

Abstract
The antimitotic anti-cancer drugs, including taxol, perturb spindle dynamics, and induce prolonged, spindle checkpoint-dependent mitotic arrest in cancer cells. These cells then either undergo apoptosis triggered by the intrinsic mitochondrial pathway or exit mitosis without proper cell division in an adaptation pathway. Using a genome-wide small interfering RNA (siRNA) screen in taxol-treated HeLa cells, we systematically identify components of the mitotic apoptosis and adaptation pathways. We show that the Mad2 inhibitor p31comet actively promotes mitotic adaptation through cyclin B1 degradation and has a minor separate function in suppressing apoptosis. Conversely, the pro-apoptotic Bcl2 family member, Noxa, is a critical initiator of mitotic cell death. Unexpectedly, the upstream components of the mitochondrial apoptosis pathway and the mitochondrial fission protein Drp1 contribute to mitotic adaption. Our results reveal crosstalk between the apoptosis and adaptation pathways during mitotic arrest.
Keywords: apoptosis, mitochondria, mitosis, mitotic slippage, the spindle checkpoint
See also: LL Fava & A Villunger (September 2014)
Introduction
The antimitotic chemotherapeutic drug taxol perturbs microtubule dynamics and causes mitotic spindle defects, including reduced tension at kinetochores (Jordan & Wilson, 2004; Kavallaris, 2010). In response to unattached kinetochores or kinetochores without proper tension, the spindle checkpoint inhibits the anaphase-promoting complex/cyclosome (APC/C), a ubiquitin ligase complex for cyclin B1, thus preventing the inactivation of cyclin-dependent kinase 1 (Cdk1) and mitotic exit (Lara-Gonzalez et al, 2012; Foley & Kapoor, 2013; Jia et al, 2013). APC/C inhibition involves the sequestration of its activator Cdc20 into the mitotic checkpoint complex (MCC) by the checkpoint proteins Mad2 and BubR1 (Fang et al, 1998; Sudakin et al, 2001; Tang et al, 2001; Fang, 2002; Luo et al, 2002; Kulukian et al, 2009; Lara-Gonzalez et al, 2011; Chao et al, 2012; Han et al, 2013). During unperturbed mitosis, when all kinetochores reach proper microtubule attachment and are under tension, the Mad2 inhibitor p31comet blocks Mad2 conformational activation and promotes MCC disassembly, leading to APC/CCdc20 activation, checkpoint silencing, cyclin B1 degradation, and cell division (Habu et al, 2002; Xia et al, 2004; Mapelli et al, 2006; Mapelli & Musacchio, 2007; Reddy et al, 2007; Yang et al, 2007; Luo & Yu, 2008; Jia et al, 2011; Teichner et al, 2011; Westhorpe et al, 2011). In the presence of taxol, however, the persistent spindle damage continually activates the spindle checkpoint, resulting in prolonged mitotic arrest.
Cancer cells have two major fates following the prolonged mitotic arrest triggered by taxol (Gascoigne & Taylor, 2008, 2009; Orth et al, 2011). Cells can die in mitosis through the activation of the intrinsic mitochondrial apoptosis pathway (Li et al, 2005; Janssen et al, 2007; Kutuk & Letai, 2008, 2010; Zhou et al, 2010). This mitochondrial pathway generally involves the antagonistic interactions among multiple members of the Bcl2 family of proteins, including the anti-apoptotic members Bcl2, Bcl-xL, and Mcl1; the pro-apoptotic members Bad, Bid, Bim, and Noxa; and the effectors Bax and Bak (Chipuk et al, 2010). Mcl1 is a key anti-apoptotic factor during mitotic arrest (Harley et al, 2010; Wertz et al, 2011). Its degradation during mitosis enables activation of Bax and Bak. Activated Bax and Bak then oligomerize on the mitochondrial outer membrane and promote mitochondrial outer membrane permeabilization (MOMP) to release cytochrome c and other factors into the cytosol, which trigger caspase activation and apoptosis (Jiang & Wang, 2004; Chipuk et al, 2010). In a second, undesirable pathway, cancer cells escape from taxol-induced mitotic arrest without proper checkpoint silencing in a process termed mitotic adaptation or slippage (Brito & Rieder, 2006; Gascoigne & Taylor, 2008, 2009). Mitotic adaptation involves the basal activity of checkpoint-inhibited APC/CCdc20, which mediates gradual cyclin B1 degradation, eventual Cdk1 inactivation, and mitotic exit (Brito & Rieder, 2006; Huang et al, 2009; Varetti et al, 2011).
There is tremendous heterogeneity in the fates of cancer cells exposed to taxol (Gascoigne & Taylor, 2008; Brito & Rieder, 2009). Apoptosis during taxol-induced mitotic arrest is the predominant fate for certain cancer cell lines, whereas mitotic adaptation is the preferred pathway for others. Even different cell populations within the same cell line can experience different outcomes. The mechanism that underlies the life and death decisions of cells under mitotic arrest is not understood, but appears to involve a competitive, molecular race between the apoptosis and adaptation pathways within a given cell (Gascoigne & Taylor, 2008, 2009; Huang et al, 2009, 2010). The final fate of the cell arrested in mitosis is thought to be determined by which of two competing pathways is executed faster kinetically. How these two pathways are coordinated is not understood. In fact, it has been argued that they may be independent of each other (Huang et al, 2010).
A better molecular understanding of mitotic apoptosis and adaptation will ultimately lead to strategies to improve the efficacy of the antimitotic drugs. Here, we perform a genome-wide siRNA screen in human cells to identify molecular components of apoptosis and adaptation following taxol-mediated mitotic arrest. We show that p31comet actively promotes mitotic adaptation through cyclin B1 degradation. Furthermore, p31comet has a separate, minor function in antagonizing apoptosis. Conversely, Noxa is a key regulator of apoptosis during mitotic arrest. Surprisingly, the upstream components of the intrinsic mitochondrial apoptosis pathway contribute to mitotic adaptation, through a mechanism that likely involves the mitochondrial fission factor Drp1. Our results establish an unanticipated crosstalk between the mitotic apoptosis and adaptation pathways and uncover a mitochondrial module and p31comet as coupler and anti-coupler of these processes, respectively.
Results
Genome-wide siRNA screen identifies regulators of mitotic apoptosis and adaptation
To identify regulators of taxol-mediated mitotic arrest, apoptosis, and adaptation in human cells, we screened an arrayed siRNA library against 21,360 annotated human genes for their effects on the cellular viability in taxol. HeLa Tet-On cells were particularly sensitive to taxol-mediated killing. After several hours of mitotic arrest, a majority of these cells underwent apoptosis. We reasoned that, by screening the siRNA library for siRNAs that increased cell viability in the presence of a lethal dose of taxol, we would identify genes involved in three cellular processes: cell cycle genes whose inactivation prevents entry into mitosis, spindle checkpoint genes that are essential for taxol-mediated mitotic arrest, and apoptotic genes required for cell death following taxol-induced mitotic arrest. In the primary screen, HeLa Tet-On cells grown in 96-well plates were transfected individually with pools of the siRNA library, with each pool containing four siRNAs targeting a single human gene (Fig1A). Each transfection was performed in triplicates. The cells were then treated with taxol for 48 h. The viability of these taxol-treated cells was determined with the CellTiter-Glo luminescent assay, which measured intracellular ATP concentrations. This primary screen identified 613 hits that increased cell viability by at least two standard deviations from the population mean and 574 hits that decreased viability by at least 1.5 standard deviations (Fig1A and B; Supplementary Tables S1 and S2).
Figure 1. Genome-wide siRNA screen identifies genes regulating cellular responses to taxol.

A Flow chart of the primary and secondary screens.
B Distribution of the cell viability values of each siRNA pool in the primary screen. Pools that increased survival by at least two standard deviations (orange line) or decreased survival at least by 1.5 standard deviations (blue line) were considered positive hits. Hits included Plk1, p31comet, and anaphase-promoting complex/cyclosome subunits (shown as blue dots) and several known spindle checkpoint genes (shown as orange dots).
C–F Functional networks identified in the screen: (C) the apoptosis network; (D) the spindle checkpoint network; (E) the DNA replication licensing gene network; and (F) the spliceosome network. Viability-increasing hits that are positive in both primary and secondary screens are colored orange. Viability-increasing hits that are positive in only the primary screen are in yellow. Viability-decreasing hits are colored blue.
Several recent studies have identified Mad2 as a common target of RNAi off-target effects (Hubner et al, 2010; Westhorpe et al, 2010; Ke et al, 2011; Sigoillot et al, 2012). Consistent with these reports, initial characterization of some top viability-increasing hits identified in our primary screen revealed that about 20 of these siRNA pools reduced Mad2 protein levels through off-target effects (Supplementary Table S1). To reduce false positives due to RNAi off-target effects, we performed a secondary screen using the same viability assay with siRNA pools from Qiagen that targeted most of the 613 initial viability-increasing hits (Fig1A). The Dharmacon and Qiagen siRNA pools were independently designed and contained different sets of siRNAs. If depletion of a given gene increased cell viability in both primary and secondary screens, it would indicate that multiple siRNAs targeting this gene could produce the same phenotype, thereby limiting off-target effects. The top 38 hits in the secondary screen were listed in Supplementary Table S3.
The top 38 hits in the secondary screen along with the initial hits from the primary screen were subjected to network analysis using Ingenuity Pathway Analysis, which revealed several functional networks each containing multiple hits. These networks included: the apoptosis network (Fig1C), the spindle checkpoint network (Fig1D), the DNA replication licensing network (Fig1E), and the spliceosome network (Fig1F). Hypergeometric distribution analysis showed that the probabilities of discovering the genes within these networks by random chance were extremely low: the apoptosis network (P = 0), the spindle checkpoint network (P = 1.02 × 10−12), the DNA replication network (P = 5.62 × 10−5), and the spliceosome network (P = 1.384 × 10−5; Whitehurst et al, 2007).
The apoptosis network contained four known components of the intrinsic mitochondrial apoptosis pathway, including Bad, Noxa (PMAIP1), Bax, and cytochrome c, confirming the involvement of this pathway in mitotic cell death (Fig1C). Our screen also isolated several known components of the spindle checkpoint, including Mad2, BubR1, Mps1, and three subunits of the chromosome passenger complex (CPC): INCENP, borealin (CdcA8), and survivin (Birc5; Fig1D). The identification of known components in both the apoptosis and spindle checkpoint networks validated our screen.
A previous study had established a requirement for the APC/C activator Cdc20 in mitotic adaptation (Huang et al, 2009). Consistently, multiple subunits of APC/C, including ANAPC1, ANAPC5, ANAPC13, CDC23, and CDC26, were among the hits that decreased cell viability in taxol in our screen (Fig1D), although Cdc20 itself was not isolated. We note that false negatives in large-scale screens are quite common. Some of the Cdc20 siRNAs might have deleterious off-target effects. Taken together, these results support a role for APC/CCdc20 in preventing adaptation and thus decreasing cell viability in taxol. In addition, Plk1 (a positive regulator of mitosis) and Mad2L1BP (p31comet, an inhibitor of Mad2) were identified in the spindle checkpoint network as hits that decreased cell viability in taxol (Fig1D).
Inactivation of the DNA replication licensing and spliceosome networks caused cell cycle defects and delayed mitotic entry (data not shown), indirectly preventing mitotic arrest and cell death and increasing cell viability in the presence of taxol. For example, we identified geminin and three subunits of the CRL4Cdt2 ligase complex (Cul4B, Ddb1, and Cdt2) in our screen (Fig1E). Inactivation of these genes is expected to cause an upregulation of Cdt1 function and DNA re-replication. Indeed, depletion of Cdt2 has been shown to cause DNA re-replication, DNA damage, and DNA damage checkpoint-dependent cell cycle arrest prior to mitosis (Jin et al, 2006). Therefore, the cell viability increase in taxol after depletion of these components is the consequence of an interphase arrest and a delay in mitotic entry.
The Mad2 inhibitor p31comet is required for cyclin B1 degradation and mitotic adaptation
p31comet prevents the conformational activation of Mad2 and promotes MCC disassembly and APC/CCdc20 activation during checkpoint silencing (Habu et al, 2002; Xia et al, 2004; Mapelli et al, 2006; Reddy et al, 2007; Yang et al, 2007; Jia et al, 2011; Teichner et al, 2011; Westhorpe et al, 2011). Depletion of p31comet sensitizes cancer cells to mitotic arrest and killing by spindle poisons (Xia et al, 2004; Ma et al, 2012). p31comet is required for adaptation in HeLa cells treated with nocodazole (Varetti et al, 2011).
The identification of p31comet as viability-decreasing hit in our genome-wide siRNA screen strongly implicates it as a major regulator of mitotic adaptation in the presence of taxol. Depletion of p31comet indeed decreased the extent of mitotic adaptation in HeLa cells (see Supplementary Fig S2D). Most control HeLa cells, however, already underwent apoptosis following prolonged mitotic arrest, with few cells experiencing mitotic adaptation. The effect of p31comet depletion on mitotic adaptation in HeLa cells was predictably small.
We thus performed live-cell imaging on U2OS cells transfected with a control siRNA targeting luciferase (siLuc), si-p31comet (siP31), or siCdc20 and treated with taxol. U2OS cells transfected with siLuc primarily underwent mitotic adaptation in the presence of taxol (Fig2A–C). Consistent with previous reports (Huang et al, 2009), depletion of Cdc20 greatly lengthened the duration of mitosis and reduced the extent of mitotic adaptation (Fig2A–C). Remarkably, p31comet depletion in U2OS cells reduced mitotic adaptation as effectively as Cdc20 depletion did (Fig2C), but was much less efficient in lengthening the duration of taxol-induced mitotic arrest compared to Cdc20 depletion (Fig2A and B). These results clearly indicate that p31comet is required for mitotic adaptation in taxol. The fact that siP31 cells undergo apoptosis sooner than siCdc20 cells suggests a role of p31comet in delaying apoptosis.
Figure 2. p31comet promotes mitotic adaptation through cyclin B1 degradation.

A Sample mitotic duration curves of U2OS cells transfected with the indicated siRNAs and treated with taxol, as determined by time-lapse microscopy. t50 is defined as the time at which 50% of cells remain in mitosis. The differences between t50 of different samples are: Δt50(siCdc20–siP31-1) = 12.7 ± 6.5 h, n = 3; and Δt50(siP31-1–siLuc) = 7.7 ± 4.1 h, n = 4; where n is the number of experiments.
B Dot plot of the phenotype and mitotic duration of each cell in (A). The bar indicates the median.
C Quantification of the terminal phenotypes of the mitotic cells in (A).
D–G U2OS cells were transfected with siControl or siP31-1, synchronized by thymidine block, and released into medium containing taxol. At 15 h after thymidine release, mitotic cells were collected by shake-off, further cultured in the taxol medium for the indicated times in the absence (D, E) or presence (F, G) of 50 μM cycloheximide (CHX), stained with MPM2 or anti-cyclin B1 antibodies and propidium iodide (PI), and subjected to FACS analysis. Representative FACS graphs of the indicated samples are shown in (D) and (F). The normalized counts of cells with 4C DNA content (as a percentage of the maximum in the histogram of each time point) were plotted against the intensities of MPM2 (top panels) or cyclin B1 (bottom panels) in log scales in (E) and (G). The MPM2 and cyclin B1 intensities of mitotic cells and the adapted cells are indicated by black and red arrows.
Even with an active spindle checkpoint, Cdc20 triggers gradual degradation of cyclin B1, leading to mitotic adaptation (Huang et al, 2009). To test whether p31comet also promoted mitotic adaptation through cyclin B1 degradation, we developed a FACS-based assay to measure the levels of endogenous cyclin B1 in individual U2OS cells during a prolonged mitotic arrest (Supplementary Fig S1). At 0 h in taxol following shake-off, mitotic cells with 4C DNA contents contained high MPM2 (which recognized phospho-epitopes generated by Cdk1) and cyclin B1 levels. As cells spent more time (2.5–7.5 h) in taxol, the intensity of MPM2 signal in most cells with 4C DNA contents did not change, indicating that these cells were still in mitosis and had high Cdk1 activity (Supplementary Fig S1A and B, top panels). In contrast, the cyclin B1 levels of these mitotic cells gradually decreased by several fold (Supplementary Fig S1A and B, bottom panels). Addition of cycloheximide, a protein translation inhibitor, accelerated the drop in cyclin B1 levels and mitotic exit (Supplementary Fig S1C and D). Consistent with previous reports (Brito & Rieder, 2006; Huang et al, 2009; Mena et al, 2010), our results indicate that cyclin B1 translation during mitosis is required to maintain a mitotic arrest due to the constant degradation of cyclin B1 that occurs even when the spindle checkpoint is active. During a prolonged mitotic arrest, the rate of cyclin B1 degradation eventually exceeds that of synthesis, leading to Cdk1 inactivation and mitotic adaptation.
A similar FACS analysis was performed on U2OS cells transfected with siControl or siP31 (Fig2D–G). Consistent with the live-cell imaging results, depletion of p31comet greatly reduced mitotic adaptation in the absence or presence of cycloheximide, as evidenced by the lack of MPM2-negative 4C cells (blue boxes; Fig2D and F). Instead, the p31comet RNAi cells had a new population of cells with slightly < 4C DNA content and MPM2 levels below that of interphase cells (red boxes). These cells likely underwent cell death in mitosis, but their DNA had not been fully fragmented, although we could not be certain that this population of cells was indeed apoptotic. Furthermore, p31comet depletion blocked the progressive decrease of cyclin B1 levels during taxol-mediated mitotic arrest (Fig2E). Even when protein synthesis was inhibited by cyclohexamide, the decrease in cyclin B1 levels was largely blocked in p31comet RNAi cells (Fig2G), suggesting that depletion of p31comet prevented cyclin B1 degradation. Thus, p31comet actively promotes cyclin B1 degradation and mitotic adaptation during taxol-induced mitotic arrest.
p31comet has a Cdc20-independent role in delaying mitotic apoptosis
Depletion of Cdc20 in U2OS cells not only reduced the extent of adaptation in the presence of taxol, but also greatly lengthened mitotic duration (Fig2A–C). According to the molecular race model (Gascoigne & Taylor, 2009; Huang et al, 2010), mitotic adaptation takes less time to execute than apoptosis does in the adaptation-prone U2OS cells. Blocking mitotic adaptation by siCdc20 forces cells to stay in mitosis and allows the slower process of apoptosis to finish. Cdc20 depletion is thus expected to lengthen taxol-induced mitotic arrest in adaptation-prone cells. However, despite having similar effects in blocking mitotic adaptation (Fig2C), depletion of p31comet was less effective than Cdc20 depletion in prolonging mitotic durations in U2OS cells (Fig2A and B). U2OS cells transfected with siP31-1 underwent apoptosis about 13 h sooner than siCdc20-treated cells did (Δt50 = 12.7 ± 6.5 h, n = 3). A similar trend was observed in non-transformed RPE1 cells (Supplementary Fig S2A and B).
Because apoptosis is already the dominant pathway in HeLa cells, further blockade of the adaptation process is not expected to change the timing of apoptosis. Indeed, depletion of Cdc20 in HeLa cells did not alter the timing of apoptosis onset during mitotic arrest by either FACS analysis (Fig3A–E) or live-cell imaging (Fig3F). In contrast, depletion of p31comet reproducibly shortened mitotic duration in taxol and accelerated cell death, as observed by FACS analysis (Fig3A–C), by live-cell imaging (Fig3F, Supplementary Fig S2C; Δt50 = 2.6 ± 0.4 h, n = 6), and by the appearance of cleaved PARP, an apoptosis marker, at earlier time points (Fig3D). The advancement of apoptosis was also observed in siP31 cells arrested in nocodazole (Supplementary Fig S2C) and could be rescued by expression of siRNA-resistant eGFP-p31comet (Supplementary Fig S2E and F). These results strongly suggest that p31comet delays the onset of mitotic apoptosis.
Figure 3. p31comet has a Cdc20-independent role in delaying mitotic apoptosis.

A HeLa cells transfected with the indicated siRNAs were synchronized by double thymidine block and released into medium with taxol. Samples were taken at different time points after thymidine release and processed for FACS. Representative FACS graphs of siControl and siP31 cells at the indicated times are shown. Mitotic cells (MPM2-positive 4C cells) are boxed and quantified.
B Quantification of the percentage of mitotic cells during the time course in (A).
C Quantification of the percentage of apoptotic cells (cells with < 2C DNA content) during the time course in (A).
D Lysates of siControl and siP31 cells in (A) were blotted with the indicated antibodies.
E Western blots of lysates of asynchronous U2OS and HeLa cells treated for 48 h (siP31-1; Mock) or 20 h (siCdc20) with the indicated siRNAs and blotted with the indicated antibodies.
F, G HeLa (F) or U2OS (G) cells transfected with the indicated siRNAs, treated with taxol, and monitored by time-lapse microscopy. Representative samples of the cumulative percentage of cells in mitosis were plotted against the time in mitosis. (F) Δt50(siLuc–siP31-1) = −2.6 ± 0.4 h, n = 6; Δt50(siCdc20–siP31-1) = −4.2 ± 2.7 h, n = 2. (G) Δt50(siCdc20–siP31-1) = 9.0 ± 1.0 h, n = 2; Δt50(siCdc20–siP31-1/siCdc20) = 6.8 ± 3.1 h, n = 2.
H Quantification of terminal phenotypes of cells in (G).
I Western blots of lysates of mitotic U2OS cells treated with the indicated siRNAs. Cells were transfected for 50 h (siP31-1, mock) and/or 20 h (siCdc20) before taxol addition. Mitotic cells were collected by shake-off at 15 h after taxol addition.
Mcl1 has recently been shown to be a key negative regulator of mitotic apoptosis (Harley et al, 2010; Wertz et al, 2011). It is degraded during early mitosis. Several ubiquitin ligases, including APC/CCdc20, have been implicated in its degradation (Harley et al, 2010; Wertz et al, 2011). We thus considered the possibility that p31comet delayed apoptosis through regulating APC/CCdc20-dependent degradation of Mcl1. If so, co-depletion of both Cdc20 and p31comet should ameliorate the advancement of apoptosis caused by p31comet depletion alone. Contrary to this expectation, the timing of apoptosis onset in cells depleted of both Cdc20 and p31comet was indistinguishable from that of cells depleted of p31comet alone (Fig3G–I). Similar results were obtained in RPE1 cells (Supplementary Fig S2A and B). Somewhat surprisingly, we did not observe accumulation of Mcl1 in siCdc20 cells in mitosis (Fig3I), perhaps due to the functional redundancy between multiple ubiquitin ligases that could mediate Mcl1 degradation. Depletion of p31comet or Cdc20 did not alter the protein levels of other apoptotic regulators during mitotic arrest in U2OS or HeLa cells (Fig3E and I). These results suggest that, in addition to promoting adaptation, p31comet might have a minor, Cdc20-independent role in suppressing apoptosis during mitotic arrest. This notion is consistent with our earlier finding that depletion of p31comet causes about 20% of HeLa cells to undergo apoptosis during an unperturbed mitosis (Jia et al, 2011). On the other hand, although Cdc20 was depleted efficiently in cells treated with both siP31 and siCdc20 (Fig3I), we cannot exclude the possibility that a small difference in Cdc20-knockdown efficiencies between the single- and double-depletion conditions accounts for the apparent dominant effects of siP31.
Noxa is a key regulator of apoptosis during mitotic arrest
The BH3-only pro-apoptotic factor Noxa was the top hit within the apoptosis network and had not previously been implicated in mitotic cell death (Ploner et al, 2008). To validate a role of Noxa in mitotic cell death, we transfected HeLa cells individually with each of the four Noxa siRNAs from the pool used in the screen and determined their ability to decrease cell death in taxol. All Noxa siRNAs reduced the Noxa mRNA levels and decreased the percentage of apoptotic cells, defined as cells with DNA contents < 2C in flow cytometry (Fig4A and B). The effect of siNoxa-1 on cell death was similar to that of the siBax-1 and siBak-1 mixture (referred to as siBaxBak; Fig4B) and was rescued by the expression of siRNA-resistant GFP-Noxa (Fig4C). We could not, however, reproducibly detect the endogenous Noxa protein by Western blotting, perhaps due to its small size (< 10 kDa) and low steady-state levels. Our results suggest that Noxa is a critical inducer of apoptosis during mitotic arrest.
Figure 4. Noxa is a key regulator of apoptosis during mitotic arrest.

- Quantification of Noxa mRNA levels by quantitative PCR in HeLa cells transfected with the indicated siRNAs for 48 h. The means and standard deviations of triplicate samples are shown.
- HeLa cells were transfected with the indicated siRNAs for 48 h, treated with taxol for 24 h, stained with PI, and subjected to FACS. The percentage of cells with < 2C DNA content (apoptotic cells) was plotted as the mean and standard deviation of triplicate experiments.
- HeLa cells were transfected with the indicated siRNAs and plasmids, treated with taxol for 15 h, and subjected to FACS. The percentage of cells with < 2C DNA content (apoptotic cells) was plotted. Average and standard deviations of three experiments are shown.
- HeLa cells transfected with siControl or siNoxa-1 were synchronized by double thymidine block and released into medium with taxol. Samples were taken at different time points after thymidine release and processed for FACS. FACS graphs at indicated times are shown. Mitotic cells (MPM2-positive 4C cells) are boxed and quantified.
- Quantification of the percentage of mitotic cells during the time course in (D).
- Quantification of the percentage of apoptotic cells (cells with < 2C DNA content) during the time course.
- HeLa cells transfected with siControl or siNoxa-1 were synchronized by thymidine block and then released into taxol, fixed at the indicated times, and stained with anti-cytochrome c and DAPI. Left panel, representative images of a mitotic cell (with cytochrome c in the mitochondria) and a cell that had undergone MOMP (with diffuse cytochrome c staining). Right panel, the percentage of cells with MOMP plotted against time after thymidine release. Average and standard deviations of triplicate experiments plotted. P-values calculated by Student's t-test are: n.s. (not significant), *P = 0.02, ***P = 0.0005.
- Representative samples from live-cell imaging of HeLa cells transfected with siLuc or siNoxa-1 and treated with taxol. Waterfall plots, each bar represents an individual cell.
- Dot plot of the phenotype and mitotic duration of each cell in (H). Cells with different fates are colored as in (H). The bar indicates the median (t50).
- Mitotic duration curves of cells in (H). The cumulative percentage of mitotic HeLa cells was plotted against their mitotic durations. Δt50(siNoxa-1–siLuc) = 11.1 ± 4.9 h, n = 4.
- Quantification of the terminal phenotypes of the mitotic cells in (H). Color scheme is the same as in (H). The means and standard deviations of two independent experiments are shown.
To explore the mechanism by which Noxa regulated apoptosis during mitotic arrest, we synchronized HeLa cells transfected with a control siRNA (siControl), siNoxa, or siBaxBak at G1/S with thymidine and released them into fresh medium containing taxol. Samples were taken at different time points and subjected to FACS and immunoblotting. Depletion of Noxa or Bax/Bak not only reduced the extent of apoptosis in taxol, but also lengthened the duration of mitosis and delayed the onset of apoptosis (Fig4D–F). The appearance of cleaved PARP was correspondingly delayed in Noxa RNAi cells, and the decrease in cyclin B1 and phospho-histone H3 levels occurred later (Supplementary Fig S3A). Noxa depletion also delayed cytochrome c release from the mitochondria, consistent with a delay in MOMP (Fig3G). These results suggest that Noxa induces mitotic cell death through the intrinsic mitochondrial pathway. In response to other stimuli, Noxa is known to sequester Mcl1 away from Bak, thus promoting Bak oligomerization and MOMP (Ploner et al, 2008). Noxa likely induces mitotic cell death through a similar mechanism.
We next monitored control and Noxa RNAi HeLa cells treated with taxol with time-lapsed microscopy (Fig4H; Supplementary Fig S3B). Compared to control cells, depletion of Noxa greatly increased the duration of taxol-induced mitotic arrest and delayed the onset of apoptosis (Δt50 = 11.1 ± 4.8 h, n = 4; Fig4H–J). A larger percentage of Noxa RNAi cells underwent mitotic adaptation (Fig4K). Similar results were obtained with siNoxa-2 (Supplementary Fig S3C and D). Furthermore, overexpression of the Noxa target, Mcl1, lengthened mitotic duration in the presence of taxol or nocodazole and increased the extent of mitotic adaptation, as determined by both FACS and live-cell imaging (Supplementary Fig S4A–E). Conversely, depletion of Mcl1 from HeLa cells shortened the duration of mitotic arrest in taxol, advanced the timing of apoptosis, and reduced mitotic adaptation (Supplementary Fig S4F and G). Collectively, our results indicate that Noxa antagonizes Mcl1 and is an initiator of the intrinsic mitochondrial apoptosis pathway during mitotic arrest.
Overexpression of Mcl1 or depletion of Noxa did not alter the behaviors of U2OS cells in the presence of taxol (Supplementary Figs S4H, S5A and B), suggesting that other Bcl2 members might be more critical for controlling apoptosis in this cell line. Interestingly, co-depletion of Noxa from p31comet RNAi cells lengthened mitotic durations and delayed the onset of apoptosis (Supplementary Fig S5A and B). Depletion of Noxa also rescued the accelerated apoptosis caused by p31comet depletion in HeLa cells (Supplementary Fig S5C and D). Taken together, these results suggest that p31comet might delay apoptosis during mitotic arrest through antagonizing Noxa. We could not reproducibly detect a physical interaction between p31comet and Noxa. The mechanism by which p31comet antagonizes Noxa-dependent mitotic apoptosis thus remains to be determined and is likely indirect.
Mitochondrial regulators contribute to mitotic adaptation
p31comet was not only required for adaptation, but also had a role in delaying apoptosis. This observation suggested that there might be crosstalk between the two competing pathways. Next, we asked whether components of the apoptosis pathway could also play a role in the adaptation process. First, we turned to the U2OS cell line, in which a majority of the cells underwent adaptation. Disabling the minor apoptosis pathway in this cell line was not expected to have a substantial effect on the timing of the major mitotic adaptation pathway, if the two pathways were independent of each other.
Surprisingly, depletion of Bax and Bak from U2OS cells not only diminished the residual apoptosis, but also lengthened the duration of taxol-induced mitotic arrest and delayed mitotic adaptation (Δt50 = 10.1 ± 3.5 h, n = 6; Fig5A–C). This phenotype requires both Bax and Bak depletion, as depletion of each individually resulted in weaker effects (Fig5D and E). Similar results were obtained in RPE1 cells (Fig5F and G). The effects of Bax/Bak depletion on the timing of mitotic adaptation cannot be readily explained by the simple competition between two independent pathways and suggest that Bax/Bak actively promote mitotic adaptation.
Figure 5. Bax/Bak contribute to mitotic adaptation.

- Western blot of lysates from asynchronous U2OS cultures transfected with the indicated siRNAs for 48 h and blotted with the indicated antibodies.
- Live-cell imaging analysis of U2OS cells transfected with the indicated siRNAs for 48 h and then treated with taxol. Cumulative percentage of mitotic cells was plotted against their mitotic durations. Δt50(siBaxBak–siLuc) = 10.1 ± 3.5 h, n = 6.
- Quantification of the terminal phenotypes of the mitotic cells in (B). Bax/Bak-deficient cells that die in mitosis without membrane blebbing are simply categorized as cell death, as opposed to apoptosis.
- Cumulative percentage of mitotic U2OS cells was plotted against their mitotic durations. t50(siBaxBak) = 12.2 h, t50(siBax-1) = 6.2 h, t50(siBak-1) = 6.7 h, t50(siLuc) = 3.9 h.
- Quantification of the terminal phenotypes in (D).
- Live-cell imaging analysis of RPE1 cells transfected with the indicated siRNAs for 48 h and then treated with taxol. Cumulative percentage of mitotic cells was plotted against their mitotic durations. Δt50(siBaxBak–siLuc) = 11.3 ± 3.4 h, n = 3.
- Quantification of the terminal phenotypes of the mitotic cells in (F).
- Live-cell imaging analysis of U2OS cells transfected with siLuc and treated with 50 μM z-VAD-fmk and taxol. Cumulative percentage of mitotic cells was plotted against their mitotic durations. t50(siLuc) = 4.2 h, t50(siLuc + z-VAD-fmk) = 4.6 h.
- Quantification of the terminal phenotypes in (H).
We next tested whether caspase activities were required for this function of Bax/Bak. Addition of 50 μM z-VAD-fmk, a caspase inhibitor, effectively reduced the residual amount of apoptosis in U2OS cells, but it had minimal effects on the mitotic duration (Fig5H and I), suggesting that the mitotic adaptation function of Bax/Bak might not require caspase activity.
The timing of adaptation is determined by the steady-state level of cyclin B1, which is influenced by both the rates of cyclin B1 synthesis and degradation. We tested whether Bax/Bak promoted adaptation through affecting cyclin B1 degradation or synthesis. As shown in Fig2D–G, depletion of Cdc20 or p31comet stabilized cyclin B1 even when protein synthesis was inhibited by cycloheximide, consistent with the known roles of Cdc20 and p31comet in cyclin B1 degradation. As expected, depletion of Cdc20 delayed mitotic adaptation of U2OS cells even in the presence of cycloheximide (Fig6A). In contrast, when protein synthesis was inhibited by cycloheximide, Bax/Bak-depleted cells underwent mitotic adaptation with kinetics similar to that of mock-treated cells (Fig6A), arguing against a major role of Bax/Bak in promoting cyclin B1 degradation. We then directly tested whether Bax/Bak regulated APC/C. APC/C was immunoprecipitated from checkpoint-arrested mitotic control and siBaxBak cells and assayed for its ability to ubiquitinate cyclin B1. The basal activity of APC/C in siBax/Bak cells was indistinguishable from that of control cells (Fig6B). Taken together, these results indicate that Bax/Bak promote mitotic adaptation independently of APC/C activity and cyclin B1 degradation. They might do so through attenuating cyclin B1 synthesis.
Figure 6. Bax/Bak promote mitotic adaptation independently of cyclin B1 degradation.
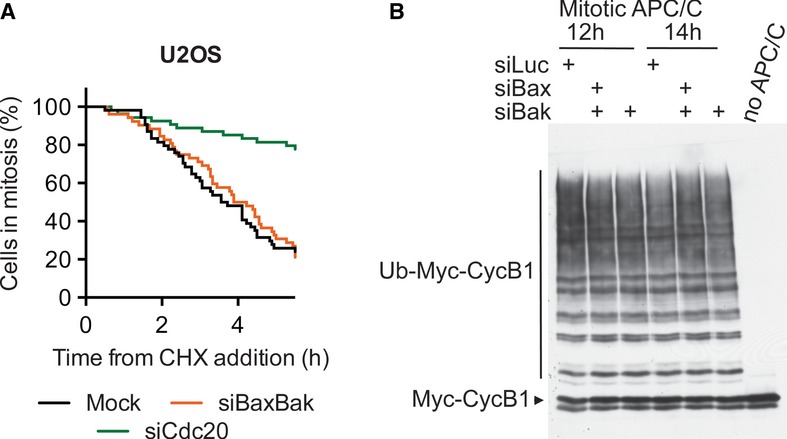
- U2OS cells transfected with the indicated siRNAs were synchronized by a thymidine block and released into taxol. Mitotic cells were collected by shake-off, incubated with cycloheximide (50 μM), and monitored by time-lapse microscopy. Cumulative percentage of cells in mitosis was plotted against their mitotic durations.
- HeLa cells transfected with the indicated siRNAs, synchronized by a thymidine block, and released into nocodazole for the indicated times. Anaphase-promoting complex/cyclosome (APC/C) was immunoprecipitated from these cells and was then assayed for its ligase activity toward cyclin B1.
The mitochondrial fission factor Drp1 contributes to mitotic adaptation
To further address the mechanism by which Bax/Bak promoted adaptation in U2OS cells, we first tested the potential involvement of mitochondrial factors that were released into the cytosol during apoptosis in a Bax/Bak-dependent manner, including cytochrome c, Smac, Omi, AIF, and Endo G. Depletion of none of these factors extended mitotic duration (data not shown). Bax/Bak have been shown to regulate the mitochondrial fission factor Drp1 (Arnoult et al, 2005; Wasiak et al, 2007). We next tested whether Drp1 was involved in mitotic adaptation. Strikingly, depletion of Drp1 from U2OS cells extended mitotic durations and delayed mitotic adaptation in taxol (Fig7A). Multiple siRNAs against Drp1 produced similar phenotypes. siDrp1-3 caused the longest delay with a Δt50 of 10.4 ± 1.3 h (n = 2), which was comparable to the delay caused by siBaxBak (10.1 ± 3.5 h, n = 6). Unlike siBaxBak, however, siDrp1 did not reduce the percentage of cells that underwent apoptosis during mitotic arrest (Fig7B), suggesting that Drp1 depletion prolonged mitosis without interfering with the apoptosis pathway.
Figure 7. The mitochondrial fission factor Drp1 contributes to mitotic adaptation.

- Cumulative percentage of mitotic U2OS cells transfected with the indicated siRNAs for 48 h and then treated with taxol was plotted against their mitotic durations determined by live-cell imaging. Δt50(siDrp1-3–siLuc) = 10.4 ± 1.3 h, n = 2.
- Quantification of the terminal phenotypes of the mitotic cells in (A).
- U2OS cells were synchronized by thymidine block and released into pre-warmed medium. Taxol was added at 12 h after release, along with DMSO or 50 μM Mdivi-1. Mitotic cells were collected 3 h later by shake-off and further cultured in the taxol medium for the indicated times in the absence (top) or presence of 50 μM cycloheximide (CHX; middle) or 5 mM sodium azide and 2 mM 2-deoxyglucose (2-DG; bottom). The cells were stained with the MPM2 antibody and PI and subjected to FACS. The normalized counts of cells with 4C DNA content (as a percentage of the maximum in the histogram of each time point) were plotted against the intensities of MPM2. The MPM2 intensities of mitotic cells and the adapted cells are indicated by black and red arrows.
- Quantification of the percentage of mitotic (MPM2+) cells at the indicated times and conditions as in (C), with averages and standard deviations of multiple experiments shown.
- U2OS cells were synchronized by thymidine block and released into pre-warmed medium. Taxol was added at 9 h after release, along with DMSO or 50 μM Mdivi-1. Mitotic cells were collected 3 h later by shake-off. ATP levels were determined by luminescence with CellTiterGlo and normalized to the number of live cells determined by trypan blue exclusion. Averages and standard deviations of three independent experiments, each with triplicate samples, are shown.
Although siDrp1 cells that entered mitosis were able to sustain the mitotic arrest longer than siLuc cells, we noticed that many siDrp1 cells failed to enter mitosis and appeared unhealthy. Indeed, Drp1 depletion has been reported to cause cell cycle defects and a G2 delay (Qian et al, 2012; Westrate et al, 2014). To avoid complications from these interphase phenotypes caused by siDrp1, we inhibited Drp1 in U2OS cells at the time of mitotic entry with the chemical inhibitor of Drp1 Mdivi-1 (Cassidy-Stone et al, 2008) and analyzed the kinetics of mitotic adaptation with FACS. An increasing percentage of cells underwent mitotic adaptation with increasing time in taxol, as evidenced by the appearance of MPM2-negative population (Fig7C; indicated by red arrows). Chemical inhibition of Drp1 with Mdivi-1 largely blocked mitotic adaptation in these cells (Fig7C and D). Addition of cycloheximide to mitotic cells treated with Mdivi-1 accelerated mitotic adaptation (Fig7C and D). The fact that cycloheximide abrogated the effect of Mdivi-1 indicated that, similar to Bax/Bak, the role of Drp1 in mitotic adaptation might involve protein synthesis.
Interestingly, the peaks of the MPM2-negative DMSO-treated cells in the FACS graphs were broader than those of cells treated with Mdivi-1 (Fig7C). The MPM2-negative DMSO cells that had undergone adaptation had variable DNA contents, from 4C to < 2C (Supplementary Fig S6A), whereas those adapted cells in Mdivi-1-treated cells had 4C DNA content. As revealed by time-lapse microscopy, the majority of U2OS cells underwent abnormal cytokinesis during adaptation in taxol, often producing daughter cells with different sizes and presumably uneven DNA contents (Supplementary Fig S6B and C). Drp1 inhibition by Mdivi-1 not only delayed mitotic adaptation, but also blocked this abnormal cytokinesis during adaptation. Similarly, Bax/Bak depletion also diminished abnormal cytokinesis during adaptation, albeit to a lesser extent than Mdivi-1 treatment (Supplementary Fig S6D). It remains to be determined whether the functions of Drp1 in promoting adaptation and in triggering aberrant cytokinesis are linked. In this experiment, we did not measure cyclin B1 levels and thus could not rule out the possibility that the increase in MPM2-positive cells caused by siBax/Bak or Mdivi-1 was due to alterations of other Cdk1 regulatory mechanisms. Nevertheless, the similarities in the phenotypes caused by Drp1 inactivation and Bax/Bak depletion are consistent with Bax/Bak and Drp1 acting through similar mechanisms.
We further explored the mechanism by which Drp1 regulated the onset of mitotic adaptation. It has been reported that energy production by the mitochondria positively correlates with the extent of mitochondrial connectivity (Mitra et al, 2009), and phosphorylation-dependent activation of Drp1 during mitosis increases mitochondrial fission and reduces both mitochondrial connectivity and presumably energy production (Taguchi et al, 2007). Acute Drp1 inactivation in human cells by siRNA or Mdivi-1 elevates ATP production in interphase (Qian et al, 2012). Strikingly, we found that Mdivi-1 treatment also elevated ATP levels in mitotic cells (Fig7E). More importantly, inhibition of ATP production with azide and 2-deoxyglucose (2-DG) abrogated the delay in mitotic adaptation caused by Mdivi-1 (Fig7C and D; Supplementary Fig S6A). Taken together, these results are consistent with the intriguing possibility that Drp1 activation in mitosis contributes to mitotic adaptation by decreasing ATP production and indirectly affecting global protein translation, including translation of cyclin B1.
Finally, we tested whether the apoptosis and adaptation pathways were the only major pathways determining cell fates after taxol-induced mitotic arrest. Depletion of p31comet from siBaxBak U2OS cells further lengthened mitotic durations (Fig8A and B). A similar synergy between p31comet depletion and Bax/Bak depletion in lengthening mitotic durations was observed in RPE1 cells (Fig8C and D). Depletion of Cdc20 also synergized with Bax/Bak depletion to prolong taxol-induced mitotic arrest in both U2OS and RPE1 cells (Fig8A–D). RPE1 cells depleted of Cdc20, and Bax/Bak were arrested in mitosis with a t50 of 54.4 h, compared to a t50 of 13.2 h in control cells. Remarkably, about 58% of the cells depleted of Cdc20 and Bax/Bak were still in mitosis at the end of the experiment. Therefore, the apoptosis and adaptation pathways are the major pathways that determine cell fates following mitotic arrest. Crippling both pathways traps cells in mitosis for exceedingly long durations.
Figure 8. Crosstalk between mitotic adaptation and apoptosis pathways.
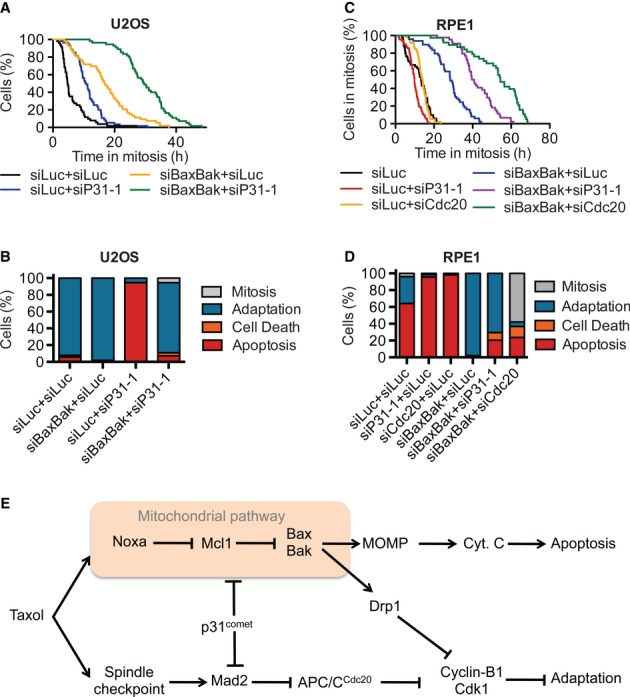
- Cumulative percentage of mitotic U2OS cells transfected with the indicated siRNAs and treated with taxol was plotted against their mitotic durations. A representative example of duplicate experiments: t50(siLuc + siLuc) = 4.7 h, t50(siLuc + siP31-1) = 10.5 h, t50(siNoxa-1 + siLuc) = 4.1 h, t50(siBaxBak + siLuc) = 16.6 h, t50(siNoxa-1 + siP31-1) = 15.3 h, t50(siBaxBak + siP31-1) = 29.3 h.
- Quantification of the terminal phenotypes in (A). The “cell death” group denotes cells that undergo death without showing the canonical membrane blebbing observed during apoptosis.
- Cumulative percentage of RPE1 cells transfected with the indicated siRNAs and treated with taxol is plotted against their mitotic durations. t50(siLuc + siLuc) = 13.3 h, t50(siLuc + siP31-1) = 9.8 h, t50(siLuc + siCdc20) = 13.7 h, t50(siBaxBak + siLuc) = 28.3 h, t50(siBaxBak + siP31-1) = 39.6 h, t50(siBaxBak + siCdc20) = 54.3 h.
- Quantification of the terminal phenotypes of the mitotic cells in (C).
- A coupled molecular race model for cell fate decisions during prolonged mitotic arrest. Apoptosis and adaptation are the two major competing pathways in this process. These two pathways are coupled upstream through a mitochondrial module consisting of Bax/Bak. Bax/Bak promote both apoptosis through MOMP and cytochrome c release and adaptation through modulation of Drp1. Because cytochrome c is not released in cells undergoing adaptation (data not shown), the bifurcation of these two pathways likely occurs upstream of MOMP. Enhanced Drp1 mitochondrial fission activity during mitosis reduces ATP production, hampers global protein translation (including cyclin B1 translation), and promotes adaptation. Small differences in the rates of the actual execution of downstream events of apoptosis or mitotic adaptation then determine the cell fates, providing a possible explanation for the heterogeneity in cellular responses to taxol. Unlike Bax/Bak which are couplers of apoptosis and adaptation, p31comet has opposing roles in the two pathways and acts as an anti-coupler. Targeting p31comet will not only block adaptation but also promote apoptosis.
Discussion
Our genome-wide siRNA screen presented herein has systematically identified molecular components in human cells that mediate apoptosis and mitotic adaptation during taxol-induced mitosis arrest. This collection of regulators is a valuable resource and enables future mechanistic studies. Our initial characterization of certain regulators, p31comet, Noxa, and Bax/Bak, has already revealed new principles in the coordination of mitotic apoptosis and adaptation. We show that p31comet has opposing functions in the two competing pathways (Fig8E). It actively promotes mitotic adaptation through APC/CCdc20-dependent degradation of cyclin B1. It also has a role in antagonizing apoptosis during mitotic arrest. Unexpectedly, Bax/Bak not only are required for mitotic apoptosis, but also contribute to mitotic adaptation, possibly through the mitochondrial fission factor Drp1.
Identification of functional networks that mediate cellular responses to taxol
Network analysis of the hits in our screen reveals four main networks that mediate cellular responses to taxol, including the apoptosis network, the spindle checkpoint network, the DNA replication licensing network, and the spliceosome network. The DNA replication licensing and spliceosome networks are required for cell cycle progression. Their inactivation results in cell cycle defects in interphase and prevents mitotic entry, thereby indirectly increasing cell viability in taxol. In addition to these networks, our screen also isolated other genes with known functions in the cell cycle, including Cdc2 (Cdk1), ribonucleotide reductases (RRM1 and RRM2), and Set8. Collectively, these results confirm that mitotic entry is required for taxol-induced apoptosis.
Although our screen isolated many known components of the spindle checkpoint, we did not uncover novel spindle checkpoint genes, confirming that the Bub and Mad genes initially identified in yeast genetic screens were indeed the major players of this important checkpoint in human cells. Inactivation of the spindle checkpoint genes increases short-term cell viability in the presence of taxol, due to mitotic exit and failure to undergo apoptosis. Conversely, multiple subunits of APC/C were isolated as viability-decreasing hits. APC/C inactivation is expected to delay mitotic progression and cause hyperactivation of the spindle checkpoint, thus blocking mitotic adaptation and sensitizing cells to taxol-mediated killing.
In our screen, we isolated multiple components of the intrinsic mitochondrial apoptosis pathway, confirming the involvement of this pathway in mitotic apoptosis. In addition to this pathway, the apoptosis network highlighted a potential role of the JAK-STAT pathway in mitotic cell death. The JAK-STAT pathway mediates cytokine signaling in metazoans and has diverse physiological functions ranging from immune response, development, differentiation, and apoptosis (Levy & Darnell, 2002). This pathway is often up-regulated in human cancers and is generally pro-survival, although certain players of this pathway have been implicated in promoting apoptosis in certain contexts. Future studies are needed to examine the involvement of this pathway in mitotic apoptosis.
Our screen did not identify other gene networks involved in processes such as necrosis or autophagy, suggesting that the apoptosis and spindle checkpoint pathways might be the only major pathways that determine cell death/survival decisions during mitotic arrest. Indeed, inactivation of the apoptosis pathway and hyperactivation of the spindle checkpoint through Cdc20 or p31comet depletion block cells in mitosis for exceedingly long durations. Thus, the duration of mitotic arrest caused by taxol is actively controlled by the apoptosis and spindle checkpoint pathways and is not solely determined by cellular metabolism.
The Noxa–Mcl1 axis in mitotic apoptosis
Our results confirm and extend the involvement of the intrinsic mitochondrial pathway in taxol-induced apoptosis. The Noxa–Mcl1 axis in this pathway is particularly important for controlling the timing of apoptosis following mitotic entry, at least in HeLa cells (Fig8E). The Mcl1 protein levels decrease during mitotic arrest. The drop in Mcl1 levels may help to tip the balance between the pro-apoptotic activities of Noxa and the anti-apoptotic activities of Mcl1, thus contributing to the timing of mitotic cell death.
Although inactivation of Noxa greatly lengthens the duration of mitotic arrest and delays the onset of apoptosis in HeLa cells, many Noxa RNAi cells still undergo apoptosis following the prolonged mitotic arrest. Furthermore, depletion of Noxa did not have any effect on mitotic apoptosis in U2OS cells. In previous studies, inactivation of other pro-apoptotic regulators in the intrinsic pathway also attenuates, but does not prevent, taxol-induced mitotic cell death (Kutuk & Letai, 2010; Moustafa-Kamal et al, 2013). Different players in this pathway are implicated as major regulators of taxol-induced cell death in these studies (Czernick et al, 2009; Kutuk & Letai, 2010; Moustafa-Kamal et al, 2013). Collectively, these results paint a complicated picture and suggest the existence of multiple, redundant mechanisms upstream of Bax/Bak for executing mitotic apoptosis. Different cell lines might preferentially use certain mechanisms.
Roles of p31comet in mitotic adaptation and in suppressing apoptosis
p31comet promotes mitotic exit in the absence of microtubule poisons (Hagan et al, 2011; Jia et al, 2011) and is required for mitotic adaptation in nocodazole (Varetti et al, 2011). Similarly, we showed that p31comet is also required for adaptation in taxol and that it does so by promoting cyclin B1 degradation. Thus, the basal APC/C activity observed during mitotic arrest is produced by p31comet-dependent inactivation of Mad2 and that low levels of inactivation occur even in the presence of an active checkpoint.
We also uncovered a minor role for p31comet in antagonizing apoptosis during mitotic arrest, suggesting that p31comet has opposing functions in the two competing pathways. Interestingly, depletion of Noxa abolishes the acceleration of mitotic apoptosis caused by p31comet depletion, suggesting that p31comet antagonizes Noxa. One intriguing possibility is that p31comet regulates apoptosis through modulation of Mcl1, which has been shown to be a substrate of APC/CCdc20 in mitosis (Harley et al, 2010). However, we did not observe a decrease in Mcl1 levels in interphase or mitotic cells after p31comet depletion. The small effects of p31comet depletion on Mcl1 degradation in Fig3D were not reproducible. Depletion of Cdc20 did not block the advancement of apoptosis timing caused by depletion of p31comet. Thus, we do not have evidence to support a model in which p31comet blocks APC/CCdc20-dependent degradation of Mcl1. In addition, we failed to detect any physical interactions between p31comet and apoptotic regulators, including Bax/Bak, Noxa, or Mcl1, either by immunoprecipitation of endogenous proteins in human cells or through in vitro binding experiments using purified p31comet, Mad2, or p31comet–Mad2 and in vitro translated Bax, Bak, Noxa, or Mcl1. These negative results suggest that the role of p31comet in suppressing apoptosis might be indirect. Regardless of the mechanism, the fact that p31comet has opposing functions in mitotic adaptation and apoptosis suggests the existence of crosstalk between the apoptosis and adaptation pathways during mitosis.
Roles of Bax/Bak and Drp1 in mitotic adaptation
In HeLa cells, which primarily undergo apoptosis during mitotic arrest, weakening of the intrinsic apoptosis pathway by Noxa inactivation extends mitotic arrest, a phenotype that can be explained by a simple molecular race between apoptosis and adaptation (Gascoigne & Taylor, 2008; Huang et al, 2010). On the other hand, we found that depletion of Bax/Bak delayed mitotic adaptation in U2OS cells in taxol. Because 90% of the cells in this line normally undergo adaptation in taxol, the molecular race model would predict that mitotic adaptation is executed faster than apoptosis in these cells. If the two pathways are truly independent of each other, disabling the slower apoptosis pathway in this cell line is not supposed to have a substantial effect on the timing of the faster mitotic adaptation pathway. Our observation that the apoptotic factors Bax/Bak contribute to mitotic adaptation thus cannot be explained by a simple competition between two independent pathways and strongly indicates that the apoptosis and adaptation pathways are coupled.
We show that Bax/Bak promote adaptation through a mechanism independent of APC/CCdc20-mediated cyclin B1 degradation. Instead, they may do so through their ability to regulate Drp1. Similar to Bax/Bak depletion, Drp1 inactivation by siRNA or its mitosis-specific inhibition by Mdivi-1 delays mitotic adaptation. Furthermore, Mdivi-1 treatment increases total ATP concentrations in mitotic cells. Inhibition of protein synthesis with cycloheximide or blocking ATP production with inhibitors of oxidative phosphorylation (azide and 2-DG) reverses the effect of Mdivi-1 and accelerates mitotic adaptation. We hypothesize that Bax/Bak and Drp1 promote mitotic adaptation indirectly through affecting mitochondrial dynamics and fitness during mitotic arrest, leading to lower ATP production that weakens global protein translation. Our evidence implicating changes in protein translation during mitotic adaptation is indirect. Additional studies are needed to directly measure the synthesis rate of cyclin B1 and other proteins in control and Bax/Bak- or Drp1-deficient cells. It is also unclear how Bax/Bak regulate Drp1 or vice versa. Finally, it will be interesting to test whether Bax/Bak and Drp1 regulate mitotic exit in the absence of spindle poisons. Because of a requirement for Bax/Bak and Drp1 in cell cycle progression prior to mitosis entry, techniques that inactivate these molecules specifically in mitosis are required to address this important question.
Coupling between mitotic apoptosis and adaptation
Our studies suggest that activation of the mitochondrial apoptosis pathway inevitably potentiates cells for mitotic adaptation. The small kinetic differences in actually executing the downstream events of apoptosis or mitotic adaptation then determine the final cell fate. The upstream coupling of two competing pathways thus provides a possible explanation for the heterogeneity in the cellular responses to taxol and for the ineffectiveness of taxol against certain tumors. Because of its opposing functions in the apoptosis and adaptation pathways, targeting p31comet is an attractive strategy to uncouple these two pathways. Chemical inhibitors of p31comet are expected to block mitotic adaptation and accelerate apoptosis during mitotic arrest and have the potential to improve the efficacy of taxol in cancer chemotherapy.
Materials and Methods
Mammalian cell culture and transfection
HeLa Tet-On, U2OS, and RPE1-hTERT cells were grown in Dulbecco's Modified Eagle Medium (Invitrogen) supplemented with 10% fetal bovine serum and 10 mM l-glutamine. siRNA transfections were performed using Lipofectamine RNAiMAX (Invitrogen) according to instructions from the manufacturer. A final concentration of 5 nM per siRNA was used, unless stated otherwise. For siBaxBak, siBax-1 and siBak-1 were mixed at a final concentration of 2.5 nM each. The siRNA sequences used in this study are:
siBax-1, GGGACGAACTGGACAGTAA; siBak-1, CAGAGAAUGCCUAUGAGUA;
siCdc20, Silencer pre-designed ID s2748 (Ambion); siControl, GGCATCTTAGCCCGTACTT;
siDrp1-2, GAAAGAAGCAGCUGAUAUG; siDrp1-3, GGAGCCAGCUAGAUAUUAA; siDrp1-5, CGUAAAAGGUUGCCUGUUA; siMad2, UACGGACUCACCUUGCUUG;
siMcl1, CGAAGGAAGUAUCGAAUUU; siNoxa-1, GCAAGAACGCUCAACCGAG;
siNoxa-2, AAACUGAACUUCCGGCAGA; siNoxa-3, GAACCUGACUGCAUCAAAA; siNoxa-4, AAUCUGAUAUCCAAACUCU; siP31-1, GGCUGCUGUCAGUUUACUU (Jia et al, 2011).
To rescue the Noxa RNAi phenotypes, HeLa Tet-On cells were first transfected with siNoxa-1 and then transfected with pCS2-eGFP or pCS2-eGFP-Noxa plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. To create the stable Myc-Mcl1 and GFP-p31-siR cell line, HeLa Tet-On cells were transfected with the pTRE2hygro-Myc-Mcl1 and pTRE2hygro-eGFP-p31-siR plasmid, respectively, using the Effectene reagent (Qiagen). Clones were selected with 300 μg/ml hygromycin (Invitrogen). Expression was induced with 2 μg/ml doxycycline (Sigma). For the p31comet rescue experiment, HeLa cells stably expressing EGFP-p31-siR and their parental cell line were transfected with siP31-1 for 30 h. Cells were then transfected with siCdc20 to avoid the premature mitotic exit caused by p31comet overexpression. Cells were synchronized 8 h after siCdc20 transfection by a single thymidine block (15 h) and then released into medium containing taxol and imaged by time-lapse microscopy.
For mitotic arrest, cells were treated with 200 nM taxol (Sigma) or 500 nM nocodazole (Sigma) for 14–16 h or as indicated. For cell cycle synchronization, cells were cultured in medium containing 2.5 mM thymidine (Sigma) for 15 h (HeLa) or 24 h (U2OS) and released into fresh medium without thymidine for the desired time. The pan-caspase inhibitor z-VAD-fmk (BD Biosciences) was used at 50 μM. For translation inhibition, freshly made cycloheximide (Sigma) was used at 50 μM.
siRNA screen
HeLa Tet-On cells were reverse transfected with siRNA pools from the Dharmacon siGENOME library targeting 21,360 unique human genes. Each siRNA pool contained four siRNAs against a given gene. Four nanomoles of each siRNA pool diluted in 20 μl of DMEM without serum were dispensed into each well of 96-well plates using a Beckman FX liquid handler. 0.25 μl of Lipofectamine RNAiMAX (Invitrogen) reagent diluted in 20 μl of DMEM without serum was added to each well. After a 30-min incubation, 5,000 or 10,000 HeLa Tet-On cells in DMEM medium containing 20% FBS were added to each well to obtain a final volume of 100 μl and a 40 nM final concentration of siRNAs. Each siRNA pool was tested in triplicates. At 24 h post-transfection, taxol was added to a final concentration of 220 nM. Cell viability was determined at 48 h after taxol addition using the CellTiter-Glo assay (Promega) that measured the intracellular ATP concentrations. Viability values were normalized to the positive controls (Mad2 siRNA) on each plate and corrected for plate variability. Hits from the primary screen were subjected to secondary screens using the same procedure as described above, except that the siRNA pools from Qiagen were used. The secondary screen was performed at two siRNA concentrations. Hits from the primary and secondary screens were analyzed using Ingenuity Pathway Analysis with manual input. All hits from the primary screen were included to generate a large enough dataset for the analysis and to facilitate the discovery of gene networks.
Flow cytometry
Cells were washed with PBS, fixed in cold 70% ethanol, washed once again with PBS, and permeabilized in PBS containing 0.25% Triton X-100. The MPM2 (Millipore; 1:40 dilution) or anti-cyclin B1 (Santa Cruz, 1:20 dilution) antibodies were diluted in PBS containing 0.25% Triton X-100 and 3% BSA (Sigma) and added to the permeabilized cells. After an incubation at room temperature for 3 h, cells were washed with PBS and incubated with anti-mouse Alexa-488 secondary antibody (Invitrogen; 1:20 dilution) in PBS containing 0.25% Triton X-100 and 3% BSA for 30 min in the dark. Cells were again washed with PBS and incubated with 200 μg/ml DNase-free RNase A (Qiagen) and 2 μg/ml propidium iodide (Sigma) in PBS. Samples were analyzed with a FACSCalibur flow cytometer (BD Biosciences). Data were processed with the FlowJo software.
Live-cell microscopy
Cells were cultured on 4- or 8-well chamber coverslips (LabTek) and transfected with the indicated siRNAs at a final concentration of 5 nM for 24–48 h and treated with taxol. Cells were then imaged with a DeltaVision system (Applied Precision) which had a humidified environmental chamber with 37°C and 5% CO2. Differential interference contrast (DIC) images were acquired with a 40× objective at 2–10 min intervals for 12–72 h. Four 6-μm z-stacks per time point were acquired. The images were manually analyzed to determine mitotic timing and cell fates and further processed in ImageJ to create time-lapse movies.
Immunofluorescence
Cells were grown on chamber slides (LabTek), fixed with 4% formaldehyde (Pierce) or cold methanol, blocked in PBS containing 3% BSA and 0.2% Triton X-100, and incubated with anti-cytochrome c (1:250; BD Biosciences) and the appropriate Alexa secondary antibodies (Invitrogen). The slides were mounted with ProLong with DAPI (Invitrogen) and analyzed with the DeltaVision system. A series of z-stack images at 0.2-μm intervals were deconvolved. Images were further processed with ImageJ and Photoshop.
Antibodies and immunoblotting
Cells were lysed in 2× SDS loading buffer. Lysates were separated by SDS–PAGE, transferred to nitrocellulose membranes, and blotted with the desired antibodies. Antibodies against Apc2, Mad2, and p31comet were described previously (Fang et al, 1998; Xia et al, 2004). The commercial antibodies used in this study include: anti-cyclin B1 (Santa Cruz), anti-PARP (Cell Signaling), anti-Mcl1 (Cell Signaling), anti-Smac (Cell Signaling), anti-Bak (Millipore), anti-Bax (BD Biosciences), and anti-phospho-H3-S10 (Millipore). Anti-rabbit immunoglobulin G (IgG) (H+L) (Dylight 800 conjugates) and anti-mouse IgG (H+L) (Dylight 680 conjugates; Cell Signaling) were used as secondary antibodies. The blots were scanned with the Odyssey Infrared Imaging System (LI-COR).
Acknowledgments
We thank Xiaodong Wang and Hui Zou for reagents, Mike White and Angelique Whitehurst for advice on the RNAi screen, and Kate Luby-Phelps for advice on live-cell imaging. L.A.D.M. is supported by a Research Supplement to Promote Diversity in the Health Sciences (R01CA125269). HY is an Investigator with the Howard Hughes Medical Institute. This work was supported in part by the Welch Foundation (I-1441) and the Cancer Prevention and Research Institute of Texas (RP110465-P3 and RP120717-P2).
Author contributions
LADM and ZK performed the siRNA screen with technical assistance from SW and supervision from MGR. LADM designed and performed most additional experiments and wrote the initial draft of the paper. RW contributed to the characterization of hits in the screen. BL performed the APC/C assay. XJX performed the statistical analysis of the functional networks. HY supervised the project, contributed to data interpretation, and edited the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP, Estaquier J, Sheng M, Blackstone C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol. 2005;15:2112–2118. doi: 10.1016/j.cub.2005.10.041. [DOI] [PubMed] [Google Scholar]
- Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16:1194–1200. doi: 10.1016/j.cub.2006.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito DA, Rieder CL. The ability to survive mitosis in the presence of microtubule poisons differs significantly between human nontransformed (RPE-1) and cancer (U2OS, HeLa) cells. Cell Motil Cytoskeleton. 2009;66:437–447. doi: 10.1002/cm.20316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao WC, Kulkarni K, Zhang Z, Kong EH, Barford D. Structure of the mitotic checkpoint complex. Nature. 2012;484:208–213. doi: 10.1038/nature10896. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czernick M, Rieger A, Goping IS. Bim is reversibly phosphorylated but plays a limited role in paclitaxel cytotoxicity of breast cancer cell lines. Biochem Biophys Res Commun. 2009;379:145–150. doi: 10.1016/j.bbrc.2008.12.025. [DOI] [PubMed] [Google Scholar]
- Fang G. Checkpoint protein BubR1 acts synergistically with Mad2 to inhibit anaphase-promoting complex. Mol Biol Cell. 2002;13:755–766. doi: 10.1091/mbc.01-09-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 1998;12:1871–1883. doi: 10.1101/gad.12.12.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley EA, Kapoor TM. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol. 2013;14:25–37. doi: 10.1038/nrm3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–122. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS. How do anti-mitotic drugs kill cancer cells? J Cell Sci. 2009;122:2579–2585. doi: 10.1242/jcs.039719. [DOI] [PubMed] [Google Scholar]
- Habu T, Kim SH, Weinstein J, Matsumoto T. Identification of a MAD2-binding protein, CMT2, and its role in mitosis. EMBO J. 2002;21:6419–6428. doi: 10.1093/emboj/cdf659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan RS, Manak MS, Buch HK, Meier MG, Meraldi P, Shah JV, Sorger PK. p31comet acts to ensure timely spindle checkpoint silencing subsequent to kinetochore attachment. Mol Biol Cell. 2011;22:4236–4246. doi: 10.1091/mbc.E11-03-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Holland AJ, Fachinetti D, Kulukian A, Cetin B, Cleveland DW. Catalytic assembly of the mitotic checkpoint inhibitor BubR1-Cdc20 by a Mad2-induced functional switch in Cdc20. Mol Cell. 2013;51:92–104. doi: 10.1016/j.molcel.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29:2407–2420. doi: 10.1038/emboj.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Mitchison TJ, Shi J. Stochastic competition between mechanistically independent slippage and death pathways determines cell fate during mitotic arrest. PLoS ONE. 2010;5:e15724. doi: 10.1371/journal.pone.0015724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Shi J, Orth JD, Mitchison TJ. Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell. 2009;16:347–358. doi: 10.1016/j.ccr.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner NC, Wang LH, Kaulich M, Descombes P, Poser I, Nigg EA. Re-examination of siRNA specificity questions role of PICH and Tao1 in the spindle checkpoint and identifies Mad2 as a sensitive target for small RNAs. Chromosoma. 2010;119:149–165. doi: 10.1007/s00412-009-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen K, Pohlmann S, Janicke RU, Schulze-Osthoff K, Fischer U. Apaf-1 and caspase-9 deficiency prevents apoptosis in a Bax-controlled pathway and promotes clonogenic survival during paclitaxel treatment. Blood. 2007;110:3662–3672. doi: 10.1182/blood-2007-02-073213. [DOI] [PubMed] [Google Scholar]
- Jia L, Kim S, Yu H. Tracking spindle checkpoint signals from kinetochores to APC/C. Trends Biochem Sci. 2013;38:302–311. doi: 10.1016/j.tibs.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Jia L, Li B, Warrington RT, Hao X, Wang S, Yu H. Defining pathways of spindle checkpoint silencing: functional redundancy between Cdc20 ubiquitination and p31comet. Mol Biol Cell. 2011;22:4227–4235. doi: 10.1091/mbc.E11-05-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006;23:709–721. doi: 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10:194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- Ke Y, Huh JW, Warrington R, Li B, Wu N, Leng M, Zhang J, Ball HL, Li B, Yu H. PICH and BLM limit histone association with anaphase centromeric DNA threads and promote their resolution. EMBO J. 2011;30:3309–3321. doi: 10.1038/emboj.2011.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulukian A, Han JS, Cleveland DW. Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev Cell. 2009;16:105–117. doi: 10.1016/j.devcel.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutuk O, Letai A. Alteration of the mitochondrial apoptotic pathway is key to acquired paclitaxel resistance and can be reversed by ABT-737. Cancer Res. 2008;68:7985–7994. doi: 10.1158/0008-5472.CAN-08-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutuk O, Letai A. Displacement of Bim by Bmf and Puma rather than increase in Bim level mediates paclitaxel-induced apoptosis in breast cancer cells. Cell Death Differ. 2010;17:1624–1635. doi: 10.1038/cdd.2010.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Gonzalez P, Scott MI, Diez M, Sen O, Taylor SS. BubR1 blocks substrate recruitment to the APC/C in a KEN-box-dependent manner. J Cell Sci. 2011;124:4332–4345. doi: 10.1242/jcs.094763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol. 2012;22:R966–R980. doi: 10.1016/j.cub.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr STATs: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Li R, Moudgil T, Ross HJ, Hu HM. Apoptosis of non-small-cell lung cancer cell lines after paclitaxel treatment involves the BH3-only proapoptotic protein Bim. Cell Death Differ. 2005;12:292–303. doi: 10.1038/sj.cdd.4401554. [DOI] [PubMed] [Google Scholar]
- Luo X, Tang Z, Rizo J, Yu H. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol Cell. 2002;9:59–71. doi: 10.1016/s1097-2765(01)00435-x. [DOI] [PubMed] [Google Scholar]
- Luo X, Yu H. Protein metamorphosis: the two-state behavior of Mad2. Structure. 2008;16:1616–1625. doi: 10.1016/j.str.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HT, Chan YY, Chen X, On KF, Poon RY. Depletion of p31comet protein promotes sensitivity to antimitotic drugs. J Biol Chem. 2012;287:21561–21569. doi: 10.1074/jbc.M112.364356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli M, Filipp FV, Rancati G, Massimiliano L, Nezi L, Stier G, Hagan RS, Confalonieri S, Piatti S, Sattler M, Musacchio A. Determinants of conformational dimerization of Mad2 and its inhibition by p31comet. EMBO J. 2006;25:1273–1284. doi: 10.1038/sj.emboj.7601033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli M, Musacchio A. MAD contortions: conformational dimerization boosts spindle checkpoint signaling. Curr Opin Struct Biol. 2007;17:716–725. doi: 10.1016/j.sbi.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Mena AL, Lam EW, Chatterjee S. Sustained spindle-assembly checkpoint response requires de novo transcription and translation of cyclin B1. PLoS ONE. 2010;5:e13037. doi: 10.1371/journal.pone.0013037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustafa-Kamal M, Gamache I, Lu Y, Li S, Teodoro JG. BimEL is phosphorylated at mitosis by Aurora A and targeted for degradation by βTrCP1. Cell Death Differ. 2013;20:1393–1403. doi: 10.1038/cdd.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R, Mitchison TJ. Analysis of mitosis and antimitotic drug responses in tumors by in vivo microscopy and single-cell pharmacodynamics. Cancer Res. 2011;71:4608–4616. doi: 10.1158/0008-5472.CAN-11-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploner C, Kofler R, Villunger A. Noxa: at the tip of the balance between life and death. Oncogene. 2008;27(Suppl 1):S84–S92. doi: 10.1038/onc.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Choi S, Gibson GA, Watkins SC, Bakkenist CJ, Van Houten B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J Cell Sci. 2012;125:5745–5757. doi: 10.1242/jcs.109769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy SK, Rape M, Margansky WA, Kirschner MW. Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature. 2007;446:921–925. doi: 10.1038/nature05734. [DOI] [PubMed] [Google Scholar]
- Sigoillot FD, Lyman S, Huckins JF, Adamson B, Chung E, Quattrochi B, King RW. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat Methods. 2012;9:363–366. doi: 10.1038/nmeth.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Chan GK, Yen TJ. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol. 2001;154:925–936. doi: 10.1083/jcb.200102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- Tang Z, Bharadwaj R, Li B, Yu H. Mad2-independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev Cell. 2001;1:227–237. doi: 10.1016/s1534-5807(01)00019-3. [DOI] [PubMed] [Google Scholar]
- Teichner A, Eytan E, Sitry-Shevah D, Miniowitz-Shemtov S, Dumin E, Gromis J, Hershko A. p31comet promotes disassembly of the mitotic checkpoint complex in an ATP-dependent process. Proc Natl Acad Sci USA. 2011;108:3187–3192. doi: 10.1073/pnas.1100023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varetti G, Guida C, Santaguida S, Chiroli E, Musacchio A. Homeostatic control of mitotic arrest. Mol Cell. 2011;44:710–720. doi: 10.1016/j.molcel.2011.11.014. [DOI] [PubMed] [Google Scholar]
- Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, Belmont LD, Kaminker JS, O'Rourke KM, Pujara K, Kohli PB, Johnson AR, Chiu ML, Lill JR, Jackson PK, Fairbrother WJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- Westhorpe FG, Diez MA, Gurden MD, Tighe A, Taylor SS. Re-evaluating the role of Tao1 in the spindle checkpoint. Chromosoma. 2010;119:371–379. doi: 10.1007/s00412-010-0261-1. [DOI] [PubMed] [Google Scholar]
- Westhorpe FG, Tighe A, Lara-Gonzalez P, Taylor SS. p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J Cell Sci. 2011;124:3905–3916. doi: 10.1242/jcs.093286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westrate LM, Sayfie AD, Burgenske DM, MacKeigan JP. Persistent mitochondrial hyperfusion promotes G2/M accumulation and caspase-dependent cell death. PLoS ONE. 2014;9:e91911. doi: 10.1371/journal.pone.0091911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, Minna JD, Michnoff C, Hao W, Roth MG, Xie XJ, White MA. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446:815–819. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- Xia G, Luo X, Habu T, Rizo J, Matsumoto T, Yu H. Conformation-specific binding of p31comet antagonizes the function of Mad2 in the spindle checkpoint. EMBO J. 2004;23:3133–3143. doi: 10.1038/sj.emboj.7600322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Li B, Tomchick DR, Machius M, Rizo J, Yu H, Luo X. p31comet blocks Mad2 activation through structural mimicry. Cell. 2007;131:744–755. doi: 10.1016/j.cell.2007.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Liu Z, Zhao Y, Ding Y, Liu H, Xi Y, Xiong W, Li G, Lu J, Fodstad O, Riker AI, Tan M. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J Biol Chem. 2010;285:21496–21507. doi: 10.1074/jbc.M109.083337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.