.1111/j.1600-065X.2009.00844.x. [DOI] [PubMed] [Google Scholar]
- Messal N, Mamessier E, Sylvain A, Celis-Gutierrez J, Thibult ML, Chetaille B, Firaguay G, Pastor S, Guillaume Y, Wang Q, Hirsch I, Nunes JA, Olive D. Differential role for CD277 as a co-regulator of the immune signal in T and NK cells. Eur J Immunol. 2011;41:3443–3454. doi: 10.1002/eji.201141404. [DOI] [PubMed] [Google Scholar]
- Miletic A, Krmpotic A, Jonjic S. The evolutionary arms race between NK cells and viruses: who gets the short end of the stick? Eur J Immunol. 2013;43:867–877. doi: 10.1002/eji.201243101. [DOI] [PubMed] [Google Scholar]
- Narni-Mancinelli E, Chaix J, Fenis A, Kerdiles YM, Yessaad N, Reynders A, Gregoire C, Luche H, Ugolini S, Tomasello E, Walzer T, Vivier E. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc Natl Acad Sci USA. 2011;108:18324–18329. doi: 10.1073/pnas.1112064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemorin JG, Laporte P, Berube G, Duplay P. p62dok negatively regulates CD2 signaling in Jurkat cells. J Immunol. 2001;166:4408–4415. doi: 10.4049/jimmunol.166.7.4408. [DOI] [PubMed] [Google Scholar]
- Niki M, Di Cristofano A, Zhao M, Honda H, Hirai H, Van Aelst L, Cordon-Cardo C, Pandolfi PP. Role of Dok1 and Dok2 in leukemia suppression. J Exp Med. 2004;200:1689–1695. doi: 10.1084/jem.20041306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes JA, Truneh A, Olive D, Cantrell DA. Signal transduction by CD28 costimulatory receptor on T cells. B7-1 and B7-2 regulation of tyrosine kinase adaptor molecules. J Biol Chem. 1996;271:1591–1598. doi: 10.1074/jbc.271.3.1591. [DOI] [PubMed] [Google Scholar]
- Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell. 2010;142:847–856. doi: 10.1016/j.cell.2010.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavuljica I, Busche A, Babic M, Mitrovic M, Gasparovic I, Cekinovic D, Markova Car E, Pernjak Pugel E, Cikovic A, Lisnic VJ, Britt WJ, Koszinowski U, Messerle M, Krmpotic A, Jonjic S. Recombinant mouse cytomegalovirus expressing a ligand for the NKG2D receptor is attenuated and has improved vaccine properties. J Clin Invest. 2010;120:4532–4545. doi: 10.1172/JCI43961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, Scalzo AA, Fremont DH, Yokoyama WM. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci USA. 2002;99:8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, Etienne A, Andre P, Romagne F, Benson D, Dombret H, Olive D. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood. 2012;120:4317–4323. doi: 10.1182/blood-2012-06-437558. [DOI] [PubMed] [Google Scholar]
- Vivier E, Nunes JA, Vely F. Natural killer cell signaling pathways. Science. 2004;306:1517–1519. doi: 10.1126/science.1103478. [DOI] [PubMed] [Google Scholar]
- Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12:239–252. doi: 10.1038/nri3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Ugolini S, Nunes JA. ADAPted secretion of cytokines in NK cells. Nat Immunol. 2013;14:1108–1110. doi: 10.1038/ni.2737. [DOI] [PubMed] [Google Scholar]
- Yagita M, Huang CL, Umehara H, Matsuo Y, Tabata R, Miyake M, Konaka Y, Takatsuki K. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia. 2000;14:922–930. doi: 10.1038/sj.leu.2401769. [DOI] [PubMed] [Google Scholar]
- Yarkoni Y, Getahun A, Cambier JC. Molecular underpinning of B-cell anergy. Immunol Rev. 2010;237:249–263. doi: 10.1111/j.1600-065X.2010.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Bundo K, Hino A, Honda K, Inoue A, Shirakata M, Osawa M, Tamura T, Nariuchi H, Oda H, Yamamoto T, Yamanashi Y. Dok1 and Dok2 are negative regulators of T cell receptor signaling. Int Immunol. 2007;19:487–495. doi: 10.1093/intimm/dxm015. [DOI] [PubMed] [Google Scholar]
- Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.