

Abstract
Breast cancer is the second leading cause of death among women in the United States. Estrogens have been implicated as major risk factors in the development of breast neoplasms. Recent epidemiologic studies have suggested a protective role of phytoestrogens in prevention of breast and other cancers. Resveratrol, a naturally occurring phytoestrogen found notably in red grapes, berries and peanuts, has been shown to possess potent anti-cancer properties. However, the poor efficacy of resveratrol has prevented its use in a clinical setting. In order to improve the efficacy of resveratrol, we have synthesized a small combinatorial library of azaresveratrol analogs and tested them for their ability to inhibit the growth of breast cancer cell lines. We have recently shown that one of the synthesized analogs, 4-(E)-{(4-hydroxyphenylimino)-methylbenzene, 1, 2-diol} (HPIMBD), has better anti-cancer properties than resveratrol. The objective of this study was to investigate the differential regulation of estrogen receptors (ERs) α and β as a potential mechanism of inhibition of breast cancer by HPIMBD. Estrogen receptors α and β have been shown to have opposing roles in cellular proliferation. Estrogen receptor α mediates the proliferative responses of estrogens while ERβ plays an anti-proliferative and pro-apoptotic role. We demonstrate that HPIMBD significantly induces the expression of ERβ and inhibits the expression of ERα. HPIMBD also inhibits the protein expression levels of oncogene c-Myc, and cell cycle protein cyclin D1, genes downstream to ERα and important regulators of cell cycle, and cellular proliferation. HPIMBD significantly induces protein expression levels of tumor suppressors p53 and p21 in MCF-7 cells. Additionally, HPIMBD inhibits c-Myc in an ERβ-dependent fashion in MCF-10A and ERβ1-transfected MDA-MB-231 cells, suggesting regulation of ERs as an important upstream mechanism of this novel compound. Molecular docking studies confirm higher affinity for binding of HPIMBD in the ERβ cavity. Thus, HPIMBD, a novel azaresveratrol analog may inhibit the proliferation of breast cancer cells by differentially modulating the expressions of ERs α and β.
Keywords: Estrogen receptors, Breast cancer, Resveratrol, Resveratrol analogs
1. Introduction
Breast cancer is a serious public health concern worldwide. In the United States, breast cancer represents the most common neoplasm and the second most frequent cause of cancer deaths among women [1–3]. Estrogens have been implicated in the development of breast cancer and studies have shown that prolonged exposure to estrogens can cause tumorigenesis in breast tissues [4–7]. In 2001, steroidal estrogens have been added to the list of known human carcinogens [8]. In the last few decades, two major hypotheses have been proposed to explain the carcinogenic effects of estrogens. The first hypothesis suggests that steroidal estrogens bind to their cognate receptors and stimulate proliferation in cells, thus leading to cancer progression [9, 10]. The other is the metabolic hypothesis which states that genotoxic products of 17β-estradiol (E2) metabolism lead to oxidative DNA damage and initiation of cancer [4, 7, 11–14]. Both the ER-dependent and estrogen metabolism-mediated oxidative stress pathways have been accepted to explain the mechanisms of estrogen-induced carcinogenesis [4, 9].
Estrogen receptors are transcription factors, which belong to the nuclear receptor superfamily, that modulate expressions of specific genes upon activation by a ligand [15]. There are two types of ERs: the classical ERα and the second, ERβ [15]. Estrogen receptor α has different biological effects than ERβ, and both receptors show different intracellular and tissue distribution patterns [15]. Both, ERα and ERβ are crucial in regulating mammary growth and development [16, 17]. Under normal physiological conditions, ERα mediates the proliferative actions of E2 which can be opposed by ERβ and together, these receptors maintain a subtle balance of estrogen signaling in cells [18]. Estrogen receptor α is expressed in only 10–20 % of human mammary epithelial cells while 80–85 % cells express ERβ [19, 20]. In contrast, expression of ERα is increased while that of ERβ is decreased in breast cancer cells [20, 21]. The expression of ERα as a measure of steroid hormone receptor status is currently an acceptable prognostic marker that predicts response to hormonal therapy [22]. However, it is generally observed that breast tumors eventually progress to an estrogen-independent growth phenotype and become resistant to anti-estrogen therapy. This resistance is usually correlated with loss of the ERs over time [22]. The role of ERβ in breast cancer, however, is not well understood [23, 24]. It has been suggested that ERβ protein levels are linked to better prognosis, increased survival and better response to anti-estrogen therapy [24]. Induction of ERβ expression in breast cancer cells has been reported to inhibit estrogen-stimulated proliferation, tumor angiogenesis, and growth in xenograft studies [25]. It has also been suggested that ERβ may function as a tumor suppressor by antagonizing the proliferative effects of ERα [17, 26]. The ratio of ERα:ERβ is now being considered as a better prognostic marker in the diagnosis and therapy of breast cancers [21, 27]. A high ERα:ERβ ratio correlates with high levels of cellular proliferation, whereas predominance of functional ERβ over ERα correlates with lower levels of proliferation [21, 27]. Thus, future directions in developing newer therapeutic molecules against breast cancer may lie in their specific regulation of ERs α and β.
Several epidemiological and experimental studies have suggested that phytoestrogens present in fruits and vegetables represent a largely untapped source of potential anticancer molecules [28, 29, 30]. Recent epidemiologic evidence has suggested a protective role of phytoestrogens in reducing the incidence of breast cancer by early exposure to high phytoestrogen containing diets [28, 29, 30]. Most of these agents, mainly belong to one of the four classes, flavonoids, isoflavonoids, coumestanes and lignans [29]. Some of these phytoestrogens have shown higher binding affinity and transcriptional activation of ERβ over ERα [31, 32]. Resveratrol (Res, 3,5,4′-trihydroxystilbene) is one such naturally occurring phytoalexin found in a wide variety of plants, notably in fruits and nuts [33]. In the last two decades, Res has been extensively studied for its anti-cancer properties. Resveratrol has been shown to inhibit the growth of breast cancer cell lines such as MCF-7 and MDA-MB-231 in xenograft studies [33, 34]. The growth inhibitory effect of Res is attributed to induction of apoptosis and cell cycle arrest in cancer cells [33, 34]. Resveratrol acts as an anti-oxidant and scavenges reactive oxygen species produced by estrogen metabolism in breast cells, and thus, may prevent DNA damage and initiation of cancer [35, 36]. Resveratrol acts as an ERα antagonist and interferes with the proliferative effects of estrogen in ER-positive tumors [37]. Resveratrol-liganded ERβ has been shown to have higher transcriptional activity than E2-liganded ERβ and an anti-proliferative effect on cells expressing ERβ [38]. However, despite protective effects of Res observed in in vitro and xenograft studies, it has been difficult to demonstrate such effects in human studies [39].
To improve the antioxidant/antitumor efficacy of Res, we have recently synthesized a combinatorial library of five azaresveratrol analogs that resemble the basic skeleton of Res, but have additional pharmacophoric groups [40]. These novel azaresveratrol analogs were characterized, purified and screened for their anti-cancer activities against several breast cancer cell lines. One analog, 4-(E)-{(4-hydroxyphenylimino)-methylbenzene, 1, 2-diol} (HPIMBD), showed better potency than Res in inhibiting the proliferation of breast cancer cell lines [40]. In the present study, we investigated the effect of HPIMBD on the regulation of ERα and β. We present evidence that HPIMBD significantly induces the mRNA and protein expression levels of ERβ and inhibits that of ERα. We hypothesize that this could be one of the mechanism(s) by which HPIMBD inhibits the proliferation of breast cancer cells. We further demonstrate that HPIMBD significantly inhibits protein expression levels of oncogenes c-Myc and cyclin D1 and induces protein expression levels of tumor suppressors p53 and p21 in MCF-7 breast cancer cell line. Taken together, our studies suggest that HPIMBD, a novel analog of Res, inhibits breast cancer cell proliferation and differentially alters the expression of ERs, which may be one of the potential mechanisms of inhibition of breast cancer cell growth.
2. Materials and Methods
2.1. Chemicals
Resveratrol was purchased from Sigma-Aldrich (St. Louis, MO). Resveratrol analog, HPIMBD was synthesized and purified by our group as reported recently [40]. Doxycycline was purchased from Clontech (Mountain View, CA). Resveratrol and HPIMBD were dissolved in dimethyl sulfoxide (DMSO) prior to treatments. Doxycycline was dissolved in sterile purified water. The concentration of DMSO in control experiments was always 1/1000th (vol/vol) of the final medium volume. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) was purchased from Sigma-Aldrich (St. Louis, MO). A stock solution of MTT reagent was prepared by dissolving MTT in sterilized PBS to a final concentration of 1 mg/ml.
2.2. Cell Culture
Non-neoplastic breast epithelial cell line MCF-10A and breast cancer cell lines MCF-7, T47D and MDA-MB-231 were purchased from ATCC (Manassas, VA). Estrogen receptor β1-transfected MDA-MB-231 and empty vector-transfected MDA-MB-231 were a gift from Dr. Leigh C. Murphy (University of Manitoba, Canada). MCF-7, T47D, MDA-MB-231, empty vector-transfected MDA-MB-231 and ERβ1-transfected MDA-MB-231 cells were cultured in DMEM/F-12 (50:50) media (Mediatech, Herndon, VA) that was supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 1% penicillin/streptomycin antibiotic (Lonza, Allendale, NJ) while MCF-10A cells were cultured in DMEM/F-12 supplemented with 5% horse serum (Fisher Scientific, Pittsburgh, PA). Cells from respective cell lines were seeded in 96-well or 6-well tissue culture plates and were grown till they reached 70% confluency. Twenty four hours prior to treatments, cancer cells were washed twice with PBS and then grown in phenol red-free DMEM/F12 (50:50) medium supplemented with 10% charcoal dextran-stripped fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) while non-tumorigenic MCF-10A cells were grown in phenol red-free media supplemented with 5% charcoal dextran-stripped horse serum (Cocalico Biologicals, Reamstown, PA). Next day, cells were treated with 50 μM doses of Res or HPIMBD for up to 72 hours. Cells from ERβ1-transfected MDA-MB-231 and empty vector-transfected MDA-MB-231 were treated with doxycycline (final concentration 1μg/ml) for 24 hours prior to treatment with Res or HPIMBD, to induce the expression of ERβ from the Tet-on inducible expression vector. After treatments, the medium was removed and cells were washed once with PBS and used for either RNA or protein isolations. All treatments were done in quadruplicate. Experiments were performed in passages 2 to 6 of cells sub-cultured from frozen stocks of respective cell lines.
2.3. MTT cell viability assay
2,000 cells per well were seeded in 96-well plates overnight. Next day, cells were treated with 50 μM Res or HPIMBD for 72 hours. As a negative control, cells were treated with vehicle (DMSO) only. After treatments, cells were incubated with 50 μl of MTT (1 mg/ml) and incubated at 37°C for 3 hours. After dissolving the formazan crystals in DMSO, plates were read in Benchmark Plus microplate spectrophotometer (Biorad, Hercules, CA) at 570 nm and 650 nm. The absorbance at 650 nm was subtracted from absorbance at 570 nm. This value was then used to calculate mean % cell viability. Percentage of viable cells were calculated compared to controls and expressed as mean + SEM.
2.4. Lactate Dehydrogenase release cytotoxicity assay
This assay was performed using a colorimetric cytotoxicity LDH assay kit from Pierce (Thermo Fisher Scientific, Rockford, IL). Cells were plated at a concentration of 2,000 cells/well in 96-well plates, the day before the experiments. Next day, cells were treated with different doses of Res or HPIMBD for up to 72 hours. After end of treatments, medium from each well was collected to measure the amount of released lactate dehydrogenase (LDH), while separate wells were exposed to lysis buffer (9% Triton X-100) and were used to measure the total amount of intracellular LDH. Absorbance was measured at 490 and 680 nm using the Benchmark Plus microplate spectrophotometer from Biorad (Hercules, CA). The absorbance at 680 nm was subtracted from absorbance at 490 nm. This value was then used to calculate mean % released LDH. The amount of cell death was expressed as a percentage of released LDH over total intracellular LDH and was calculated as the mean + SEM of three different experiments.
2.5. BrdU Incorporation Assay
The Bromodeoxyuridine (BrdU) incorporation assay for normal breast epithelial and breast cancer cell lines was carried out using the FITC BrdU Flow Kit (BD Biosciences, San Jose, CA). Cells were plated at a concentration of 5 × 105 cells/well in 6-well plates. Next day, cells were treated with 50 μM Res or HPIMBD for 48 hours. Cells were incubated with BrdU (10 μM) for 20 hours. BrdU incorporating cells (cells in synthesis (S)-phase of cell cycle) were analyzed following manufacturer’s instructions. Briefly, cells were harvested, followed by fixation and permeabilization using BD Cytofix/Cytoperm Buffer. After one hour incubation with DNase at 37°C, cells were stained with FITC-conjugated anti-BrdU monoclonal antibody for 20 mins and analyzed by Flow Cytometry using BDFACS Canto II flow cytometer (BD Biosciences, San Jose, CA). Sample data were acquired using BD FACSDiva V6.1.1 operating software (BD Biosciences, San Jose, CA). The results are expressed as the % of cells that incorporated BrdU and are calculated as the mean + SEM of three different experiments.
2.6. Reverse transcription and Real-time PCR
Real-time PCR was used to quantify mRNA expression levels of ERα and ERβ. After treatments with Res or HPIMBD, total RNA from cultured cells was isolated using TRI reagent (Molecular Research Center, Inc., Cincinnati, OH) according to the supplier’s recommendations. Five μg total RNA was reverse transcribed using the superscript II reverse transcription system and an oligo-dT18 primer (Invitrogen, Carlsbad, CA). Real-time PCR was performed in duplicate 25 μl reactions using human specific ERα (QT00044492) and ERβ (QT00060641) primers (Qiagen, Valencia, CA) and using i-Cycler Real-time PCR System (Biorad, Hercules, CA). The expression of cyclophilin, a housekeeping gene, was used for quantification and standardization purposes, as reported previously [41, 42]. The expression levels of ERα or β relative to cyclophilin were determined by dividing the number of cDNA molecules for the gene of interest by the number of cyclophilin cDNA molecules. Standards were created for each gene and a standard curve was run on each plate to allow for accurate quantification of cDNA, as reported previously [13, 41, 43].
2.7. Western Blot analysis
Forty microgram total protein, isolated from quadruplicates of control or treated cells, was size fractionated on a 12% SDS-polyacrylamide gel, and transferred onto PVDF membranes under standard conditions [5, 13, 43–45]. Membranes were blocked in 5% dry non-fat milk/PBS/0.05% Tween-20 at four degrees in a refrigerator overnight. Affinity purified rabbit polyclonal antibodies against ERα (Santa Cruz, sc-543), ERβ (Santa Cruz, sc-8974), c-Myc (Santa Cruz, sc-788), cyclin D1 (Santa Cruz, sc-753), p53 (Santa Cruz, sc-55476) and p21 (Santa Cruz, sc-6264) were diluted in PBS/0.05% Tween-20 and used for immune-detection. Estrogen receptor β antibody was diluted 1:8000 while all other antibodies were diluted 1:2000. After incubation for six hours at room temperature with the respective primary antibodies, membranes were washed three times (8 minutes per wash) using PBS/0.05% Tween-20. Horse radish peroxidase conjugated anti-rabbit IgG (Santa Cruz, sc-2004) or anti-mouse IgG (Santa Cruz, sc-2005) was diluted in PBS/0.05% Tween-20 and used as a secondary antibody. After incubation for one hour at room temperature with respective secondary antibodies for each protein, the membranes were washed again as described above. Chemiluminescent detection was performed using the BM Chemiluminescence Detection kit (Roche, Indianapolis, IN) and the FluorChem HD2 Imaging system (Alpha Innotech Corporation, San Leandro, CA), with AlphaEaseFC Image Analysis software (Alpha Innotech Corporation, San Leandro, CA). Membranes probed for ERα, ERβ, c-Myc, cyclin D1, p53 and p21 were washed twice in PBS/0.05% Tween-20, stripped with Restore Western blot stripping buffer (Thermo Scientific, Rockford, IL) and re-incubated overnight at room temperature with α-tubulin mouse monoclonal antibody (Santa Cruz, sc-53030) diluted 1:2000 in PBS/0.05% Tween-20. Horse radish peroxidase-conjugated anti-rat IgG antibody (Santa Cruz, sc-2006) was diluted 1:2000 in PBS/0.05% Tween-20 and used as a secondary antibody for α-tubulin detection. Secondary antibody was incubated with the membrane for one hour at room temperature prior to chemiluminescent detection using the method described above.
2.8. RNA interference
Small interfering RNAs (siRNAs) for ERβ (sc-35325) and scrambled control siRNA-A (sc-37007) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). MCF-10A, ERβ1-transfected MDA-MB-231, MCF-7 and T47D cells were transfected with 1 nmol/l siERβ using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA). Scrambled siRNA (1 nmol/l) transfected cells were used as negative controls as described recently [5, 13]. Cells transfected with siERβ were treated with 50 uM HPIMBD for 12 hours (ERβ1-transfected MDA-MB-231), 24 hours (MCF-10A) and 48 hours (MCF-7 and T47D) and used for western blot analyses.
2.9. Molecular Docking
All the docking studies were carried out by using AutoDock Vina software [46] to evaluate the ligand interactions with crystal structure binding site of ERβ obtained from PDB ID (1YY4) [47]. The previous ligand in the active site was removed and the 3-D grid box generated with grid center co-ordinates and 40 × 40 × 40 point size considering active site residues included within it. All the docking studies were performed on default parameters of the Autodock Tools. Prior to docking, total Kollman and Gasteiger charges were added to the protein. Lamarckian GA was utilized for the search of best conformations. About 10 conformations for Res and HPIMBD were generated. Later, the most stable docking conformation of each compound was selected based upon best scores predicted by the scoring function and the lowest binding energy and visualized with Pymol [48].
2.10. Statistical analysis
Statistical analyses were performed by using Sigma Plot 11.0 software (Systat Software, San Jose, CA) and IBM SPSS Statistics 19 software (IBM, Armonk, NY). All cell culture treatments were done in triplicate or quadruplicate. Student’s t-test and one-way analysis of variance (ANOVA) with a least significant difference (LSD) post-hoc test were used to compare ERα and ERβ mRNA expression changes and ERα, ERβ, c-Myc, cyclin D1, p53 and p21 protein expression changes in treated cells compared to respective vehicle controls. These tests were also used to compare the changes in percent proliferation of cells, percent release of LDH and percent BrdU incorporation in treatments compared to respective vehicle controls. A p-value < 0.05 was considered significant.
3. Results
3.1. HPIMBD significantly inhibits proliferation of breast cancer cell lines
We have previously reported the effect of Res and HPIMBD on cellular proliferation of non-neoplastic breast epithelial cell line MCF-10A and breast cancer cell lines MDA-MB-231 and T47D using MTT assay [40]. As mentioned earlier [40], compound 3e, named HPIMBD in the current study, exhibited most potent activity against MDA-MB-231 cells (with 65–75% inhibition of cellular proliferation) and T47D cells (with 40–60% inhibition), while Res induced only 40% inhibition in both cell lines tested [40]. Also, HPIMBD did not affect the growth of MCF-10A cells at 50 μM concentration suggesting its non-toxicity towards normal breast cells and selectivity towards cancer cells [40]. In the present study, we evaluated the growth inhibitory effect of Res and HPIMBD on three other breast cancer cell lines, namely, MCF-7, vector-transfected and ERβ1-transfected MDA-MB-231 cell lines. Resveratrol treatment resulted in about 40% inhibition of cellular proliferation in MCF-7 and MDA-MB-231 vector-transfected cell lines while it inhibited 50% of cells in the ERβ1-transfected MDA-MB-231 cell line (Fig. 1a). HPIMBD treatment resulted in about 60% inhibition in MCF-7 cells and more than 70% inhibition in the vector-transfected and ERβ1-transfected MDA-MB-231 cells (Fig. 1a). These studies suggest that HPIMBD may be more effective in inhibiting the proliferation of breast cancer cells compared to the parent compound Res.
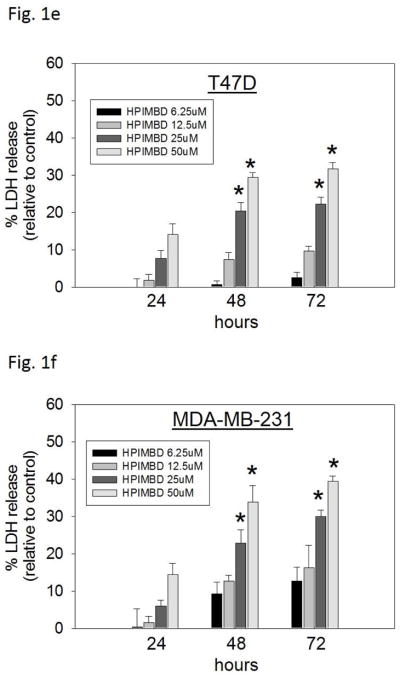
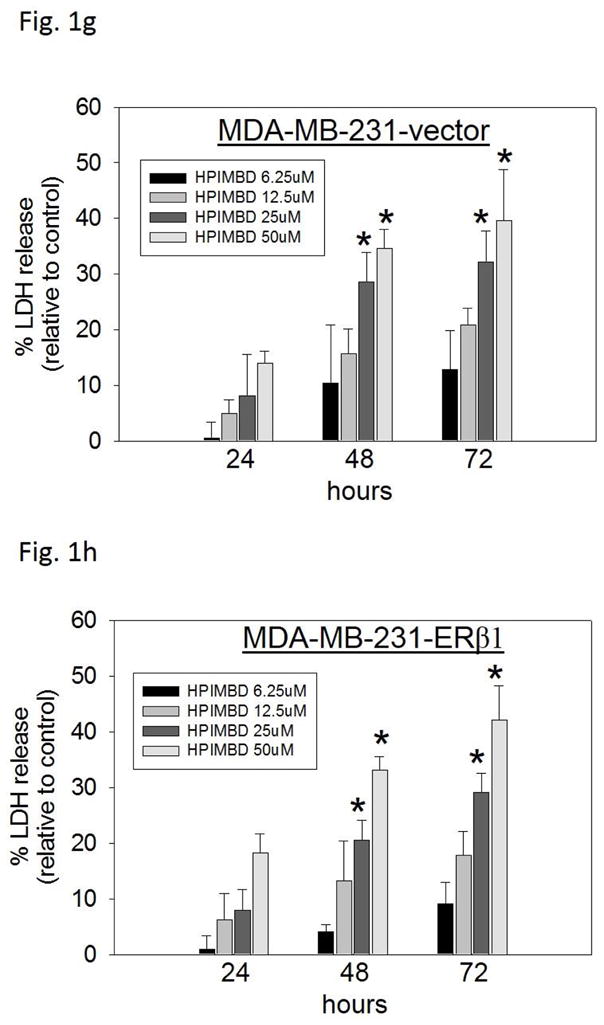
Fig. 1. HPIMBD significantly inhibits the proliferation of breast cancer cell lines and shows a dose- and time-dependent cytotoxicity.

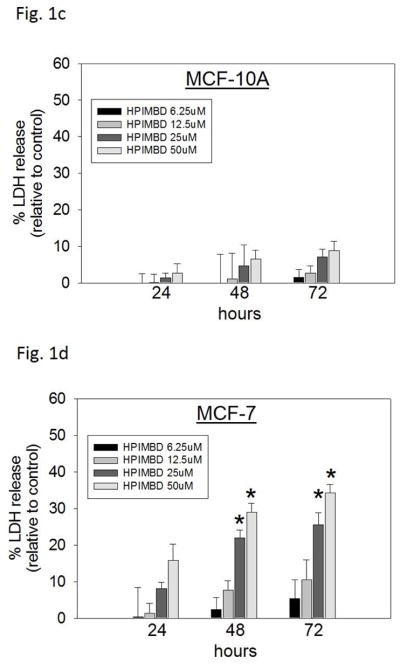

a) Non-neoplastic breast epithelial cell line MCF-10A and breast cancer cell lines MCF-7, T47D, MDA-MB-231, vector-transfected and ERβ1-transfected MDA-MB-231 were treated with vehicle (DMSO), 50 μM Res or HPIMBD for 72 hours and MTT assays were performed. Percentage proliferation was determined by dividing the absorbance in the HPIMBD- or Res-treated cells by that in vehicle-treated cells X 100. Each experiment was performed in quadruplicate and data are expressed as percentage proliferation + SEM relative to respective vehicle-treated controls.
b) LDH release assays were performed on non-neoplastic breast epithelial and breast cancer cell lines as described above. Percentage increase in LDH release was determined by dividing the difference of absorbances between HPIMBD- or Res-treated and vehicle-treated cells to the difference of absorbances between total intracellular LDH and vehicle-treated cells X 100. Each experiment was performed in triplicate and data are expressed as percentage LDH release + SEM relative to respective vehicle-treated controls (taken as 0%).
c–h) Non-tumorigenic breast epithelial cell line and breast cancer cell lines were treated with vehicle (DMSO) or graded doses of HPIMBD for up to 72 hours and LDH release assays were performed. Percentage increase in LDH release was determined as described above.
i) Non-neoplastic breast epithelial cell line and breast cancer cell lines were treated with vehicle (DMSO), 50 μM Res or HPIMBD for 48 hours and BrdU incorporation assays were performed. Each experiment was performed in triplicate and the data are expressed as percentage BrdU incorporation + SEM.
(*) indicates a P value <0.05 compared to controls.
3.2. Dose and time-dependent cytotoxicity induced by Res analog HPIMBD in breast cancer cell lines
Breast cancer cell lines MCF-7, T47D, MDA-MB-231, vector-transfected and ERβ1-transfected MDA-MB-231 were treated with different doses of HPIMBD (0, 6.25, 12.5, 25 and 50μM) and 50 μM Res for up to 72 hours. The extent of cell death was measured after 24, 48 and 72 hours of treatment by LDH release assays. The amount of LDH released from dying cells is directly proportional to cell death. HPIMBD at 50μM dose significantly caused about 30% increase in LDH release in MCF-7 and T47D cells and about 40% increase in the triple-negative MDA-MB-231, vector-transfected and ERβ1-transfected MDA-MB-231 cell lines after 72 hours of treatment (Fig. 1b). Resveratrol treatment also resulted in about 20% increase in LDH release in all the above mentioned breast cancer cell lines after 72 hours (Fig. 1b). To determine the effect of HPIMBD on cell death of normal breast epithelial cells, MCF-10A cells were treated with different doses of HPIMBD (0, 6.25, 12.5, 25 and 50 μM). HPIMBD up to 50 μM dose did not have any cytotoxic effect on non-neoplastic breast epithelial cells MCF-10A (Fig. 1c). A time-dependent study showed that HPIMBD did not cause significant cytotoxicity after 24 hours of treatment but significantly increased cytotoxicity at 48 hours which further increased after 72 hours in all the tested breast cancer cell lines (Fig. 1d–h). A dose-dependent study suggested that increasing doses of HPIMBD increased the cytotoxicity in all the breast cancer cell lines tested, with significant cytotoxic effects seen at 25 and 50 μM doses (Fig. 1d–h). These results suggest that HPIMBD can induce dose- and time-dependent cytotoxic effects in breast cancer cell lines.
3.3. HPIMBD significantly decreased BrdU incorporation in breast cancer cell lines
To assess the effect of HPIMBD on cellular proliferation, non-neoplastic breast epithelial cell line MCF-10A and breast cancer cell lines MCF-7, T47D, MDA-MB-231, vector-transfected and ERβ1-transfected MDA-MB-231 were treated with 50 μM Res or HPIMBD for 48 hours and BrdU incorporation was assessed. Resveratrol and HPIMBD did not affect the cellular proliferation of MCF-10A cells at 50 μM dose (Fig. 1i). However, both Res and HPIMBD caused a significant decrease in the percentage of breast cancer cell lines incorporating BrdU (Fig. 1i). The vehicle treated control cells of all the cell lines tested showed about 20–30% of BrdU incorporation (Fig. 1i). Resveratrol treatment resulted in about 11–17% of cancer cells incorporating BrdU, which amounts to about 40–50% decrease in BrdU incorporation compared to respective controls (Fig. 1i). HPIMBD was better than Res in inhibiting cellular proliferation. HPIMBD treatment resulted in about 4–7% of cancer cells incorporating BrdU, which amounts to 75–85% decrease in BrdU incorporation compared to respective controls (Fig. 1i). While Res treatment resulted in about ~2-fold decrease in the number of cells incorporating BrdU, HPIMBD treatment resulted in 4- to 6-fold decrease in BrdU incorporation in breast cancer cell lines (Fig. 1i). Taken together, our studies suggest that HPIMBD significantly inhibits the proliferation of breast cancer cells and does so more potently than that of Res.
3.4. HPIMBD significantly induces mRNA and protein expression levels of ERβ in breast cancer cells
Breast epithelial and breast cancer cell lines MCF-10A, MCF-7, T47D, MDA-MB-231 as well as ERβ1-transfected MDA-MB-231 and empty vector-transfected MDA-MB-231 were treated with 50 μM Res or HPIMBD for up to 72 hours. HPIMBD significantly induced ERβ mRNA levels in all cell lines tested compared to Res or control treated cells (Fig. 2a). MCF-7 and MDA-MB-231 cell lines showed ~4-fold whereas T47D cell line showed >2-fold increase in ERβ mRNA on treatment with HPIMBD whereas Res did not have any significant effect on expression levels of ERβ mRNA (Fig. 2a). HPIMBD significantly induced protein expression levels of ERβ, ranging from ~2 to ~4-fold, in all the breast cancer cell lines tested and modestly increased ERβ protein expression in MCF-10A cells by 1.3-fold (Fig. 2b). A time-course study was performed to determine time-dependent changes in expression levels of ERβ in MCF-7, T47D and MDA-MB-231 cell lines after treatment with HPIMBD or Res for up to 72 hours. HPIMBD treatment resulted in a maximal induction of ERβ 48 hours post-treatment, in both MCF-7 and T47D cell lines (Fig. 2c). However, a maximal induction of ERβ was observed at 12 hours post-treatment with HPIMBD in MDA-MB-231 cells (Fig. 2c). Time-course study with Res showed a significant induction of ERβ protein expression in T47D cell line about 2-fold at 24, 48 and 72 hours and in MDA-MB-231 cell line of about 2-fold only at 12 hours (Fig. 2c). However, Res did not induce ERβ expression in MCF-7 cell line (Fig. 2c). A dose-response study was performed to determine the effect of dose of HPIMBD on ERβ expression. MDA-MB-231 cells were treated with 25, 50 and 100 μM doses of HPIMBD and ERβ protein expression was assessed (Fig. 2d). HPIMBD induced protein expression levels of ERβ in a dose-dependent fashion with a maximal induction observed at a dose of 100 μM (Fig. 2d). There was only a slight difference in ERβ induction between 50 and 100 μM doses of HPIMBD. Therefore, a dose of 50μM was chosen for all protein expression studies.
Fig. 2. HPIMBD induces mRNA and protein expression levels of ERβ.
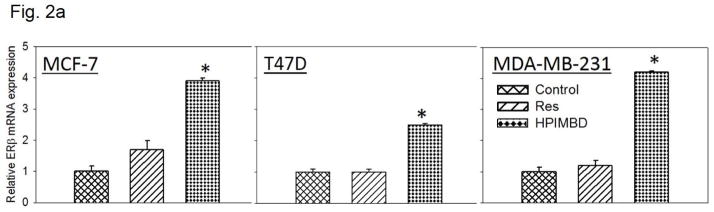


a) Breast cancer cell lines MCF-7, T47D and MDA-MB-231 were treated with vehicle (DMSO), 50 μM Res or HPIMBD for 24 hours. Total RNA was isolated and reverse transcribed to cDNA. Real-time quantitative PCR was performed to analyze the expression of cyclophilin and ERβ mRNA. Expression of ERβ mRNA was determined by dividing the number of cDNA molecules of ERβ by the number of cDNA molecules of cyclophilin, a housekeeping gene. Fold change was determined by dividing the expression of ERβ in the HPIMBD- or Res-treated cells by the expression of ERβ in vehicle-treated cells. Each experiment was performed in quadruplicate and the data are expressed as fold change + SEM relative to control.
b) Non-neoplastic breast epithelial MCF-10A and breast cancer cell lines MCF-7, T47D, MDA-MB-231, vector-transfected and ERβ1-transfected MDA-MB-231 were treated with vehicle (DMSO), 50 μM doses of Res or HPIMBD for up to 72 hours. Proteins were isolated and western blot analyses were performed as described in the Materials and Methods section. Intensities of the bands were quantified and normalized to α-tubulin. Fold changes in ERβ protein expression (Mean + SEM) in respective cell lines treated with Res or HPIMBD compared to vehicle-treated controls were calculated from four individual experiments and are given at the top of each blot. Representative western blots are shown for each cell line mentioned above at the time point of maximal induction of ERβ in that cell line (MCF10A = 24 hours; MDA-MB-231, vector-transfected and ERβ1-transfected MDA-MB-231 = 12 hours; MCF-7 and T47D = 48 hours).
c) MCF-7, T47D and MDA-MB-231 cell lines were treated with vehicle (DMSO), 50 μM Res or HPIMBD up to 72 hours and a time-course study was performed. Intensities of the bands were quantified and normalized to α-tubulin. Fold changes compared to vehicle-treated controls were calculated from four individual experiments. The bar graph represents fold change in ERβ protein expression (Mean + SEM) in respective cell lines treated with Res or HPIMBD compared to vehicle-treated controls.
d) MDA-MB-231 cells were treated with vehicle or 25 to 100 μM doses of HPIMBD and a dose-response study was performed. Intensities of the bands were quantified and normalized to α-tubulin. Fold changes in ERβ protein expression (Mean + SEM) compared to vehicle-treated control were calculated from four individual experiments and are given at the top of each blot.
(*) indicates a P value <0.05 compared to controls.
3.5. HPIMBD significantly inhibits mRNA and protein expression levels of ERα in breast cancer cells
MCF-7 and T47D breast cancer cells express ERα while MDA-MB-231 belongs to the class of triple negative breast cancer cell line and does not express ERα [49]. MCF-7 and T47D cells were treated with 50 μM Res or HPIMBD for up to 72 hours. HPIMBD significantly inhibited mRNA expression of ERα in MCF-7 and T47D cell lines compared to respective controls (Fig. 3a). HPIMBD also significantly inhibited protein expression levels of ERα in both MCF-7 and T47D cell lines (Fig. 3b). The protein inhibition was ~70% in T47D and ~50% in MCF-7 cell lines respectively (Fig. 3b). Resveratrol inhibited mRNA and protein expression levels of ERα in MCF-7 cells by 80% but had no significant effect on mRNA or protein expression levels of ERα in T47D cell line (Fig. 3a, b). A time-course study, performed in MCF-7 and T47D cell lines showed maximal inhibition of ERα protein expression levels at 48 hours post-HPIMBD treatment (Fig. 3c). Time-course study with Res showed a significant inhibition of ERα at 24 and 48 hours in MCF-7 cells but did not show any effect on ERα expression levels in T47D cell line (Fig. 3c). A dose-response study was also performed in T47D cell line after treatment with 25, 50 and 100 μM doses of HPIMBD. HPIMBD showed a dose-dependent inhibition of ERα expression levels with maximal inhibition of up to 90% with 100 μM dose (Fig. 3d).
Fig. 3. HPIMBD inhibits mRNA and protein expression levels of ERα.


a) Breast cancer cell lines MCF-7 and T47D were treated with vehicle (DMSO), 50 μM Res or HPIMBD for 24 hours. Total RNA was isolated and reverse transcribed to cDNA. Real-time quantitative PCR was performed to analyze the expression of cyclophilin and ERα mRNA. Expression of ERα mRNA was determined as described in the Materials and Methods section. Each experiment was performed in quadruplicate and the data are expressed as fold change + SEM relative to control.
b) Breast cancer cell lines MCF-7 and T47D were treated with vehicle (DMSO), 50 μM Res or HPIMBD for up to 72 hours. Proteins were isolated and western blot analyses were performed. Fold changes in ERα (Mean + SEM) compared to vehicle-treated controls were calculated from four individual experiments and are given at the top of each blot. Representative western blots are shown for MCF-7 and T47D cell lines after 48 hours of treatments, which is the time point of maximal inhibition of ERα in both the cell lines post-HPIMBD treatment.
c) MCF-7 and T47D cell lines were treated with vehicle (DMSO), 50 μM Res or HPIMBD for up to 72 hours and a time-course study was performed. Proteins were isolated and western blot analyses were performed. Intensities of the bands were quantified and normalized to α-tubulin. Fold changes compared to vehicle-treated controls were calculated from four individual experiments. The bar graph represents fold change in ERα protein expression (Mean + SEM) in respective cell lines treated with Res or HPIMBD compared to vehicle-treated controls.
d) T47D cells were treated with vehicle or 25 to 100 μM doses of HPIMBD for 48 hours and a dose-response study was performed. Proteins were isolated and western blot analyses were performed. Intensities of the bands were quantified and normalized to α-tubulin. Fold changes in ERα protein expression (Mean + SEM) compared to vehicle-treated controls were calculated from four individual experiments and are given at the top of each blot.
(*) indicates a P value <0.05 compared to controls.
The ratio of ERβ:ERα was determined as a ratio of fold protein expressions compared to respective controls, in MCF-7 and T47D cells after 48 hours of treatment using 50 μM Res or HPIMBD. HPIMBD showed a significant increase in ERβ:ERα ratio up to 7.5- and 9.6-fold as compared to 5- and 2.1-fold increase by Res in MCF-7 and T47D cell lines, respectively (Table 1).
Table 1.
Ratio of ERβ to ERα protein expressions in MCF-7 and T47D cell lines treated with Res or HPIMBD for 48 h. MCF-7 and T47D breast cancer cell lines were treated with vehicle (DMSO), 50 μM Res or HPIMBD for 48 h, which is the time point of maximal induction of ERβ and maximal inhibition of ERα in these cell lines. Proteins were isolated and western blot analyses were performed. Intensities of the bands were quantified and normalized to α-tubulin. Fold changes in ERβ or ERα protein expression (Mean ± SEM) treated with Res or HPIMBD compared to vehicle-treated controls were calculated from four individual experiments and ratios of ERβ:ERα were determined
| Cell line | Treatment | ERβ (fold change vs. control) | ERα (fold change vs. control) | Ratio (ERβ/ERα) |
|---|---|---|---|---|
| MCF-7 | Resveratrol | 1 | 0.2 | 5 |
| HPIMBD | 3.8 | 0.5 | 7.5 | |
| T47D | Resveratrol | 1.9 | 0.9 | 2.1 |
| HPIMBD | 2.9 | 0.3 | 9.6 |
3.6. HPIMBD inhibits protein expression levels of c-Myc and cyclin D1 in MCF-7 breast cancer cells
In order to determine the effect of induction of ERβ and inhibition of ERα by HPIMBD, on cellular proliferation, protein expression levels of markers of cellular proliferation c-Myc and cyclin D1 were investigated. MCF-7 cells were treated with 50μM HPIMBD or Res for 48 hours, the time point that showed a maximal induction of ERβ and maximal inhibition of ERα, respectively. HPIMBD significantly inhibited protein expression levels of c-Myc and cyclin D1 while Res did not have any significant effect at 50μM dose after 48 hours of treatment (Fig. 4a).
Fig. 4. HPIMBD inhibits protein expression levels of c-Myc and cyclin D1, induces p53 and p21 in MCF-7 cells.

a) Breast cancer cell line MCF-7 was treated with vehicle, 50 μM Res or HPIMBD for 48 hours. Proteins were isolated and western blot analyses were performed. Intensities of the bands were quantified and normalized to α-tubulin. Fold changes in c-Myc or cyclin D1 protein expression (Mean + SEM) treated with Res or HPIMBD compared to vehicle-treated controls were calculated from four individual experiments and are given at the top of each blot.
b) Breast cancer cell line MCF-7 was treated with vehicle, 50 μM Res or HPIMBD for 12 hours. Proteins were isolated and western blot analyses were performed. Intensities of the bands were quantified and normalized to α-tubulin. Fold changes in p53 or p21 protein expression (Mean + SEM) treated with Res or HPIMBD compared to vehicle-treated controls were calculated from four individual experiments and are given at the top of each blot.
(*) indicates a P value <0.05 compared to controls.
3.7. HPIMBD induces protein expression levels of tumor suppressor genes, p53 and p21, in MCF-7 breast cancer cells
Resveratrol is known to induce apoptosis by induction of tumor suppressor genes p53 and p21 in breast cancer cells [50]. In order to assess the effect of Res analog HPIMBD on p53 and p21, MCF-7 cells were treated with 50 μM HPIMBD or Res and, protein expression levels of p53 and p21 were determined. MCF-7 cell line was chosen for this study as this is the only cell line among those tested that expresses wild-type p53 [51]. Both Res and HPIMBD significantly induced protein expression levels of both p53 and p21 after 12 hours of treatments (Fig. 4b). HPIMBD showed a slightly higher induction of proteins p53 and p21 compared to Res (Fig. 4b).
3.8. HPIMBD inhibits protein expression of oncogene c-Myc via an ER-dependent pathway
HPIMBD treatment significantly decreased protein expression levels of oncogene c-Myc in MCF-7 breast cancer cells (Fig. 4a). We also determined this inhibitory effect on c-Myc expression by HPIMBD in other breast cancer cell lines. MCF-10A, T47D, MDA-MB-231, vector-transfected and ERβ1-transfected MDA-MB-231 cells were treated with either vehicle control or 50 μM doses of either Res or HPIMBD. HPIMBD significantly inhibited the protein expression levels of oncogene c-Myc in all the breast cancer cell lines tested, by 40–70% in MCF-10A, MDA-MB-231, vector-transfected and ERβ1-transfected MDA-MB-231 cells after 24 hours of treatment and by 60% in MCF-7 and T47D cells after 48 hours of treatment (Fig. 5a). Resveratrol modestly inhibited c-Myc expression in all the tested breast cancer cell lines (Fig. 5a). However, Res significantly inhibited the expression of c-Myc in the above tested breast cancer cells at later time points (data not shown). Next, we investigated whether HPIMBD inhibits c-Myc in an ERβ-dependent fashion. MCF10A, ERβ1-transfected MDA-MB-231, MCF-7 and T47D cell lines were transfected with siERβ for 48 hours, followed by treatment with HPIMBD for 12 hours (ERβ1-transfected MDA-MB-231), 24 hours (MCF-10A) or 48 hours (MCF-7 and T47D). Treatment time points were chosen based on maximal induction of ERβ in respective cell lines after HPIMBD treatment. Silencing ERβ significantly upregulated c-Myc protein expression in MCF-10A and ERβ1-transfected MDA-MB-231 cells but not in ERα-positive MCF-7 and T47D cells (Fig. 5b). Treatment with HPIMBD following siERβ transfection failed to inhibit c-Myc protein expression in MCF-10A and ERβ1-transfected MDA-MB-231 cells, suggesting that HPIMBD inhibits c-Myc via an ERβ-dependent pathway in these two cell lines (Fig. 5b). However, treatment with HPIMBD following siERβ transfection did not interfere with the ability of HPIMBD in inhibiting c-Myc protein expression in MCF-7 and T47D cells, suggesting that HPIMBD inhibits c-Myc in these two cell lines in an ERβ-independent fashion (Fig. 5b) most likely via the inhibition of ERα which we have shown to be down-regulated by HPIMBD at both mRNA and protein levels (Fig. 3a and b). It has already been reported that ERα regulates c-Myc expression in breast cells [52, 53].
Fig. 5. HPIMBD inhibits the expression of oncogene c-Myc in breast cancer cell lines.


a) Non-neoplastic breast epithelial cell line MCF-10A and breast cancer cell lines T47D, MDA-MB-231, vector-transfected and ERβ1-transfected MDA-MB-231 cells were treated with vehicle (DMSO), 50 μM Res or HPIMBD. Proteins were isolated and western blot analyses were performed. Intensities of the bands were quantified and normalized to α-tubulin. Fold changes in c-Myc protein expression (Mean + SEM) treated with Res or HPIMBD compared to vehicle-treated controls were calculated from four individual experiments and are given at the top of each blot.
b) MCF10A, ERβ1-transfected MDA-MB-231, MCF-7 and T47D cells were transfected with either 1 nmol/l of scrambled small interfering RNA or siERβ for 48 hours, and subsequently treated with 50 μM HPIMBD for 12 hours (ERβ1-transfected MDA-MB-231), 24 hours (MCF-10A) or 48 hours (MCF-7 and T47D). The treatment time points are based on maximal induction time of ERβ for respective cell lines following HPIMBD treatment. Proteins were isolated and western blot analyses were performed. Intensities of the bands were quantified and normalized to α-tubulin. Fold changes in ERβ or c-Myc protein expression (Mean + SEM) treated with scrambled, siERβ or HPIMBD compared to vehicle-controls were calculated from four individual experiments and are given at the top of each blot.
(*) indicates a P value <0.05 compared to controls.
3.9. HPIMBD binds ERβ with high affinity
Docking studies were carried out with Res and HPIMBD using protocols as reported previously [40]. Both Res and HPIMBD were found to dock into the active site of ERβ protein cavity with good binding energy and hydrogen bonding interactions (Figure 6 and Table 2). HPIMBD exhibited lowest binding energy (−8.6 Kcal/mol) and hence more stability as compared to Res (−8.2 Kcal/mol). The vicinal hydroxyl groups of HPIMBD were found to interact with GLY472 (2.5 Å) and HIS475 (2.4 Å) via hydrogen bonding, while other hydroxyl group forms hydrogen bonds with ARG346 (2.5 Å) and LEU339 (2.4 Å). Resveratrol exhibited hydrogen bonding with GLY472 (2.0 Å), GLU305 (2.2 and 2.3 Å) and ARG346 (2.3 Å) respectively. One hydroxyl group among 3,5-hydroxyl groups of Res was unable to interact with protein cavity, whereas adjacent 3,4-hydroxyl group of HPIMBD stabilized the compound in the protein cavity of ERβ, suggesting the importance of the adjacent 3,4-hydroxy group of HPIMBD over 3,5-hydroxyl group of Res in binding to ERβ (Fig. 6). Hence, the present docking study suggests that HPIMBD is better than Res for interacting with ERβ.
Fig. 6. Binding of Res and its analog HPIMBD into the active site of ERβ, as assessed by computer modeling studies.

Table 2.
Docking results and consensus scores of resveratrol and its analog HPIMBD in ERβ protein cavity
| Chemical | Binding energy (Kcal/mole) | Hydrogen bonding residues | Bond distance (Å) | No. of hydrogen bonds |
|---|---|---|---|---|
| Resveratrol | GLY472 | 2.0 | ||
| −8.2 | GLU305 | 2.2 and 2.3 | 4 | |
| ARG346 | 2.3 | |||
| HPIMBD | ARG346 | 2.5 | ||
| −8.6 | LEU339 | 2.4 | 4 | |
| GLY472 | 2.5 | |||
| HIS475 | 2.4 |
4. Discussion
Estrogen signaling has been extensively studied for the last 20 years, and the complexity of this classical signal transduction pathway is only beginning to be unraveled. It is well known that expression of ERα is increased and that of ERβ is decreased in early breast cancers [20, 21]. Loss of ERβ in cancer compared to normal tissue has led to the hypothesis that ERβ may function as a tumor suppressor [17, 26]. It is also postulated that ERα and ERβ have opposite biological actions, demonstrating a yin-yang relationship [18]. Recent evidence suggests that ERβ may negatively regulate cellular proliferation and have a protective role in normal breast and that it is the ratio of ERβ relative to ERα that may be important in determining the susceptibility of a tissue to estrogen-induced carcinogenesis and thus the outcome of anti-cancer therapy [17, 25, 26, 27].
The therapeutic potential of ERα has been utilized in the treatment of breast cancer for decades with the use of ERα antagonist tamoxifen [22, 54]. However, prolonged treatment with tamoxifen results in acquired resistance in almost one-third of the breast cancer patients [22]. Accumulating evidence indicates that ERβ can also be exploited for cancer prevention and therapy [24]. In the majority of clinical studies conducted, ERβ expression indicates a favorable response to adjuvant tamoxifen therapy, and patients with ERβ positive tumors appear to respond better to endocrine therapy and show an increased overall and disease-free survival, reduced disease progression and a lower risk of relapse [24]. Thus, ERβ has emerged as potential marker for predicting response to endocrine therapy [24].
With the identification of a second estrogen receptor subtype, ERβ in 1996, selectively targeting ERs for cancer treatment has evolved into a new field of identifying subtype selective ligands that are highly selective and potent for ERβ with minimal induction of ERα activity. Ligands with selectivity for ERβ show promise as cancer treatments given the anti-proliferative role of ERβ in many tissues [55]. ERβ is not yet targeted clinically for cancer treatment, but ERβ agonists hold therapeutic promise in cancers [55]. Such compounds could promote ERβ-mediated growth inhibition while avoiding proliferative effects of ERα [55]. Selective ERβ agonists may be used to stimulate the tumor suppressor function of ERβ [55]. Both natural and synthetic ERβ-selective ligands have been described. It has been suggested that selective ERβ agonists such as diarylpropionitrile (DPN) should be explored for the development of better hormone replacement therapy regimens to reduce or eradicate the risk of breast cancer [55].
Phytoestrogens are natural and dietary compounds that exhibit differential activation of ERs [29, 31, 32]. Unlike E2, which binds both ERβ and ERα with similar affinity, some phytoestrogens display a substantially higher affinity for ERβ [29, 31, 32]. The differential affinities of phytoestrogens for ERβ and ERα suggest that physiologic concentrations of phytoestrogens may be enough to activate ERβ but not ERα, implying that rather than acting via the classical ERα pathway, phytoestrogens may activate ERβ and induce its antiproliferative effects [29, 31, 32]. The greater affinity of some phytoestrogens for ERβ is thought to be of significant value in cancer prevention, as ERβ has been proposed to exert a protective role against cancer [24, 26, 27]. Resveratrol is one such unique phytoestrogen that is able to mimic the activity and effects of endogenous estradiol, which would seem to contradict the beneficial effects of Res on the breast. However, it has been shown that Res acts as a mixed agonist/antagonist on the ER and exhibits higher transcriptional activity when bound to ERβ than to ERα [38]. Moreover, Res seems to have antagonist activity with ERα but not with ERβ [37, 38]. The role of ERβ in breast cancer is not well understood and confounding factors such as ERα and ERβ homo and heterodimers and ERβ splice variants makes interpretation of results difficult [18, 23, 24].
A logical approach to improving bioavailability and thereby potentially enhancing the chemopreventive effect of Res, has been to make analogs of Res. Various analogs with substitutions like hydroxy-, methoxy-, fluoro-, acetate-, methyl- and methyl ether- on the parent Res backbone have been synthesized [56]. Methylated derivatives of Res have shown better bioavailability while polyhyroxyl substitutions have shown improved cytotoxicity and apoptotic ability [40, 56].
We have synthesized a few novel analogs of Res by including the aza functionality group in the conjugated system of Res stilbene [40]. Among these, one analog, HPIMBD, demonstrated better potency than Res in selectively inhibiting the proliferation of breast cancer cells [40]. In this study, we have demonstrated that HPIMBD significantly induces mRNA and protein expression levels of ERβ and inhibits that of ERα. It thus increased the ERβ to ERα ratio in breast cancer cell lines (Table 1). This may shift the balance of estrogen signaling in these cells predominantly from ERα-mediated proliferation to ERβ-mediated inhibition. This shift may have a significant effect on the proliferation of breast cancer cells. The ERβ1-transfected MDA-MB-231 cell line also showed induction of ERβ1 upon HPIMBD treatment. ERβ1 is the most abundant wild-type variant of ERβ [24]. The ERβ1 transgene is under the rTA-regulated promoter and not its natural promoter. The increase in mRNA and protein expression levels of ERβ could be because of increased stability of RNA or protein or as a result of transcriptional activation. These mechanisms need to be evaluated and will be examined in future studies. HPIMBD inhibits protein expression levels of genes downstream to ERα (Fig. 4). Proto-oncogene c-Myc is a well characterized target gene of ERα, a transcription factor that plays an important role in cellular proliferation in response to estrogens and mutations in this gene have been implicated in several cancers [52, 53]. Cyclin D1 is a cell cycle protein, a regulatory subunit of the cyclin-dependent protein kinases and functions mainly to regulate cell cycle progression [57]. Activation of cyclin D1 in response to an appropriate signal can rescue cells from the arrested phase and prevent them from going into apoptosis [57]. About 50–80 % of breast cancers overexpress cyclin D1 [58]. Cyclin D1 expression has been consistently correlated to ER-positivity in breast cancer cells and studies suggest an opposing effect of ERα and ERβ on cyclin D1 expression [17]. P53 is a well-known tumor suppressor and is often termed as the guardian of the genome [59]. P53 is a transcription factor that regulates apoptosis and DNA repair by regulating a number of genes in response to oncogenic insults and DNA damage [59]. P21, a downstream target of p53 and a cyclin dependent kinase inhibitor, is also involved in the regulation of cell cycle and apoptosis [59]. Inhibition of c-Myc and cyclin D1, and induction of p53 and p21 by HPIMBD suggests that HPIMBD may regulate cell cycle and apoptosis in breast cancer cells. However, more mechanistic studies need to be performed in order to dissect the exact mechanistic pathway of cell death induced by HPIMBD in breast cancer cell lines.
c-Myc is a well characterized target of ERα and, ERβ inhibits estrogen signaling by opposing the actions of ERα [17, 26, 52, 53]. Thus, ERβ may exert a repressive effect on c-Myc in breast cancer cells [19, 26]. Silencing of ERβ resulted in overexpression of c-Myc (Fig. 5b). HPIMBD was not able to inhibit the protein expression of c-Myc in the absence of ERβ (Fig. 5b) suggesting that ERβ may play an important role in inhibition of c-Myc and thus breast cancer cell proliferation by HPIMBD. However, this effect of ERβ was observed in MCF-10A and ERβ1-transfected MDA-MB-231 cell lines but not in ERα expressing MCF-7 and T47D cells (Fig. 5b). We hypothesize that this regulatory effect of ERβ on c-Myc is abrogated in the presence of the more dominant receptor ERα in MCF-7 and T47D cell lines. Therefore, even if ERβ is present in these cell lines, the co-expression of ERα may maintain the expression of c-Myc as c-Myc is a well-known target gene of ERα [52, 53]. Since silencing ERβ does not interfere with the ability of HPIMBD in inhibiting c-Myc expression, we suggest that HPIMBD may be inhibiting the expression of c-Myc as a direct effect of inhibition of ERα in ERα-expressing cell lines MCF-7 and T47D, as we have observed in the present study (Fig. 3b). Taken together, we conclude that HPIMBD may inhibit the proliferation of breast cancer cells by differential regulation of expressions of both ERs α and β.
In our previous report, we have shown that Res and HPIMBD docked efficiently into the protein cavity of ERα [40]. HPIMBD interacted with the protein cavity of ERα forming four hydrogen bonds with amino acids HIS524, ARG394 and GLU353, compared to Res which formed only one hydrogen bond with ARG394. The study suggested that hydroxyl groups of HPIMBD favor more hydrogen bonding in the protein cavity of ERα. However, the binding energy of Res in the ERα cavity (−8.6 Kcal/Mol) was slightly more than that of HPIMBD (−8.2 Kcal/Mol). In the present study, docking of HPIMBD and Res in ERβ protein cavity was investigated. HPIMBD showed a lower binding energy (−8.6 Kcal/mol) compared to Res (−8.2 Kcal/Mol), suggesting slightly more stable docking in ERβ cavity. HPIMBD also showed additional hydrogen bonding interactions (ARG346, LEU339, GLY472 and HIS475) that may confer more stability compared to Res (ARG346, GLY472 and GLU305) in ERβ protein cavity (Table 2). Also, the binding energy of HPIMBD was lower for ERβ (−8.6 Kcal/mol) compared to ERα (−8.2 Kcal/Mol), indicating that it may have higher affinity for binding with ERβ compared to ERα. Taken together, the docking results are in agreement with our experimental results. In summary, our current studies indicate that, HPIMBD, a novel analog of Res differentially affects ERs α and β in breast cancer cells and this may be one of the potential mechanisms for inhibiting the proliferation of breast cancer cell lines by HPIMBD. HPIMBD may, thus be useful as a therapeutic strategy in breast cancer treatment or prevention.
Highlights.
Novel resveratrol analog HPIMBD inhibits growth of breast cancer cells
HPIMBD induces protein expression levels of ERβ and inhibits that of ERα
HPIMBD inhibits c-Myc and cyclin D1, and induces p53 and p21
HPIMBD suppresses c-Myc in an ER-dependent fashion
Acknowledgments
This work was supported by the National Institutes of Health Grant (CA 109551), the University of Missouri Research Board Grant, and financial support from the School of Pharmacy, University of Missouri-Kansas City (HKB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Shinohara MM, Tozbikian G, Wolfe JT, Shin SJ, Mies C, Elenitsas R. Cutaneous metastatic breast carcinoma with clear cell features. J Cutan Pathol. 2013;40:753–757. doi: 10.1111/cup.12162. [DOI] [PubMed] [Google Scholar]
- 3.DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64:52–62. doi: 10.3322/caac.21203. [DOI] [PubMed] [Google Scholar]
- 4.Bhat HK, Calaf G, Hei TK, Loya T, Vadgama JV. Critical role of oxidative stress in estrogen-induced carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:3913–3918. doi: 10.1073/pnas.0437929100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh B, Bhat HK. Superoxide dismutase 3 is induced by antioxidants, inhibits oxidative DNA damage and is associated with inhibition of estrogen-induced breast cancer. Carcinogenesis. 2012;33:2601–2610. doi: 10.1093/carcin/bgs300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mense SM, Remotti F, Bhan A, Singh B, El-Tamer M, Hei TK, Bhat HK. Estrogen-induced breast cancer: alterations in breast morphology and oxidative stress as a function of estrogen exposure. Toxicol Appl Pharmacol. 2008;232:78–85. doi: 10.1016/j.taap.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yager JD, Liehr JG. Molecular mechanisms of estrogen carcinogenesis. Annu Rev Pharmacol Toxicol. 1996;36:203–232. doi: 10.1146/annurev.pa.36.040196.001223. [DOI] [PubMed] [Google Scholar]
- 8.Nelson R. Steroidal oestrogens added to list of known human carcinogens. Lancet. 2002;360:2053. doi: 10.1016/S0140-6736(02)12045-9. [DOI] [PubMed] [Google Scholar]
- 9.Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L. Cracking the estrogen receptor’s posttranslational code in breast tumors. Endocr Rev. 2011;32:597–622. doi: 10.1210/er.2010-0016. [DOI] [PubMed] [Google Scholar]
- 10.Singh B, Bhat NK, Bhat HK. Partial inhibition of estrogen-induced mammary carcinogenesis in rats by tamoxifen: balance between oxidant stress and estrogen responsiveness. PLoS One. 2011;6:e25125. doi: 10.1371/journal.pone.0025125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cavalieri EL, Rogan EG. A unifying mechanism in the initiation of cancer and other diseases by catechol quinones. Ann N Y Acad Sci. 2004;1028:247–257. doi: 10.1196/annals.1322.029. [DOI] [PubMed] [Google Scholar]
- 12.Patel MM, Bhat HK. Differential oxidant potential of carcinogenic and weakly carcinogenic estrogens: Involvement of metabolic activation and cytochrome P450. J Biochem Mol Toxicol. 2004;18:37–42. doi: 10.1002/jbt.20005. [DOI] [PubMed] [Google Scholar]
- 13.Singh B, Bhat NK, Bhat HK. Induction of NAD(P)H-quinone oxidoreductase 1 by antioxidants in female ACI rats is associated with decrease in oxidative DNA damage and inhibition of estrogen-induced breast cancer. Carcinogenesis. 2012;33:156–163. doi: 10.1093/carcin/bgr237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh B, Mense SM, Remotti F, Liu X, Bhat HK. Antioxidant butylated hydroxyanisole inhibits estrogen-induced breast carcinogenesis in female ACI rats. J Biochem Mol Toxicol. 2009;23:202–211. doi: 10.1002/jbt.20281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 16.Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- 17.Bartella V, Rizza P, Barone I, Zito D, Giordano F, Giordano C, Catalano S, Mauro L, Sisci D, Panno ML, Fuqua SA, Ando S. Estrogen receptor beta binds Sp1 and recruits a corepressor complex to the estrogen receptor alpha gene promoter. Breast Cancer Res Treat. 2012;134:569–581. doi: 10.1007/s10549-012-2090-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matthews J, Gustafsson JA. Estrogen signaling: a subtle balance between ER alpha and ER beta. Mol Interv. 2003;3:281–292. doi: 10.1124/mi.3.5.281. [DOI] [PubMed] [Google Scholar]
- 19.Fox EM, Davis RJ, Shupnik MA. ERbeta in breast cancer--onlooker, passive player, or active protector? Steroids. 2008;73:1039–1051. doi: 10.1016/j.steroids.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roger P, Sahla ME, Makela S, Gustafsson JA, Baldet P, Rochefort H. Decreased expression of estrogen receptor beta protein in proliferative preinvasive mammary tumors. Cancer Res. 2001;61:2537–2541. [PubMed] [Google Scholar]
- 21.Leygue E, Dotzlaw H, Watson PH, Murphy LC. Altered estrogen receptor alpha and beta messenger RNA expression during human breast tumorigenesis. Cancer Res. 1998;58:3197–3201. [PubMed] [Google Scholar]
- 22.Kuukasjarvi T, Kononen J, Helin H, Holli K, Isola J. Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy. J Clin Oncol. 1996;14:2584–2589. doi: 10.1200/JCO.1996.14.9.2584. [DOI] [PubMed] [Google Scholar]
- 23.Palmieri C, Cheng GJ, Saji S, Zelada-Hedman M, Warri A, Weihua Z, Van Noorden S, Wahlstrom T, Coombes RC, Warner M, Gustafsson JA. Estrogen receptor beta in breast cancer. Endocr Relat Cancer. 2002;9:1–13. doi: 10.1677/erc.0.0090001. [DOI] [PubMed] [Google Scholar]
- 24.Murphy LC, Leygue E. The role of estrogen receptor-β in breast cancer. Semin Reprod Med. 2012;30:5–13. doi: 10.1055/s-0031-1299592. [DOI] [PubMed] [Google Scholar]
- 25.Hartman J, Lindberg K, Morani A, Inzunza J, Strom A, Gustafsson JA. Estrogen receptor beta inhibits angiogenesis and growth of T47D breast cancer xenografts. Cancer Res. 2006;66:11207–11213. doi: 10.1158/0008-5472.CAN-06-0017. [DOI] [PubMed] [Google Scholar]
- 26.Williams C, Edvardsson K, Lewandowski SA, Strom A, Gustafsson JA. A genome-wide study of the repressive effects of estrogen receptor beta on estrogen receptor alpha signaling in breast cancer cells. Oncogene. 2008;27:1019–1032. doi: 10.1038/sj.onc.1210712. [DOI] [PubMed] [Google Scholar]
- 27.Shaaban AM, O’Neill PA, Davies MP, Sibson R, West CR, Smith PH, Foster CS. Declining estrogen receptor-beta expression defines malignant progression of human breast neoplasia. Am J Surg Pathol. 2003;27:1502–1512. doi: 10.1097/00000478-200312000-00002. [DOI] [PubMed] [Google Scholar]
- 28.Adlercreutz H. Phytoestrogens: epidemiology and a possible role in cancer protection. Environ Health Perspect. 1995;103(Suppl 7):103–112. doi: 10.1289/ehp.95103s7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mense SM, Hei TK, Ganju RK, Bhat HK. Phytoestrogens and breast cancer prevention: possible mechanisms of action. Environ Health Perspect. 2008;116:426–433. doi: 10.1289/ehp.10538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adlercreutz H. Phytoestrogens and breast cancer. J Steroid Biochem Mol Biol. 2002;83:113–118. doi: 10.1016/s0960-0760(02)00273-x. [DOI] [PubMed] [Google Scholar]
- 31.An J, Tzagarakis-Foster C, Scharschmidt TC, Lomri N, Leitman DC. Estrogen receptor beta-selective transcriptional activity and recruitment of coregulators by phytoestrogens. J Biol Chem. 2001;276:17808–17814. doi: 10.1074/jbc.M100953200. [DOI] [PubMed] [Google Scholar]
- 32.Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B, Gustafsson JA. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology. 1998;139:4252–4263. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 33.Athar M, Back JH, Kopelovich L, Bickers DR, Kim AL. Multiple molecular targets of resveratrol: Anti-carcinogenic mechanisms. Arch Biochem Biophys. 2009;486:95–102. doi: 10.1016/j.abb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garvin S, Ollinger K, Dabrosin C. Resveratrol induces apoptosis and inhibits angiogenesis in human breast cancer xenografts in vivo. Cancer Lett. 2006;231:113–122. doi: 10.1016/j.canlet.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 35.Cavalieri EL, Rogan EG. Unbalanced metabolism of endogenous estrogens in the etiology and prevention of human cancer. J Steroid Biochem Mol Biol. 2011;125:169–180. doi: 10.1016/j.jsbmb.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh B, Shoulson R, Chatterjee A, Ronghe A, Bhat NK, Dim DC, Bhat HK. Resveratrol inhibits estrogen-induced breast carcinogenesis through induction of NRF2-mediated protective pathways. Carcinogenesis. 2014;35:1872–1880. doi: 10.1093/carcin/bgu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhat KP, Lantvit D, Christov K, Mehta RG, Moon RC, Pezzuto JM. Estrogenic and antiestrogenic properties of resveratrol in mammary tumor models. Cancer Res. 2001;61:7456–7463. [PubMed] [Google Scholar]
- 38.Bowers JL, Tyulmenkov VV, Jernigan SC, Klinge CM. Resveratrol acts as a mixed agonist/antagonist for estrogen receptors alpha and beta. Endocrinology. 2000;141:3657–3667. doi: 10.1210/endo.141.10.7721. [DOI] [PubMed] [Google Scholar]
- 39.Walle T. Bioavailability of resveratrol. Ann N Y Acad Sci. 2011;1215:9–15. doi: 10.1111/j.1749-6632.2010.05842.x. [DOI] [PubMed] [Google Scholar]
- 40.Siddiqui A, Dandawate P, Rub R, Padhye S, Aphale S, Moghe A, Jagyasi A, Venkateswara Swamy K, Singh B, Chatterjee A, Ronghe A, Bhat HK. Novel Aza-resveratrol analogs: synthesis, characterization and anticancer activity against breast cancer cell lines. Bioorg Med Chem Lett. 2013;23:635–640. doi: 10.1016/j.bmcl.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Bhat HK, Epelboym I. Quantitative analysis of total mitochondrial DNA: competitive polymerase chain reaction versus real-time polymerase chain reaction. J Biochem Mol Toxicol. 2004;18:180–186. doi: 10.1002/jbt.20024. [DOI] [PubMed] [Google Scholar]
- 42.Bhat HK, Hacker HJ, Bannasch P, Thompson EA, Liehr JG. Localization of estrogen receptors in interstitial cells of hamster kidney and in estradiol-induced renal tumors as evidence of the mesenchymal origin of this neoplasm. Cancer Res. 1993;53:5447–5451. [PubMed] [Google Scholar]
- 43.Mense SM, Chhabra J, Bhat HK. Preferential induction of cytochrome P450 1A1 over cytochrome P450 1B1 in human breast epithelial cells following exposure to quercetin. J Steroid Biochem Mol Biol. 2008;110:157–162. doi: 10.1016/j.jsbmb.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh B, Chatterjee A, Ronghe AM, Bhat NK, Bhat HK. Antioxidant-mediated up-regulation of OGG1 via NRF2 induction is associated with inhibition of oxidative DNA damage in estrogen-induced breast cancer. BMC Cancer. 2013;13:253. doi: 10.1186/1471-2407-13-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh B, Ronghe AM, Chatterjee A, Bhat NK, Bhat HK. MicroRNA-93 regulates NRF2 expression and is associated with breast carcinogenesis. Carcinogenesis. 2013;34:1165–1172. doi: 10.1093/carcin/bgt026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z, Li Y, Ai C, Wang Y. In silico prediction of estrogen receptor subtype binding affinity and selectivity using statistical methods and molecular docking with 2-arylnaphthalenes and 2-arylquinolines. Int J Mol Sci. 2010;11:3434–3458. doi: 10.3390/ijms11093434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24:417–422. doi: 10.1007/s10822-010-9352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhat HK, Vadgama JV. Role of estrogen receptor in the regulation of estrogen induced amino acid transport of System A in breast cancer and other receptor positive tumor cells. Int J Mol Med. 2002;9:271–279. [PubMed] [Google Scholar]
- 50.Zhang S, Cao HJ, Davis FB, Tang HY, Davis PJ, Lin HY. Oestrogen inhibits resveratrol-induced post-translational modification of p53 and apoptosis in breast cancer cells. Br J Cancer. 2004;91:178–185. doi: 10.1038/sj.bjc.6601902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Polotskaia A, Hoffman S, Krett NL, Shanmugam M, Rosen ST, Bargonetti J. 8-Amino-adenosine activates p53-independent cell death of metastatic breast cancers. Mol Cancer Ther. 2012;11:2495–2504. doi: 10.1158/1535-7163.MCT-12-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang C, Mayer JA, Mazumdar A, Fertuck K, Kim H, Brown M, Brown PH. Estrogen induces c-myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP-1 transcription factor. Mol Endocrinol. 2011;25:1527–1538. doi: 10.1210/me.2011-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dadiani M, Seger D, Kreizman T, Badikhi D, Margalit R, Eilam R, Degani H. Estrogen regulation of vascular endothelial growth factor in breast cancer in vitro and in vivo: the role of estrogen receptor alpha and c-Myc. Endocr Relat Cancer. 2009;16:819–834. doi: 10.1677/ERC-08-0249. [DOI] [PubMed] [Google Scholar]
- 54.Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med. 1998;339:1609–1618. doi: 10.1056/NEJM199811263392207. [DOI] [PubMed] [Google Scholar]
- 55.Shanle EK, Xu W. Selectively targeting estrogen receptors for cancer treatment. Adv Drug Deliv Rev. 2010;62:1265–1276. doi: 10.1016/j.addr.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fulda S. Resveratrol and derivatives for the prevention and treatment of cancer. Drug Discov Today. 2010;15:757–765. doi: 10.1016/j.drudis.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 57.Musgrove EA, Lee CS, Buckley MF, Sutherland RL. Cyclin D1 induction in breast cancer cells shortens G1 and is sufficient for cells arrested in G1 to complete the cell cycle. Proc Natl Acad Sci U S A. 1994;91:8022–8026. doi: 10.1073/pnas.91.17.8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weinstat-Saslow D, Merino MJ, Manrow RE, Lawrence JA, Bluth RF, Wittenbel KD, Simpson JF, Page DL, Steeg PS. Overexpression of cyclin D mRNA distinguishes invasive and in situ breast carcinomas from non-malignant lesions. Nat Med. 1995;1:1257–1260. doi: 10.1038/nm1295-1257. [DOI] [PubMed] [Google Scholar]
- 59.Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network. J Cell Sci. 2003;116:4077–4085. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]