

Abstract
Restricted fetal growth is associated with postnatal mortality and morbidity and may be directly related to alterations in the capacity of the placenta to supply nutrients. We proposed previously that imprinted genes can regulate nutrient supply by the placenta. Here, we tested the hypothesis that the insulin-like growth factor 2 gene (Igf2) transcribed from the placental-specific promoter (P0) regulates the development of the diffusional permeability properties of the mouse placenta. Using mice in which placental-specific Igf2 had been deleted (P0), we measured the transfer in vivo of three inert hydrophilic solutes of increasing size (14C-mannitol, 51CrEDTA, and 14C-inulin). At embryonic day 19, placental and fetal weights in P0 conceptuses were reduced to 66% and 76%, respectively, of wild type. In P0 mutants, the permeability·surface area product for the tracers at this stage of development was 68% of that of controls; this effect was independent of tracer size. Stereological analysis of histological sections revealed the surface area of the exchange barrier in the labyrinth of the mouse placenta to be reduced and thickness increased in P0 fetuses compared to wild type. As a result, the average theoretical diffusing capacity in P0 knockout placentas was dramatically reduced to 40% of that of wild-type placentas. These data show that placental Igf2 regulates the development of the diffusional exchange characteristics of the mouse placenta. This provides a mechanism for the role of imprinted genes in controlling placental nutrient supply and fetal growth. Altered placental Igf2 could be a cause of idiopathic intrauterine growth restriction in the human.
Restricted fetal growth increases significantly the risk of mortality, neurodevelopmental handicap, and other morbidities in the neonatal period and in childhood (1, 2). Furthermore, small size at birth in human population studies is associated with increased risk of high blood pressure and abnormal glucose tolerance in adulthood (3). These associations can be reproduced in animal models of restricted in utero growth (4). A full understanding of the control of fetal growth would therefore make a significant impact on the burden of common diseases.
Fetal growth rate is determined by a number of factors, including maternal environment and nutrition, hormonal milieu, and maternal and paternal genotypes (5). Imprinted genes, those expressed from either the maternal or paternal allele, have a major influence on fetal growth. In particular, we proposed that imprinted genes expressed in the placenta would have significant roles in modulating nutrient supply to the fetus (6). The insulin-like growth factor 2 gene (Igf2) is expressed from the paternal allele and promotes fetal growth. Mouse fetuses in which the Igf2 gene has been completely deleted weigh ≈60% of wild-type fetuses (7). There are several transcripts of the Igf2 gene arising from the use of alternative promoters (P0, P1, P2, P3); one of these, the P0 transcript, is expressed only in the labyrinthine cell layers, which form the exchange barrier of the mouse placenta (8, 9). We have shown recently that knockout of this P0 transcript results in placental growth restriction, with mutants 68% of wild-type weight and fetal growth restriction with mutants 69% of wild type at birth (9). Remarkably, a large part of the phenotypic effect of complete Igf2 deletion can therefore be attributed to the lack of the placental transcript. Fetal growth restriction in knockout mice in which placental-specific Igf2 has been deleted (P0) is directly related to a decrease in the transfer capacity of the placenta, because transfer of an inert hydrophilic tracer 51CrEDTA was reduced per gram of placenta in mutants as compared to wild type. One explanation for this is that the diffusional permeability of the knockout placentas is reduced, impairing the transfer capacity of the placentas even further than would be expected from their reduced size.
Passive diffusion makes a quantitatively significant contribution to fluxes of all solutes across the placenta (10–12). In the human, up to 50% of the unidirectional flux of, for example, ions is via diffusion (12), and therefore any decrease in the passive permeability of the placenta to hydrophilic nutrients will have a significant impact on total transfer capacity and therefore fetal growth potential. Despite its importance in overall placental transfer capacity, however, it is not known how placental permeability is regulated. Passive permeability may be measured by using uncharged hydrophilic solutes, large enough for their transfer to be relatively unaffected by blood flow and unable to cross the placenta by any other mechanism. Fick's law of diffusion (see Methods) shows that regulation can theoretically result from alterations in the surface area and/or thickness of the exchange barrier. Additionally, a change in the width of the pore through which diffusion of the solutes occurs, altering the degree of steric hindrance, would also affect permeability (13). Therefore, there could be differential effects on molecules that depend on their size.
Here we test the hypothesis that labyrinthine Igf-II regulates the development of the diffusional permeability properties of the mouse placenta and ask which parameters of diffusion might be affected. We analyzed the transfer across the P0 mutant placenta of three inert hydrophilic solutes of increasing size (14C-mannitol, 51CrEDTA, and 14C-inulin) and showed that permeability is reduced in these animals. Stereology revealed a reduction in surface area and an increase in thickness of the exchange barrier in the labyrinth; this finding is consistent with the physiological measurements. Therefore, labyrinthine Igf2 expression regulates the development of the normal diffusional exchange characteristics of the mouse placenta.
Methods
Mice. P0 mice (9) were maintained in a C57BL/6J genetic background. For this study, C57BL/6J female mice were bred with heterozygous P0 males. Pregnant females were used at embryonic day (E)16 and E19 (E was defined as the day of vaginal plug detection). Mutant embryos were distinguished from wild types with a Southern blotting-based assay, as shown (9).
Measurement of Permeability·Surface Area Product (P·S). In the absence of an electrical gradient the rate of transfer of any solute by diffusion (Jnet) is described by Fick's law of diffusion (10):
 |
where A is the surface area of the exchange barrier, Dw is the diffusion coefficient in water at 37°C for the solute in question (inversely proportional to molecular size), l is the thickness of the exchange barrier, and Cm and Cf are the mean concentrations of the solute in the maternal and fetal plasma, respectively, across the length of the exchange barrier. For uncharged hydrophilic solutes for which transfer is relatively unaffected by blood flow and which cannot cross the placenta by any other mechanism, 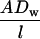 is equivalent to P·S with the units volume/time per gram of placenta (12). P·S is calculated by rearrangement of Fick's law and by using data from experiments where Jnet and (Cm–Cf) are measured. Determinations of Jnet and (Cm–Cf) are based usually on methodology first described by Flexner et al. (14) and which we have previously applied to the rat placenta (12, 15). Fetal accumulation of a tracer is measured at times after its injection into the maternal circulation when transfer is essentially unidirectional, i.e., there is no backflux (15), and Cf in Fick's equation can be ignored. The amount accumulated by the fetus (equivalent to the unidirectional maternofetal flux) is divided by the mean maternal plasma concentration over the course of the experiment, per unit time and per gram of placenta.
is equivalent to P·S with the units volume/time per gram of placenta (12). P·S is calculated by rearrangement of Fick's law and by using data from experiments where Jnet and (Cm–Cf) are measured. Determinations of Jnet and (Cm–Cf) are based usually on methodology first described by Flexner et al. (14) and which we have previously applied to the rat placenta (12, 15). Fetal accumulation of a tracer is measured at times after its injection into the maternal circulation when transfer is essentially unidirectional, i.e., there is no backflux (15), and Cf in Fick's equation can be ignored. The amount accumulated by the fetus (equivalent to the unidirectional maternofetal flux) is divided by the mean maternal plasma concentration over the course of the experiment, per unit time and per gram of placenta.
In the experiments reported here, mice at E16 or E19 (term is day 20/21) were anesthetized with fentanyl/fluanisone and midazolam. A neck incision was made and the jugular vein identified. A 100-μl bolus of saline containing 3.5 μCi 14C-mannitol (NEN NEC314; specific activity 53.7 mCi·mmol) or 70 μCi 51CrEDTA (NEN NEZ147; specific activity 324.7 mCi·mg) or 70 μCi 14C-inulin-carboxyl (ICN; specific activity 2.4 mCi·g) was then injected into the jugular vein via a short length of tubing attached to a 27-gauge needle; in some experiments, 14C-mannitol and 51CrEDTA were injected together into the same animal. At times up to 4 min after injection of tracer (studies showed there was insignificant tracer backflux at this time; data not shown), laparotomy was performed, a sample of maternal arterial blood taken, and mothers and fetuses killed. Fetuses and placentas were removed and separately weighed and the former then lysed overnight at 55°C in 2 ml (E16 fetuses) or 4 ml (E19 fetuses) of Biosol (National Diagnostics). Fractions of maternal plasma and fetal samples were then added to scintillation liquid (Bioscint, National Diagnostics) for β counting (Packard Tri-Carb 1900) or to appropriate tubes for γ counting (Packard Cobra 5005). For those samples that contained both 51Cr and 14C, time was allowed for the former to decay before β counting. Placental tissue was used for genotyping.
P·S for each tracer was calculated as:
 |
where Nx = counts in fetus taken at time x, when mother was killed; (AUC0-x) = area under curve, from time 0 to time of killing mother, derived from graph of maternal arterial counts vs. time, where each time point is given by the single sample from an individual mouse; W is the wet weight of the placenta.
Morphometry. Placentas from E19 mice (four litters; two wild type and two P0 from each) were weighed, hemisected, and corresponding halves fixed and embedded for generating paraffin wax or resin sections. The wax blocks were exhaustively sectioned at 7 μm. Measurements were carried out blind to genotype. Absolute placental volume was determined by point counting (see Fig. 3) on 10 systematic uniform random paraffin sections (1.25 × objective magnification) and applying the Cavalieri Principle, V(obj) = t·a(p)·ΣP, in which V(obj) is the estimated placental volume, t is the mean thickness between the sections (number of intervening sections multiplied by section thickness), a(p) is the area associated with each point, and ΣP is the sum of points falling on the sections (16). Tissue shrinkage was assessed by measuring the diameter of 100 randomly selected erythrocytes and comparing this value to that obtained from fresh maternal erythrocytes (17). Placental volume was corrected accordingly. Further point counting was done by using ×10 objective magnification to determine the absolute volume of the labyrinth zone.
Fig. 3.

Photomicrographs of a wild-type placenta (A) and a P0 knockout placenta (B) selected to illustrate thin and thick examples of the exchange barrier (between maternal blood space and fetal capillary). Also illustrated are grids used in stereological analysis. (A) Cycloid arcs and a line grid have been superimposed as used for the estimation of surface densities and harmonic mean thickness respectively. (B) A grid of test points has been superimposed as used for the estimation of volume fractions. MBS, maternal blood space; FC, fetal capillary; T, trophoblast. (Bar = 10 μm.)
A corresponding 1-μm resin section was taken across the center of each placenta perpendicular to the chorionic plate. Twelve fields were randomly selected within the labyrinth zone and viewed by using a ×100 objective lens. For fetal capillary and maternal blood space surface areas, a grid of cycloid arcs (see Fig. 3) was used to count intersects with the different component boundaries (18, 19). The harmonic mean labyrinth exchange barrier (consisting of three trophoblast layers, basement membrane, and fetal capillary endothelium, and sometimes called the interhemal membrane) thickness was measured by using orthogonal intercept lengths: starting points were determined by superimposed lines of random orientation (similar to those in Fig. 3) that intersect the fetal boundary of the barrier (19, 20). The shortest distance to the nearest maternal blood space boundary was measured and the harmonic mean thickness then calculated (19, 21). Tissue shrinkage for resin embedding was found to be <2%, so no corrections were applied. The theoretical diffusing capacity of the exchange barrier was determined by multiplying the mean area of the fetal and maternal surfaces by Krogh's diffusion coefficient for oxygen (17.3 × 10–8 cm2·min·kPa) (22) and dividing the result by the harmonic mean thickness.
Data Presentation and Statistical Analysis. Data were analyzed on a litter and genotype basis and are presented as mean ± SEM. A single average P·S value for mutants and a single average P·S value for wild types in each litter were calculated and the number of litters used in the calculation of SEM. P·S data were statistically analyzed by using Student's t test (paired or unpaired as appropriate) or by ANOVA. Morphometric variables were analyzed by ANOVA followed by the Fisher Protected Least Significant Difference post hoc test.
Results and Discussion
Placental and fetal weights for animals used in this study were (mean ± SEM): 0.074 ± 0.006 g and 0.411 ± 0.017 g, respectively, for P0 (85 conceptuses from 23 litters); 0.103 ± 0.008 g and 0.437 ± 0.017 g, respectively, for wild type (90 conceptuses from 23 litters) at E16; 0.061 ± 0.004 g and 0.947 ± 0.033 g, respectively, for P0 (95 conceptuses from 25 litters); and 0.092 ± 0.005 g and 1.245 ± 0.037 g, respectively, for wild type (89 conceptuses from 25 litters).
Systematic measurements of the permeability of the placenta have been made in several species, including sheep, human, and rat, but not in mouse. We therefore needed to establish the diffusional characteristics of the normal mouse placenta in vivo to determine which parameters are altered in the P0 placentas. Fig. 1 shows P·S values plotted against Dw for the three inert hydrophilic tracers in wild-type fetuses at E16 and E19. P·S was higher at E19 compared to E16. This was significant for the two larger tracers (51CrEDTA and 14C-inulin) and was proportional to the tracer size: 31-fold for 14C-inulin, 5-fold for 51CrEDTA, and 1.8-fold for 14C-mannitol. From classic pore theory (13), if all three tracers diffused across the exchange barrier (labyrinth trophoblast, basement membrane, and fetal endothelial layers of the mouse placenta) through extracellular water-filled channels (or pores) much wider than the molecules themselves, so there is no steric hindrance, then P·S normalized to Dw should be a constant. As shown in Table 1 P·S/Dw was significantly different between the three tracers at E16 and inversely related to size, whereas there was no significant difference at E19. Together, these data suggest that the permeability of the mouse placenta to hydrophilic solutes increases during normal pregnancy due at least partially to an increase in the radius of the extracellular “paracellular” diffusional pathway, therefore reducing restriction to diffusion.
Fig. 1.

P·S for wild-type mice plotted against Dw for 14C-mannitol (n = 7 litters at E16 and 7 litters at E19), 51CrEDTA (n = 19 litters at E16 and 18 litters at E19) and 14C-inulin (n = 7 litters at both gestations) at E16 (squares) and E19 (triangles). Data are shown as mean ± SEM (obscured by symbol for some points). **, P < 0.001 vs. respective E16 data (Student's t test).
Table 1. P·S/Dw values (cm/g of placenta) for wild-type and placental-specific (P0) Igf2 knockout placentas.
| P·S/Dw, cm/g placenta
|
|||||
|---|---|---|---|---|---|
| Dw (cm2/s × 106) | Wild type day 16 | P0 day 16 | Wild type day 19 | P0 day 19 | |
| 14C-mannitol | 9.9 | 32.7 ± 5.1 (7) | 24.6 ± 4.8 (7) | 57.3 ± 12.2 (6) | 39.1 ± 6.8 (6) |
| 51CrEDTA | 7.0 | 10.7 ± 2.0* (19) | 8.1 ± 1.8* (19) | 53.7 ± 5.4 (18) | 34.5 ± 3.9 (18) |
| 14C-inulin | 2.6 | 2.5 ± 0.3* (7) | 1.4 ± 0.2* (7) | 77.8 ± 14.9 (7) | 55.7 ± 12.6 (7) |
Dw, see ref. 15. Mean ± SEM is shown (number of litters). *, P < 0.001 vs. respective d16 mannitol (ANOVA followed by Bonferroni t test).
This work reports previously undescribed measurements of P·S for the mouse placenta. Previous studies have shown that hemochorial placentas such as the rat and human have P·S values for hydrophilic tracers one or two orders of magnitude higher than in epitheliochorial placentas such as the sheep (11). Our P·S values reported here for the hemochorial mouse placenta are higher but of the same order of magnitude as those for rat and human (12). There are small differences in technique that might account for the higher values here, such as the use of a maternal plasma isotope decay curve based on single values from individual mice rather than on multiple samples from the same animal as used in the rat (15); the small size of the mouse precluded the latter approach. However, the higher mouse P·S values could also represent a real species difference, so that passive diffusion makes an even greater contribution to placental transfer capacity than in the human or rat.
We next compared P·S values in P0 animals with wild-type animals for all three tracers at both gestations. As detailed in Methods, placental weights in P0 animals were 72% and 66% of wild type at E16 and E19, and fetal weights in mutants were 94% and 76% of wild type at the two gestations. These measurements are similar to those reported previously (9) with placental weight significantly lower in the P0 group at both gestations (P < 0.001) but fetal weight significantly lower only at E19. Deletion of the placental-specific Igf-2 transcript resulted in a significantly reduced P·S for 14C-inulin at E16 and in a significant reduction for all three tracers at E19 (Fig. 2). The P0 mice show a similar increase in P·S over gestation as the wild-type mice. Furthermore, at E16, the difference in P·S/Dw values between the three tracers in P0 mice is similar to that observed for wild-type mice (Table 1). At E19, there is no difference in P·S/Dw values for P0 mice, again as seen in the wild-type animals. These data together show that deletion of the P0 transcript results in a decreased permeability of the mouse placenta to hydrophilic solutes, and that this is due to a decrease in exchange barrier surface area or an increase in its thickness rather than any change in the radius of the paracellular diffusional pathway.
Fig. 2.

P·S for wild-type (wt) and P0 knockout mice. Note different scales of axes. Data are shown as mean + SEM, n as shown in Table 1. *, P < 0.05; **, P < 0.01; ***, P < 0.001 vs. respective wild-type group (paired Student's t test between wt and P0 groups at each gestation).
Stereological techniques were therefore used on histological sections to assess the association between the reduction in weight and P·S in the P0 knockout mice at E19 and possible changes in placental morphology. The morphology of the mouse placenta in wild-type and P0 animals is illustrated in Fig. 3. The absolute volume estimate, determined using the Cavalieri principle, for the P0 knockout placentas was 0.065 ± 0.005 cm3, a reduction in volume to 58% of wild-type littermates that averaged 0.112 ± 0.007 cm3 (P < 0.001). The maternal surface area of the labyrinth exchange barrier in the P0 placentas was 52% of that in wild type (P < 0.001): 15.38 ± 1.22 cm2 and 29.43 ± 1.66 cm2, respectively. For the fetal side-surface area, mutant placentas were reduced to 48% of wild type (P < 0.001): 14.88 ± 1.48 cm2 and 30.57 ± 2.56 cm2, respectively. The harmonic mean thickness of the labyrinth exchange barrier reported here emphasizes the presence of the thin areas of the exchange barrier that will contribute most to passive diffusion. In P0 knockout placentas, the value was 4.24 ± 0.13 μm compared with 3.33 ± 0.14 μm (P < 0.001) in their wild-type littermates, an increase in thickness of the mutant placentas to 128% of wild type.
The theoretical diffusing capacity of a membrane can be estimated from the available surface area for exchange and the distance (thickness) through which diffusible substances must travel to reach the opposite side of a given exchange barrier. The derived value does not equate with true placental diffusing capacity, because it does not take into account such factors as oxygen dissociation from the maternal erythrocytes, uptake by the fetal erythrocytes, and diffusion across the relevant plasma interfaces (21, 23). Theoretical diffusing capacity also reflects only structural parameters and so does not take into account changes in blood flow. Nonetheless, it is useful for comparison of the structural capabilities of exchange organs such as the placenta. Average theoretical diffusing capacity in P0 knockout placentas was 0.0063 ± 0.0006 cm2/min/kPa, a dramatic reduction in capacity to 40% of wild-type littermates whose diffusing capacity was 0.0158 ± 0.0013 cm2/min/kPa (P < 0.0001).
The genetic, physiological, and morphometric techniques used here have allowed us to show that the placental-specific transcript of Igf2 has a major role in the normal functional development of the mouse placenta and therefore in determining fetal size at birth. At E19, deletion of the P0 transcript resulted in a decrease to 66% of normal placental weight and a further decrease in permeability per gram of placenta to 68% of normal (derived from the data in Table 1), resulting in a total decrease in permeability of the mutant placenta to 45% of normal. These data are in striking agreement with the decrease to 40% of wild type, in theoretical diffusion capacity calculated from morphometric data. Together, the data show that placental-specific Igf2 is required for the attainment of normal placental size and of normal surface area and thickness of the labyrinthine layer where solute exchange takes place in the mouse. Because the decrease in placental permeability caused by deleting placental-specific Igf2 will apply equally to both maternal–fetal and fetal–maternal unidirectional fluxes, the overall effect on net flux for any particular solute will depend on the electrochemical gradient between maternal and fetal plasma. However, P0 fetuses are growth restricted at birth, demonstrating the importance of the decrease in placental diffusion capacity for the ability of the fetus to accumulate nutrients and grow. We conclude that maintenance of the normal diffusion capacity of the placenta, through correct development of the surface area and thickness of the exchange barrier, is a key mechanism by which Igf2 controls fetal growth. How precisely the placental Igf2 transcript affects growth of the placenta and its exchange barrier architecture remains to be determined in future studies, but for the time being, our observations serve to emphasize further the importance of imprinted genes for development in utero.
There have been no measurements of P·S for human placentas from fetuses that suffer intrauterine growth restriction (IUGR). The human placenta differs morphologically from that of the mouse but is of the same hemochorial type (24). It is therefore important to note that a reduction in placental volume and surface area of the exchange barrier has been reported in cases of IUGR (25, 26), proportionally similar to that found in the P0 placentas here. Although measurements of the harmonic mean thickness of the exchange barrier are not available for the human in IUGR, estimates based on the arithmetic mean thickness indicate that the diffusing capacity is reduced by ≈50% in these placentas (26). By analogy to our mice model, this would be a significant cause of the placental restriction of fetal growth in these cases. We propose that decreased expression and activity of trophoblast-specific Igf2 is a major cause of this structural defect and therefore of the decreased transfer capacity of the placenta that leads to idiopathic IUGR. Such a conclusion is consistent with recent data showing that low early-pregnancy levels of pregnancy-associated plasma protein A, a protease specific for IGF -binding proteins, are significantly associated with risk of IUGR (27).
Acknowledgments
This study was supported by grants from the Biotechnology and Biological Sciences Research Council and Medical Research Council. M.C. is a Babraham Career Progression Fellow. P.M.C. is funded by the Anatomical Society.
Abbreviations: Pn, Igf2 promoter n; P0, mice in which placental-specific Igf2 has been deleted; En, embryonic day n; P·S, permeability·surface area product; IUGR, intrauterine growth restriction.; Dw, diffusion coefficient in water at 37°C; IGF, insulin-like growth factor.
References
- 1.Hack. M.& Merkatz, I. R. (1995) New Engl. J. Med. 333, 1773–1774. [DOI] [PubMed] [Google Scholar]
- 2.Kjellmer, I., Liedholm, M., Sultan, B., Wennergren, M., Wallin Götborg, C. & Thordstein, M. (1997) Acta Paediatr. Suppl. 422, 83–84. [DOI] [PubMed] [Google Scholar]
- 3.Barker, D. J. P., Ericsson, J. G., Forsén, T. & Osmond, C. (2002) Int. J. Epidemiol. 31, 1235–1239. [DOI] [PubMed] [Google Scholar]
- 4.Bertram, C. E. & Hanson, M. A. (2001) Br. Med. Bull. 60, 103–121. [DOI] [PubMed] [Google Scholar]
- 5.Sparks. J. W., Ross, J. C. & Cetin, I. (1998) in Fetal and Neonatal Physiology Volume, eds. Polin, R. A., Fox, W. W. (Saunders, Philadelpia), 2nd Ed., pp. 267–289.
- 6.Reik, W., Constância, M., Fowden, A., Anderson, N., Dean, W., Ferguson-Smith, A., Tycko, B. & Sibley, C. (2003) J. Physiol. 547, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeChiara, T. M., Robertson, E. J. & Efstratiadis, A. (1991) Cell 64, 849–859. [DOI] [PubMed] [Google Scholar]
- 8.Moore, T., Constância, M., Zubair, M., Bailleul, B., Feil, R., Sasaki, H. & Reik, W. (1997) Proc. Natl. Acad. Sci. USA 94, 12509–12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Constância, M., Hemberger, M., Hughes, J., Dean, W., Ferguson-Smith, A., Fundele, R., Stewart, F., Kelsey, G., Fowden, A., Sibley, C. & Reik, W. (2002) Nature 417, 945–948. [DOI] [PubMed] [Google Scholar]
- 10.Sibley, C. P. & Boyd, R. D. H. (1988) in Oxford Reviews of Reproductive Biology, ed. Clarke, J. (Oxford Univ. Press, Oxford), Vol. 10, pp. 382–435. [PubMed] [Google Scholar]
- 11.Bain, M. D., Copas, D. K., Taylor, A., Landon, M. J. & Stacey, T. E. (1990) J. Physiol. 431, 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sibley, C. P. (1994) Placenta 15, 675–691. [DOI] [PubMed] [Google Scholar]
- 13.Stulc, J. (1989) Placenta 10, 113–119. [DOI] [PubMed] [Google Scholar]
- 14.Flexner, L. B. & Pohl, H. A. (1941) J. Cell Comp. Physiol. 18, 49–59. [Google Scholar]
- 15.Atkinson, D. E., Robinson, N. R. & Sibley, C. P. (1991) Am. J. Physiol. 261, R1462–R1464. [DOI] [PubMed] [Google Scholar]
- 16.Gundersen, H. J. G. & Osterby, R. (1981) J. Microsc. 12, 65–73. [DOI] [PubMed] [Google Scholar]
- 17.Burton, G. J. & Palmer, M. E. (1988) Placenta 9, 327–332. [DOI] [PubMed] [Google Scholar]
- 18.Baddeley, A. J., Gundersen, H. J. G. & Cruz-Orive L. M. (1986) J. Microsc. 142, 259–276. [DOI] [PubMed] [Google Scholar]
- 19.Coan, P. M., Ferguson-Smith, A. C. & Burton, G. J. (2004) Biol. Reprod., 70, in press. [DOI] [PubMed]
- 20.Jensen, E., Gundersen, H. J. G. & Osterby, R. (1979) J. Microsc. 115, 19–33. [DOI] [PubMed] [Google Scholar]
- 21.Burton, G. J. & Feneley, M. R. (1992) J. Dev. Physiol. 17, 39–45. [PubMed] [Google Scholar]
- 22.Laga, E. M., Driscoll, S. G. & Munro, H. N. (1973) Biol. Neonate 23, 231–259. [DOI] [PubMed] [Google Scholar]
- 23.Mayhew, T. M., Jackson, M. R. & Haas, J. D. (1986) Placenta 7, 121–131. [DOI] [PubMed] [Google Scholar]
- 24.Georgiades, P., Ferguson-Smith, A. C. & Burton, G. J. (2002) Placenta 23, 3–19. [DOI] [PubMed] [Google Scholar]
- 25.Jackson, M. R., Walsh, A. J., Morrow, R. J., Mullen, J. B. M., Lye, S. J. & Ritchie, J. W. K. (1995) Am. J. Obstet. Gynecol. 172, 518–525. [DOI] [PubMed] [Google Scholar]
- 26.Mayhew, T. M., Ohadike, C., Baker, P. N., Crocker, I. P., Mitchell, C., Ong, S. S. (2003) Placenta 24, 219–226. [DOI] [PubMed] [Google Scholar]
- 27.Smith, G. C., Stenhouse, E. J., Crossley, J. A., Aitken, D. A., Cameron, A. D. & Connor, J. M. (2002) J. Clin. Endocrinol. Metab. 87, 1762–1767. [DOI] [PubMed] [Google Scholar]