

The budding yeast kinase Hrr25 regulates two selective autophagy–related pathways by phosphorylating degradation target receptors and thereby promoting their interaction with Atg11 and the formation of autophagosomal membrane.
Abstract
In selective autophagy, degradation targets are specifically recognized, sequestered by the autophagosome, and transported into the lysosome or vacuole. Previous studies delineated the molecular basis by which the autophagy machinery recognizes those targets, but the regulation of this process is still poorly understood. In this paper, we find that the highly conserved multifunctional kinase Hrr25 regulates two distinct selective autophagy–related pathways in Saccharomyces cerevisiae. Hrr25 is responsible for the phosphorylation of two receptor proteins: Atg19, which recognizes the assembly of vacuolar enzymes in the cytoplasm-to-vacuole targeting pathway, and Atg36, which recognizes superfluous peroxisomes in pexophagy. Hrr25-mediated phosphorylation enhances the interactions of these receptors with the common adaptor Atg11, which recruits the core autophagy-related proteins that mediate the formation of the autophagosomal membrane. Thus, this study introduces regulation of selective autophagy as a new role of Hrr25 and, together with other recent studies, reveals that different selective autophagy–related pathways are regulated by a uniform mechanism: phosphoregulation of the receptor–adaptor interaction.
Introduction
Macroautophagy (hereafter, autophagy) is a highly conserved degradation pathway in which cellular constituents are sequestered by a double-membrane vesicle called the autophagosome and delivered to the lysosome (in mammalian cells) or the vacuole (in yeast and plant cells; Nakatogawa et al., 2009; Yang and Klionsky, 2010; Mizushima et al., 2011). Autophagy can occur in both nonselective and selective manners. Under starvation conditions, the autophagosome randomly sequesters a portion of the cytoplasm, and the degradation products are supplied as essential nutrient sources. On the other hand, when deleterious materials, such as protein aggregates, damaged mitochondria, or invasive bacteria arise within cells, the autophagosome enwraps them exclusively for elimination. These latter types of autophagy, termed selective autophagy, have attracted attention in recent years as a result of their direct relevance to human diseases (Deretic and Levine, 2009; Johansen and Lamark, 2011; Mizushima and Komatsu, 2011; Weidberg et al., 2011).
Studies in yeast have identified ∼40 autophagy-related (Atg) proteins, among which 15 proteins play fundamental roles in the formation of the autophagosomal membrane in any type of autophagy and are thus considered to be the core machinery. Some of the other Atg proteins regulate the core machinery and determine the targets of autophagosomal sequestration in selective autophagy. In the budding yeast Saccharomyces cerevisiae, the cytoplasm-to-vacuole targeting (Cvt) pathway is a selective autophagy–related pathway that mediates the transport of enzymes that function in the vacuolar lumen (Lynch-Day and Klionsky, 2010). Although the Cvt pathway is in essence a biosynthetic pathway, studies of this pathway have established the conceptual framework of target recognition during selective autophagy. In the Cvt pathway, the vacuolar aminopeptidase Ape1 self-assembles in the cytoplasm to form an aggregate-like structure that is recognized by the receptor protein Atg19 (Fig. 1 A; Kim et al., 1997; Scott et al., 2001). Atg19 also binds other vacuolar enzymes such as the aspartyl aminopeptidase Ape4 and the α-mannosidase Ams1, thereby incorporating them into the assembly (Hutchins and Klionsky, 2001; Shintani et al., 2002; Yuga et al., 2011). Atg19 then recruits the adaptor protein Atg11, which associates the assembly with the vacuolar membrane and recruits the core Atg proteins to initiate membrane formation (Shintani et al., 2002).
Figure 1.

Hrr25 is involved in the Cvt pathway. (A) Schematic model for the initiation of the Cvt pathway (see Introduction for details). (B) Yeast cells grown to mid–log phase (OD600 = ∼2) were treated with or without IAA for 30 min and examined by immunoblotting using anti-AID and anti-GFP antibodies. Pgk1 served as a loading control. (C) Cells grown to an OD600 of ∼1.0 were treated with or without IAA. The values of OD600 at each time point were plotted to draw the growth curve. The data shown are from a single representative experiment out of multiple experiments performed under similar conditions. (D, left) Cells expressing Ams1-GFP were grown to late log phase (OD600 = ∼4), treated with or without IAA for 2 h, and examined by immunoblotting using antibodies against Ape1 and GFP. GFP′ indicates GFP fragments produced by the vacuolar cleavage of Ams1-GFP. (right) The intensities of the bands corresponding to pApe1 and mApe1 were measured, and the ratios of mApe1 to total Ape1 are shown as the efficiencies of Ape1 maturation (percentages). Error bars represent standard deviations (n = 3). (E) Cells grown to mid–log phase were treated with or without IAA, further grown to post–log phase (OD600 = ∼6.0), and examined by immunoblotting using antibodies against Ape1. (F) Cells grown to mid–log phase were treated with IAA for 30 min. Ape1 maturation at the indicated time periods after the addition of 50 µg/ml cycloheximide was examined by immunoblotting using anti-Ape1 antibodies. WT, wild type.
Mitophagy and pexophagy are selective-autophagy pathways that target mitochondria and peroxisomes, respectively, and recent studies identified Atg32 and Atg36 as specific receptors for these pathways in S. cerevisiae (Kanki et al., 2009b; Okamoto et al., 2009; Motley et al., 2012). Both mitophagy and pexophagy use Atg11 as an adaptor; as with Atg19 in the Cvt pathway, Atg32 and Atg36 interact with Atg11 to trigger the corresponding pathways. In light of the increasing number of other examples, it is now generally accepted that autophagic receptors recruit the core Atg proteins to the appropriate targets, either via adaptors or directly, to initiate most selective autophagy pathways in organisms ranging from yeast to humans (Kirkin et al., 2009; Johansen and Lamark, 2011; Weidberg et al., 2011; Suzuki, 2013).
Under normal conditions, most of the protein and organelle targets of selective autophagy play essential roles in various cellular activities. Therefore, regulation of the initiation of their sequestration is critical; however, the mechanisms underlying these regulatory events remain elusive. In mitophagy and pexophagy, expression levels of Atg32 and Atg36 are increased under conditions that induce the corresponding pathway (Okamoto et al., 2009; Motley et al., 2012). This may be an aspect of the mechanism that regulates initiation of these pathways. However, the forced expression of the receptors under noninducing conditions does not lead to degradation of the target organelles significantly (Kanki et al., 2009b; Okamoto et al., 2009; Motley et al., 2012). This observation implied the existence of additional regulatory mechanisms that activate these pathways under appropriate conditions. Indeed, Atg32 and Atg36 are phosphorylated under mitophagy- and pexophagy-inducing conditions, respectively (Aoki et al., 2011; Motley et al., 2012). It was proposed that the phosphorylation of these receptors is important for the induction of each pathway, but the responsible kinases have remained unknown.
Hrr25 is a homologue of CK1δ (casein kinase 1δ) that phosphorylates Ser or Thr residues in proteins and is involved in a wide range of cellular functions, including the DNA damage response, chromosome segregation, ribosome biogenesis, and membrane traffic (Hoekstra et al., 1991; DeMaggio et al., 1992; Petronczki et al., 2006; Schäfer et al., 2006; Lord et al., 2011). Here, we show that Hrr25 phosphorylates Atg19 and Atg36 to initiate the Cvt pathway and pexophagy, respectively, by promoting their interactions with Atg11.
Results
Hrr25 is required for the Cvt pathway but not for starvation-induced autophagy
A global analysis of yeast kinase interactions indicated that Hrr25 interacts with Cvt pathway–related proteins, including Atg19 and Atg11 (Breitkreutz et al., 2010). Therefore, we investigated whether Hrr25 is involved in the Cvt pathway. Because Hrr25 is essential for yeast growth, we used the auxin-inducible degron (AID) system to conditionally knock down its expression. In this system, a protein fused with the AID tag is rapidly degraded by the ubiquitin–proteasome system, when cells are treated with the plant hormone auxin (Nishimura et al., 2009). Hrr25 was efficiently knocked down when cells expressing C-terminally AID-tagged Hrr25 (hrr25-aid cells) were treated with the auxin indole-3-acetic acid (IAA; Fig. 1 B). Under these conditions, the cells continued to grow at a slower rate (Fig. 1 C), which should be supported by a residual amount of the protein (Fig. 1 B). Using this strain, we examined the involvement of Hrr25 in the Cvt pathway. In wild-type cells, vacuolar transport of Ape1 via the Cvt pathway results in the removal of the propeptide from Ape1: specifically, Ape1 is converted from the proform (pApe1) to the mature form (mApe1) in the vacuole (Fig. 1 D). In cells lacking Atg19, Ape1 maturation was completely blocked, as reported previously (Scott et al., 2001). We found that hrr25-aid cells treated with IAA showed a remarkable defect in the maturation of Ape1 (Fig. 1 D). A milder but significant defect was also observed in hrr25-aid cells without IAA treatment, indicating that AID tagging itself affects the function of Hrr25 to some extent. Whereas we analyzed cells grown to late log phase in these experiments, the Cvt pathway is more active in later stages of growth; a larger proportion of Ape1 was matured in wild-type cells in post–log phase (Fig. 1 E; also see the following paragraph). We showed that a severe defect was also observed in Hrr25-depleted cells in this growth phase. In addition, we performed cycloheximide chase experiments to examine the kinetics of Ape1 maturation (Fig. 1 F). Most pApe1 was converted to the mature form during the chase period in wild-type cells, but this conversion was significantly retarded in Hrr25-depleted cells. We also monitored the vacuolar transport of another Cvt target, Ams1, by detecting GFP fragments generated by the cleavage of Ams1-GFP in the vacuole (Fig. 1 D). The results showed that depletion of Hrr25 also severely impaired vacuolar transport of Ams1. These results suggested that Hrr25 is required for the Cvt pathway.
We also examined the involvement of Hrr25 in starvation-induced autophagy. First, we observed the vacuolar transport of GFP-Atg8 (Suzuki et al., 2001; Shintani and Klionsky, 2004). Because Atg8 is transported into the vacuole via autophagy, GFP fluorescence in the vacuole increases in wild-type cells expressing GFP-Atg8 upon treatment with the autophagy-inducer rapamycin (Fig. 2 A). When the core Atg protein Atg2 was knocked down using the AID system, the vacuolar transport of GFP-Atg8 was strongly blocked. In contrast, knockdown of Hrr25 did not affect vacuolar transport of Atg8. The vacuolar cleavage of GFP-Atg8 results in the production of GFP fragments (GFP′), which are detected by immunoblotting using anti-GFP antibodies (Fig. 2, B and C; and Fig. S1, A and B). In agreement with the results of fluorescence microscopy, no significant decrease in the production of GFP fragments was observed in hrr25-aid cells treated with IAA, although Hrr25 was also efficiently knocked down in the presence of rapamycin. We also performed an alkaline phosphatase (ALP) assay to assess autophagic activity of cells (i.e., bulk degradation of the cytoplasm; Noda et al., 1995). Whereas knockdown of Atg2 significantly reduced autophagic activity induced by nitrogen starvation or rapamycin treatment, that of Hrr25 had no effect (Figs. 2 D and S1 C). These results suggest that Hrr25 is not required for starvation-induced, nonselective autophagy.
Figure 2.

Starvation-induced, nonselective autophagy normally occurs in Hrr25-depleted cells. (A) Cells expressing GFP-Atg8 were treated with rapamycin (rap) in the presence or absence of IAA for 3 h followed by fluorescence microscopy. Arrowheads indicate vacuoles. BF, bright field. Bars, 5 µm. (B) Cells used in A were also examined by immunoblotting using antibodies against GFP and AID. GFP′ indicates GFP fragments produced by the vacuolar degradation of GFP-Atg8. (C) The intensities of the bands in B were quantified, and GFP′ intensity was divided by the sum of GFP′ and GFP-Atg8 intensities to calculate processed GFP (percentages). Error bars represent standard deviations (n = 3). (D) Cells expressing Pho8Δ60 were incubated in nitrogen starvation medium in the presence or absence of IAA for the indicated time periods, and ALP activities in cell lysates were measured. The values for wild-type cells starved for 4 h in the absence of IAA were set as 100. Error bars represent standard deviations (n = 3). AU, arbitrary unit; WT, wild type.
Nitrogen starvation and rapamycin treatment promote vacuolar transport of Ape1, which largely depends on Atg19 and Atg11 as well as that under normal conditions (i.e., the Cvt pathway). We showed that Hrr25 is also important for Ape1 transport in cells treated with rapamycin (Fig. S1 D).
Hrr25 promotes Atg11 targeting to the Ape1–Atg19 complex
We next sought to determine which step in the Cvt pathway (Fig. 1 A) was affected by Hrr25 depletion. The self-assembly of Ape1 can be observed under a fluorescence microscope as the formation of dots of mRFP-Ape1; this process was not affected by the knockdown of Hrr25 (Fig. 3 A). In wild-type cells, the receptor protein Atg19 localizes to Ape1 dots, and this step was also unaffected by Hrr25 knockdown (Fig. 3 A). In the following step, the adaptor protein Atg11 is recruited to the Ape1–Atg19 complex and thus colocalizes with Ape1 dots in wild-type cells (Fig. 3 B). In Hrr25-depleted cells, this step was significantly impaired, and Atg11 was dispersed throughout the cytoplasm.
Figure 3.

Hrr25 promotes Atg11 targeting to the Ape1–Atg19 complex by strengthening the Atg19–Atg11 interaction. (A and B) Yeast cells coexpressing mRFP-Ape1 and either Atg19-GFP (A) or Atg11-GFP (B) were treated with or without IAA for 2 h and observed under a fluorescence microscope. Ape1 dots colocalized with Atg19 or Atg11 were counted, and their percentages relative to the total numbers of Ape1 dots are shown with standard deviations (n = 3). BF, bright field. Bars, 5 µm. (C) Cells expressing Atg19-GFP and Atg11-3×HA were treated with or without IAA for 2 h. The cell lysates (total) were subjected to immunoprecipitation using the anti-GFP antibody, and the immunoprecipitates (IP) were analyzed by immunoblotting using antibodies against Atg19, HA, and Ape1. Open arrowheads indicate phosphorylated Atg19. WT, wild type.
The recruitment of Atg11 is a prerequisite for the recruitment of core Atg proteins, such as Atg1 and Atg8, and as expected, the localization of these proteins to Ape1 dots was also compromised in Hrr25-depleted cells (Fig. S2, A–C). A previous study reported that the Ape1–Atg19 complex is detached from the vacuole in the absence of Atg11 (Shintani et al., 2002). Consistent with that finding, knockdown of Hrr25 caused detachment of Ape1 dots from the vacuole, similar to the situation in atg11Δ cells, although it did not affect the total amount of Atg11 (Fig. S2, D and E). Collectively, these data suggested that Hrr25 depletion impairs Atg11 targeting to the Ape1–Atg19 complex, resulting in the impairment of the Cvt pathway.
Hrr25 phosphorylates the COP II component Sec23 to regulate vesicle traffic from the ER to the Golgi (Lord et al., 2011), and both the Cvt pathway and starvation-induced autophagy are defective in sec23 mutant cells (Ishihara et al., 2001). Therefore, we investigated whether the depletion of Hrr25 indirectly affected the Cvt pathway via a defect in Sec23 function. To this end, we knocked down Sec23 using the AID system and confirmed that both the Cvt pathway and starvation-induced autophagy were impaired in Sec23-depleted cells (Fig. S3, A and B). However, in contrast to the situation in Hrr25 knockdown cells, Atg11-GFP substantially localized to mRFP-Ape1 dots in Sec23 knockdown cells (Fig. S3 C). Thus, defective Atg11 targeting to the Ape1–Atg19 complex in Hrr25-depleted cells cannot be attributed to compromised Sec23 function.
Hrr25-mediated Atg19 phosphorylation promotes the Atg19–Atg11 interaction
Because Atg11 recruitment to the Ape1–Atg19 complex is mediated by the direct interaction between Atg19 and Atg11 (Shintani et al., 2002), we next asked whether the knockdown of Hrr25 would affect this interaction. Immunoprecipitation of Atg19 coprecipitated Atg11 as well as pApe1 in wild-type cells (Fig. 3 C). Hrr25 knockdown markedly reduced the coprecipitation of Atg11 without affecting that of pApe1, suggesting that Hrr25 is required for the Atg19–Atg11 interaction. In these experiments, additional bands were observed above the major bands of both wild-type Atg19 and Atg19-GFP in wild-type cells but not in hrr25-aid cells treated with IAA (Fig. 3 C). These additional bands disappeared when the immunoprecipitates were treated with protein phosphatase (Fig. 4 A), suggesting that they represented phosphorylated forms of Atg19 and that Hrr25 is involved in Atg19 phosphorylation. We expressed GST-fused Atg19 and Hrr25 in Escherichia coli and subjected the purified proteins to an in vitro kinase assay. Recombinant Hrr25 phosphorylated GST-Atg19 but not GST alone (Fig. 4 B). In addition, the phosphorylation of GST-Atg19 was not observed when the kinase-dead mutant of Hrr25 (K38A; Hoekstra et al., 1991; Murakami et al., 1999) was used. Collectively, these results suggest that Hrr25 directly phosphorylates Atg19 in the Cvt pathway.
Figure 4.

Hrr25-mediated phosphorylation in the Atg11-binding region of Atg19 promotes the Atg19–Atg11 interaction. (A) Immunoprecipitates obtained from yeast cells expressing Atg19-GFP using anti-GFP antibody were treated with λ protein phosphatase in the presence or absence of phosphatase inhibitors and analyzed by immunoblotting using anti-Atg19 antibodies. (B) Recombinant GST and GST-Atg19 were incubated with wild-type Hrr25 (WT) or the K38A mutant (KA) in the presence of ATP and visualized by immunoblotting using anti-GST antibody. Protein phosphorylation was detected using the phosphate-binding reagent Phos-tag. The asterisk indicates a nonspecific band that appeared independently of Hrr25 activity in the Phos-tag staining. (C) The amino acid sequence of the Atg11-binding region in Atg19. Ser and Thr residues are colored blue. (D) Ape1 maturation in atg19Δ cells carrying single-copy Atg19 plasmids or the empty vector was monitored as described in Fig. 1 D. (E) ATG11-3×HA atg19Δ cells carrying single-copy Atg19-GFP plasmids were subjected to immunoprecipitation (IP) as described in Fig. 3 C. (F and G) mRFP-APE1 atg19Δ cells carrying single-copy Atg19-GFP plasmids (F) or Atg11-GFP mRFP-APE1 atg19Δ cells carrying single-copy Atg19 plasmids (G) were observed under a fluorescence microscope. Bars, 5 µm. (H) hrr25-aid atg1Δ atg19Δ cells expressing Atg19 mutants from single-copy plasmids were treated with or without IAA for 2 h, and Atg19 phosphorylation was analyzed by SDS-PAGE using the Anderson gel system and immunoblotting using anti-Atg19 antibodies. Open arrowheads indicate phosphorylated Atg19. (I) In vitro kinase assay was performed using GST-Atg19 mutants and wild-type Hrr25 as described in B; the reaction mixtures were incubated for 30 min. (J) In vitro kinase assay was performed using GST-Atg11 and wild-type Hrr25 as described in B. WT, wild type.
We further investigated Hrr25-mediated phosphorylation sites on Atg19. A previous study showed that deletion of eight residues in the C-terminal region of Atg19 (amino acids 388–395) abolishes its binding to Atg11, indicating that Atg19 interacts with Atg11 via this region (Fig. 4 C; Shintani et al., 2002). We hypothesized that Hrr25 phosphorylates a Ser or Thr residue in this region and that this modification increases the affinity of Atg19 for Atg11. Ser and Thr residues in this region were replaced with Ala individually, and the maturation of Ape1 in cells expressing these Atg19 mutants was examined (Fig. 4 D). We found that the S391A and S394A mutations caused the severe defect in Ape1 maturation. Immunoprecipitation analysis showed that both of these mutants were defective in the interaction with Atg11 (Fig. 4 E). Consistent with these results, localization of Atg11 to Ape1 dots was significantly decreased in both of the mutants, although the mutant proteins themselves efficiently localized to Ape1 dots (Fig. 4, F and G). To analyze the phosphorylation state of the nontagged Atg19 mutants, we used the Anderson gel system (Anderson et al., 1973), which allowed the clear separation of their phosphorylated and unphosphorylated forms (Fig. 4 H). Both Ser391 and Ser394 are within the motif for phosphorylation by CK1 (Xue et al., 2008). However, the S391A mutation, but not S394A, attenuated the Hrr25-dependent phosphorylation of Atg19. The phosphorylation state of the S391A S394A double mutant (S391/S394A) was similar to that of the S391A single mutant. In addition, an in vitro kinase assay showed that Hrr25 phosphorylated the S391A mutant to a lesser degree, whereas the S394A mutant was phosphorylated comparably to the wild-type protein (Fig. 4 I). Introducing the S394A mutation did not decrease the phosphorylation level of the S391A mutant. Collectively, these data suggest that Hrr25 phosphorylates Ser391 in the Atg11-binding region of Atg19, thereby increasing its affinity for Atg11 and promoting Atg11 targeting to the Ape1–Atg19 complex. Ser394 is important for the Atg19–Atg11 interaction but may not be phosphorylated. It should be noted that Hrr25-dependent phosphorylation was still observed in the S391A mutant (Fig. 4, H and I), suggesting that Hrr25 also phosphorylates other residues of Atg19 in addition to Ser391.
Because a previous mass spectrometric analysis identified not only Atg19 but also Atg11 in immunoprecipitates of Hrr25 (Breitkreutz et al., 2010), we examined the possibility that Hrr25 phosphorylates Atg11. In the immunoblotting analysis of yeast cell lysates, a band shift that might represent Atg11 phosphorylation was not observed (Fig. 3 C). In addition, an in vitro analysis showed that Hrr25 scarcely phosphorylated GST-Atg11 in contrast to GST-Atg19 (Fig. 4 J). These results suggest that Hrr25 does not phosphorylate Atg11. Our immunoblotting analysis of the immunoprecipitate of Hrr25 detected Atg19 but not Atg11 (Fig. S4). Atg11 indirectly bound to Hrr25 via Atg19 might be identified in the previous analysis.
Atg19 phosphorylation by Hrr25 activates the Cvt pathway in late growth phases
The Cvt pathway has often been described as constitutively active. However, in fact, the mature form of Ape1 increases as the culture approaches the end of logarithmic growth (Figs. 5 A and 1 C). We also performed cycloheximide chase experiments to examine the kinetics of the vacuolar transport of Ape1 in cells in different growth phases (Fig. 5 B). When wild-type cells were grown to later phases, the expression level of pApe1 increased, and the amount of pApe1 converted to mApe1 during the chase period also increased (Fig. 5 B, right graph). Although the initial amount of pApe1 (at time 0) in early log phase (OD600 = ∼1) was lower than that in late log phase (OD600 = ∼4), its conversion to mApe1 was not significantly faster than that in the latter phase (Fig. 5 B, left graph). These results suggest that Ape1 transport activity is enhanced in later stages of growth in wild-type cells. Knockdown of Hrr25 severely retarded Ape1 transport in all the growth phases (Fig. 5 B). We also showed that Atg19 phosphorylation increased in a manner similar to the stimulation of Ape1 transport (Fig. 5 A). Phosphorylated Atg19 accumulated more evidently when the Cvt pathway was blocked by ATG1 deletion, consistent with the notion that phosphorylated Atg19 is an active form, which is preferentially incorporated into the Cvt vesicle and degraded in the vacuole. The expression of Atg19 increased as cells were grown to later phases (Fig. 5 A). This obscured whether Atg19 phosphorylation by Hrr25 is constitutive or regulated. To solve this problem, we replaced the promoter of ATG19 on the chromosome with that of the constitutively expressing gene CET1. In this strain, the level of Atg19 was almost constant among different growth phases, and the up-regulation of Hrr25-dependent Atg19 phosphorylation in later growth phases was clearly observed (Fig. 5 C). Collectively, these results suggest that Hrr25 is required for the basal activity of the Cvt pathway and also up-regulates the pathway as yeast cell culture proceeds to later growth phases (also see Discussion).
Figure 5.

Hrr25 activates the Cvt pathway in late growth phases via Atg19 phosphorylation. (A, left) Yeast cells were grown to different OD600 and subjected to immunoblotting using antibodies against Ape1, Atg19, and Pgk1. A sample prepared from Hrr25-depleted cells (Hrr25 knockdown [KD]) served to distinguish between phosphorylated and unphosphorylated Atg19. (middle and right) The intensities of the bands corresponding to pApe1, mApe1, and phosphorylated (open arrowheads) and unphosphorylated (closed arrowheads) Atg19 were measured, and the efficiencies of Ape1 maturation (percentages) and Atg19 phosphorylation (percentages) are shown with error bars (standard deviations; n = 3). (B) Cells grown to the indicated OD600 were treated with 50 µg/ml cycloheximide, and Ape1 maturation at different time points was examined by immunoblotting using anti-Ape1 antibodies. The ratios of mApe1 to total Ape1 (percentages) at each time point were shown with error bars representing standard deviations (n = 3). The amounts of Ape1 transported into the vacuole during the chase period were also estimated as follows. The mApe1/total Ape1 ratios at time 0 were set as 0 by subtracting the intensities of mApe1 at time 0 as background. Then, these values were multiplied by the intensities of pApe1 at time 0, and the values for wild-type cells grown to an OD600 of ∼1 were set as 100. Error bars represent standard deviations (n = 3). AU, arbitrary unit. (C) Yeast cells were grown to different OD600 and subjected to immunoblotting using antibodies against GFP and Pgk1. hrr25-aid cells in mid–log phase were treated with IAA and grown to an OD600 of ∼8 to prepare the Hrr25 knockdown sample. (D) Yeast cells were grown to mid–log phase and shifted to a nitrogen starvation (nitro. starv.) medium containing IAA. WT, wild type.
Using yeast cells constitutively expressing Atg19, we showed that the phosphorylation of Atg19 by Hrr25 was also promoted by nitrogen starvation (Fig. 5 D). Hrr25 was important for vacuolar transport of Ape1 in the presence of rapamycin that mimics nitrogen starvation (Fig. S1 D). Hrr25 may also contribute to the stimulation of Ape1 transport under starvation conditions by phosphorylating Atg19.
Hrr25 is required for pexophagy
We investigated whether Hrr25 is involved in other selective-autophagy pathways. The cytoplasmic aldehyde dehydrogenase Ald6 is preferentially degraded via autophagy under nitrogen starvation conditions (Onodera and Ohsumi, 2004). We assessed autophagic degradation of Ald6 by detecting GFP fragments generated from Ald6-GFP in the vacuole; the results showed that Hrr25 is not required for this pathway (Fig. S5 A). We also examined ribophagy, autophagic degradation of ribosomes under starvation conditions, by monitoring vacuolar processing of GFP-fused ribosomal proteins (Kraft et al., 2008); knockdown of Hrr25 had no effect on ribophagy (Fig. S5 B). We next examined the involvement of Hrr25 in mitophagy. To this end, we used two different constructs, the mitochondrial matrix protein Idh1 fused with GFP (Kanki et al., 2009a) and mitochondrial matrix-targeted dihydrofolate reductase (mtDHFR)–mCherry (Kondo-Okamoto et al., 2012). The vacuolar processing of these proteins was examined under mitophagy-inducing conditions. The results suggested that Hrr25 is not important for mitophagy (Fig. S5, C and D). We also showed that the mitophagy-specific receptor Atg32 fused with GST was only slightly phosphorylated by Hrr25 in contrast to GST-Atg19 in an in vitro kinase assay (Fig. S5 E). Finally, we examined pexophagy under Hrr25-depleted conditions. Yeast cells expressing the peroxisomal protein Pex11 fused with GFP were cultured in an oleate medium to cause proliferation of peroxisomes and then shifted to a glucose medium without a nitrogen source to induce pexophagy (Motley et al., 2012). GFP fragments resulting from the vacuolar degradation of Pex11-GFP accumulated in wild-type cells but not in mutant cells lacking the pexophagy receptor Atg36, and knockdown of Hrr25 almost completely blocked Pex11-GFP degradation (Fig. 6 A). As in the case of the Cvt pathway, hrr25-aid cells exhibited a significant defect in pexophagy even in the absence of IAA treatment (Figs. 1 D and 6 A). Pexophagy is also induced by prolonged culture of yeast cells in an oleate medium (Motley et al., 2012). hrr25-aid cells not treated with IAA exhibited a severe defect in pexophagy under this alternative condition (Fig. 6 B). These results suggest that Hrr25 is necessary not only for the Cvt pathway but also for pexophagy.
Figure 6.

Hrr25 is required for pexophagy and responsible for Atg36 phosphorylation. (A, left) Yeast cells expressing Pex11-GFP were grown in an oleate medium and then incubated in nitrogen starvation (nitro. starv.) medium in the presence or absence of IAA and examined by immunoblotting using anti-GFP antibodies. GFP′, GFP fragments produced by the vacuolar degradation of Pex11-GFP. (right) GFP′ intensity was divided by the sum of GFP′ and Pex11-GFP intensities to calculate Pex11-GFP degradation (deg.; percentages). Error bars represent standard deviations (n = 3). (B) Cells expressing Pex11-GFP were grown in an oleate medium without IAA for the indicated time periods, and pexophagy was examined by immunoblotting using anti-GFP antibodies. (C) Cells expressing Atg36-GFP were incubated for 2 h under pexophagy-inducing conditions as described in A in the presence of IAA and subjected to immunoprecipitation using anti-GFP antibody. The immunoprecipitates were treated with λ protein phosphatase in the presence or absence of phosphatase inhibitors and analyzed by immunoblotting using anti-GFP antibodies. (C–E) The square brackets indicate phosphorylated Atg36-GFP (C and D) or GST-Atg36 (E). (D) Cells were incubated under pexophagy-inducing conditions as described in A and analyzed by immunoblotting with antibodies against GFP, AID, and Pgk1. (E) In vitro kinase assay was performed using GST-Atg36 as described in Fig. 4 B. The asterisk indicates a nonspecific band that appeared in the Phos-tag staining independently of Hrr25 activity. (F) Yeast cells were grown to different OD600 and analyzed by immunoblotting as described in Fig. 5 C. (G) Yeast cells were grown to mid–log phase and shifted to a nitrogen starvation medium containing IAA. (F and G) Open arrowheads indicate phosphorylated Atg36-GFP. KD, knockdown; WT, wild type.
Hrr25 is responsible for Atg36 phosphorylation
Atg36 is phosphorylated by an unknown kinase under pexophagy-inducing conditions (Motley et al., 2012). In light of our finding that Hrr25 is a kinase required for pexophagy, we investigated the possibility that Hrr25 phosphorylates Atg36. To test this idea, we used cells lacking Atg1 to block Atg36 degradation via pexophagy. As reported previously, Atg36-GFP was detected as multiple bands under pexophagy-inducing conditions, which were downshifted to a single band by phosphatase treatment, suggesting that these bands represented phosphorylated Atg36 (Fig. 6 C; Motley et al., 2012). Phosphorylation of Atg36 was markedly decreased by the depletion of Hrr25 (Fig. 6, C and D). Moreover, an in vitro kinase assay revealed that Hrr25 directly phosphorylates Atg36 (Fig. 6 E). Collectively, these results suggest that Hrr25 phosphorylates Atg36 during pexophagy.
A previous study reported that the phosphorylation of Atg36 was promoted when yeast cells were subjected to nitrogen starvation after successive culture in a glucose medium and an oleate medium, during which expression of Atg36 increased and its phosphorylation also occurred to some extent (Motley et al., 2012). To more clearly investigate conditions that stimulate Atg36 phosphorylation, we constructed yeast cells that constitutively expressed Atg36 under the control of the CET1 promoter. We found that Hrr25-dependent phosphorylation of Atg36 was suppressed in the cells grown to mid–log phase (OD600 = ∼2) in a glucose medium and remarkably induced in later growth phases (Fig. 6 F). We also showed that Atg36 phosphorylation by Hrr25 was stimulated when the cells grown to mid–log phase in a glucose medium were directly subjected to nitrogen starvation without culture in an oleate medium (Fig. 6 G). These results suggest that as in the case of Atg19 (Fig. 5, C and D), Hrr25-dependent Atg36 phosphorylation is stimulated in late growth phases and under nitrogen starvation conditions.
Hrr25 controls Atg11 targeting to Atg36 on the peroxisome
Atg36 is anchored to the peroxisome by its association with the peroxisomal protein Pex3 and also interacts with Atg11 for its recruitment (Motley et al., 2012). Because this molecular mechanism is analogous to that of the Cvt pathway, we hypothesized that Hrr25-mediated phosphorylation of Atg36 promotes its interaction with Atg11. Indeed, immunoprecipitation analysis showed that knockdown of Hrr25 weakened the interaction of Atg36 with Atg11, whereas it did not affect its interaction with Pex3 (Fig. 7, A and B). Consistent with these results, Atg11 failed to localize to the peroxisome in Hrr25-depleted cells, although Atg36 localized normally to the peroxisome (Fig. 7, C and D). These results suggest that Hrr25 promotes Atg11 targeting to the peroxisome by strengthening the Atg36–Atg11 interaction.
Figure 7.

Hrr25 targets Atg11 to the peroxisome by promoting the Atg36–Atg11 interaction. (A and B) atg1Δ (Hrr25 knockdown [KD], −) and hrr25-aid atg1Δ cells (Hrr25 knockdown, +) coexpressing Atg11-3×HA (A) or Pex3-9×Myc (B) with Atg36-GFP or Atg36 fused with the tandem affinity purification (TAP) tag were incubated under pexophagy-inducing conditions in the presence of IAA for 2 h as described in Fig. 6 A. The cell lysates (total) were subjected to immunoprecipitation using anti-GFP antibody, and the immunoprecipitates (IP) were analyzed by immunoblotting using antibodies against GFP, HA, and Myc. Atg36-TAP was detected using HRP-conjugated anti–mouse IgG. (C and D) Cells coexpressing Pex11-mCherry and either Atg36-GFP (C) or Atg11-GFP and Atg36-ECFP (see Materials and methods; D) were treated with IAA for 2 h under pexophagy-inducing conditions and observed under a fluorescence microscope. Pex11 dots colocalized with Atg36 or Atg11 were counted, and their percentages relative to the total numbers of Pex11 dots are shown with standard deviations (n = 3). White and black arrowheads indicate Pex11 dots with and without clear Atg11 signals, respectively. Bars, 5 µm. BF, bright field.
Analysis of Atg36 residue phosphorylated by Hrr25
It appears that Atg36 is phosphorylated at multiple residues and that Hrr25 is responsible for most of these modifications (Fig. 6 D). We sought to determine a phosphorylation site in Atg36 involved in pexophagy regulation by Hrr25. A recent study proposed a consensus sequence for Atg11 binding (D/SILSSS) in Atg32, Atg36, and PpAtg30 (a pexophagy receptor in the methylotrophic yeast Pichia pastoris; Farré et al., 2013), and this motif is also found in Atg19 (Fig. 8 A). It is remarkable that Ser391 of Atg19 (Fig. 4), Ser114 of Atg32, and Ser112 of PpAtg30 (Farré et al., 2008; Aoki et al., 2011), whose phosphorylation is important for the interaction of these receptors with Atg11, are all located within the motif. Moreover, it was shown that a mutation at Ser97 of Atg36, which is at the same position as the phosphorylated Ser residues in Atg32 and PpAtg30, caused severe defects in the Atg36–Atg11 interaction and pexophagy (Fig. 8, B and C; Farré et al., 2013). We further showed that Atg11 was barely recruited to peroxisomes in the Atg36 mutant (Fig. 8, D and E). Moreover, Atg36 phosphorylation was partially but significantly decreased by the S97A mutation (Fig. 8 F). We also examined the phosphorylation of this mutant in vitro. Because Hrr25 intensely phosphorylated Atg36 in vitro, we used a truncated form of Atg36 (Asp91-Gln147), in which background phosphorylation was eliminated (Fig. 8 G). When this form of Atg36 was incubated with Hrr25, its phosphorylation was detected as a single band with a slower mobility on SDS-PAGE, and this phosphorylation was totally abolished by the S97A mutation. Collectively, these results suggest that Ser97 is one of the Atg36 residues phosphorylated by Hrr25, which is important for the Atg36–Atg11 interaction and thus for pexophagy.
Figure 8.

Analysis of the Atg36 S97A mutant. (A) Amino acid sequences of Atg11-binding regions in autophagic receptors. Ser and Thr residues are colored blue. Arrowheads indicate experimentally determined or predicted phosphorylation sites. (B) ATG11-3×HA atg1Δ atg36Δ cells carrying single-copy Atg36-GFP plasmids or the empty vector were subjected to immunoprecipitation (IP) as described in Fig. 7 A. (C) Pexophagy in PEX11-GFP atg36Δ cells carrying single-copy Atg36-3×FLAG plasmids or the empty vector was examined as described in Fig. 6 A. nitro. starv., nitrogen starvation; GFP′, GFP fragments produced by the vacuolar degradation of Pex11-GFP. (D) PEX11-mCherry atg1Δ atg36Δ cells carrying single-copy Atg36-GFP plasmids were subjected to pexophagy-inducing conditions for 2 h and observed under a fluorescence microscope. Bars, 5 µm. (E) ATG11-GFP PEX11-mCherry atg1Δ atg36Δ cells expressing wild-type Atg36 or the S97A mutant fused with a nonfluorescent mutant of GFP (see Materials and methods) from single-copy plasmids were analyzed as described in D. White and black arrowheads indicate Pex11 dots with and without clear Atg11 signals, respectively. Bars, 5 µm. (F) atg1Δ atg36Δ cells carrying single-copy Atg36-GFP plasmids or the empty vector were incubated under pexophagy-inducing conditions for 2 h and examined by immunoblotting using antibodies against GFP and Pgk1. The square bracket indicates phosphorylated Atg36-GFP. (G) In vitro kinase assay was performed using 3 µM GST-Atg36 (91–147) variants and 0.1 µM wild-type Hrr25 as described in Fig. 4 B. White and black arrowheads indicate phosphorylated and unphosphorylated GST-Atg36 (91–147), respectively. WT, wild type.
Discussion
In this study, we investigated the role of Hrr25 in selective autophagy. We first showed that Hrr25 phosphorylates Atg19 to regulate the initiation of Cvt vesicle formation. Our results suggest that Atg19 phosphorylation by Hrr25 is required for both the basal activity of the Cvt pathway and its stimulation in late growth phases. The expression of pApe1 and Atg19 is induced under those growth conditions (Fig. 5). This is also likely to enhance the Cvt pathway, the contribution of which may be represented as the difference in the Ape1 transport activity of Hrr25-depleted cells in early and late log phases (Fig. 5 B, right graph, open and closed triangles). However, the degree was much smaller than the contribution of Hrr25, which may be represented as the difference between wild-type and Hrr25-depleted cells in late log phase (Fig. 5 B, right graph, closed circles and triangles). These results suggest that Hrr25-mediated Atg19 phosphorylation plays a predominant role in the stimulation of the Cvt pathway in late growth phases. The active transport of degradative enzymes via the Cvt pathway would increase vacuolar degradation capacity and therefore be important for cells to adapt to environmental changes under those growth conditions.
We also showed that Hrr25 is a kinase responsible for the phosphorylation of the pexophagy receptor Atg36. The finding that the same kinase regulates two distinct selective autophagy–related pathways was unexpected. However, both the Cvt pathway and pexophagy are activated under similar conditions, including growth-saturating and nitrogen starvation conditions. A common signal triggered by both of these conditions may stimulate Hrr25 to phosphorylate Atg19 and Atg36. Pexophagy, which disposes of superfluous peroxisomes, is strongly induced under the aforementioned conditions after culture in a peroxisome-proliferating medium, in which the expression of Atg36 is elevated. In contrast, pexophagy occurs only moderately under normal growth conditions even if Atg36 is forcibly expressed (Motley et al., 2012). Therefore, an appropriate combination of multiple distinct signals that induce Atg36 expression and promote its phosphorylation by Hrr25 may be required for initiation of pexophagy.
Hrr25-mediated phosphorylation of Atg19 and Atg36 increased the interactions of both of these receptors with the adaptor Atg11. Thus, Hrr25 regulates the Cvt pathway and pexophagy by a uniform mechanism (Fig. 9). During the course of this study, another group has reported similar results showing the involvement of Hrr25 in the Cvt pathway (Pfaffenwimmer et al., 2014). Phosphorylation of several Atg19 residues, including Ser391, was detected by mass spectrometry. It was also suggested that Hrr25 phosphorylates some of these residues to promote the Atg19–Atg11 interaction. In addition, it has recently been reported that the mitophagy receptor Atg32 is phosphorylated by casein kinase 2, which allows Atg32 to interact with Atg11 (Kanki et al., 2013). Moreover, it was shown that PpAtg30 is also phosphorylated, and this phosphorylation is important for the binding of PpAtg30 to Atg11 (Farré et al., 2013). Although the kinase that phosphorylates PpAtg30 is still unknown, these observations suggest that the initiation of these selective autophagy–related pathways is regulated by conceptually identical mechanisms, i.e., phosphoregulation of the receptor–adaptor interaction.
Figure 9.
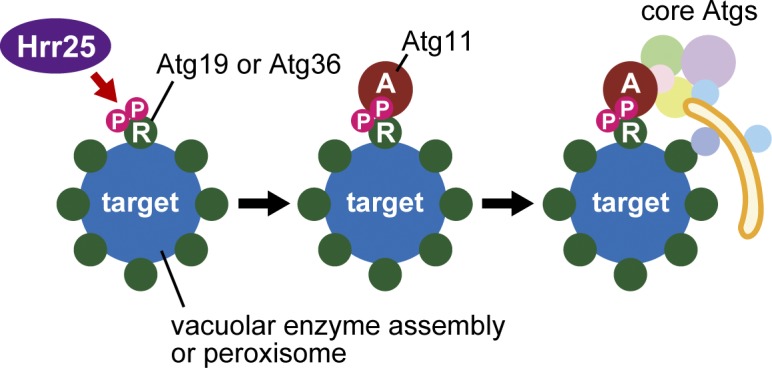
Model for the regulation of selective autophagy–related pathways by Hrr25. Hrr25 regulates the initiation of the Cvt pathway and pexophagy by conceptually identical mechanisms. Hrr25 phosphorylates Atg19 and Atg36 (R, receptor), which, respectively, recognize the assembly of vacuolar enzymes and the peroxisome (target), to recruit Atg11 (A, adaptor), which subsequently recruits the core Atg proteins that mediate membrane formation. P, phosphorylated.
Phosphorylation of receptors can also regulate their interactions with degradation targets (Matsumoto et al., 2011) or core Atg proteins (Wild et al., 2011; Liu et al., 2012; Farré et al., 2013). Furthermore, a recent study has provided the case that methylation of Arg residues in degradation targets promotes their interactions with an adaptor (Li et al., 2013). Thus, the initiation of selective autophagy is regulated at multiple steps by different protein modifications. This indicates the significance of the regulation of selective autophagy, for which other types of mechanisms may also operate in cells.
Hrr25 is a CK1δ homologue that plays pivotal roles in a variety of cellular events, some of which are conserved across species. This study revealed a new role for this kinase, the regulation of selective autophagy, raising a series of related questions: Are CK1δ homologues also involved in selective autophagy in other organisms? How does Hrr25/CK1δ regulate diverse cellular processes under different conditions? What is the relationship between selective autophagy and other cellular processes that are regulated by Hrr25/CK1δ? Future studies should address these important issues.
Materials and methods
Yeast strains
The yeast strains used in this study are listed in Table S1. Gene disruption and tagging were performed by a PCR-based method (Janke et al., 2004; Nakatogawa et al., 2012). The strains for the AID-based depletion of proteins (Nishimura et al., 2009) were similarly constructed by adding the AID tag sequence to the C termini of Hrr25 and Atg2 or the N terminus of Sec23. The strains expressing mRFP-Ape1 were constructed by integrating pPS128 (donated by D. Klionsky, University of Michigan, Ann Arbor, MI), which expresses the fusion protein from the APE1 promoter (Strømhaug et al., 2004), into the APE1 locus on the chromosome. Peroxisomes form a few clusters in a cell when Atg36 is C-terminally tagged with GFP (Motley et al., 2012; also seen in Fig. 7 C); otherwise, they are dispersed in the cytoplasm as multiple dots. Because Atg11 also forms a considerable number of dots under pexophagy-inducing conditions, it was difficult to assess whether their colocalization was significant in cells expressing Atg11-GFP and Pex11-mCherry. Therefore, ECFP was fused to the C terminus of Atg36 in those cells to allow examination of Atg11 localization to the peroxisome (Fig. 7 D). As a result of the same reason, the plasmids expressing Atg36 fused with a nonfluorescent mutant of GFP were used in Fig. 8 E to observe the peroxisomal localization of Atg11-GFP in the Atg36 S97A mutant.
Plasmids
The plasmids to express the Atg19 mutants in yeast (used in Fig. 4) were constructed as follows. Oligonucleotide sequences used in this study are given in Table S2. The ATG19 gene with its own promoter and terminator regions was amplified by PCR from BY4741 (Brachmann et al., 1998) genomic DNA using the primers XhoI-ATG19-forward and ATG19-BamHI-reverse and ligated into the single-copy vector pRS313 (Sikorski and Hieter, 1989) using appropriate restriction enzymes. Similarly, the ATG19-GFP and ATG36-GFP genes with their own promoters were amplified by PCR using genomic DNA from CTY275 and YNH803 and the pairs of oligonucleotides XhoI-ATG19-forward and PGKter-SacI-reverse and KpnI-ATG36-forward and PGKter-SacI-reverse to construct pRS313-ATG19-GFP (used in Fig. 4) and pRS316-ATG36-GFP. A DNA region containing the ATG36-GFP gene in pRS316-ATG36-GFP was subcloned into pRS315 using PvuII to construct pRS315-ATG36-GFP (used in Fig. 8). To construct the plasmids for expression of GST-fused proteins in E. coli, the open reading frames of ATG36 and HRR25 were amplified by PCR from BY4741 genomic DNA using the primers BamHI-ATG36-forward and ATG36-SalI-reverse or BamHI-HRR25-forward and HRR25-SalI-reverse and ligated into the pGEX-6P-1 vector (GE Healthcare) using appropriate restriction enzymes. pGEX-6P-ATG36 (Asp91-Gln147) and that containing the S97A mutation were similarly constructed using the primers BamHI-ATG36-91-forward/BamHI-ATG36-91-S97A-forward and ATG36-147-SalI-reverse. pGEX-6P-ATG19 (Noda et al., 2008), pGEX-6P-ATG11 (donated by N. Noda, Institute of Microbial Chemistry, Tokyo, Japan), and pGEX-6P-ATG32 (2–388) (donated by K. Okamoto, Osaka University, Osaka, Japan) were used to express GST-fused Atg19, Atg11, and Atg32 (2–388) in E. coli, respectively. Site-directed mutagenesis was performed using the QuikChange kit (Agilent Technologies), and appropriate pairs of oligonucleotides were used to construct the Atg19, Atg36, and Hrr25 mutant plasmids. pRS315-ATG36-3×FLAG and pRS315-ATG36–nonfluorescent GFP (used in Fig. 8 E) were also constructed by site-directed mutagenesis using pRS315-ATG36-GFP as a template and the pairs of oligonucleotides Atg36-GFP>3×FLAG-forward and Atg36-GFP>3×FLAG-reverse and GFP-inactivation-forward and GFP-inactivation-reverse, respectively.
Media
Yeast cells were cultured in synthetic dextrose (SD) + casamino acids (CA) + ATU medium (0.17% yeast nitrogen base [YNB] without amino acids and ammonium sulfate [YNB w/o aa and as], 0.5% ammonium sulfate, 2% glucose, 0.5% CA, 0.002% adenine sulfate, 0.002% tryptophan, and 0.002% uracil) at 30°C, except that cells carrying pRS313- and pRS315-derived plasmids were cultured in SD + dropout medium (0.17% YNB w/o aa and as, 0.5% ammonium sulfate, and 2% glucose supplemented with 0.002% adenine sulfate, 0.002% uracil, and appropriate amino acids) lacking histidine and leucine, respectively. To induce nonselective autophagy, cells were treated with 0.2 µg/ml rapamycin or incubated in nitrogen starvation medium SD-N (0.17% YNB w/o aa and as and 2% glucose). Pexophagy was induced as described previously (Motley et al., 2012). In brief, yeast cultures grown in SD + CA + ATU or SD + dropout medium for 24 h were diluted 10-fold with SO + CA + ATU medium (0.17% YNB w/o aa and as, 0.5% ammonium sulfate, 0.12% oleate, 0.2% Tween 40, 0.1% glucose, 1% CA, 0.1% yeast extract, 0.002% adenine sulfate, 0.002% tryptophan, and 0.002% uracil) and grown for 18 h, and the medium was replaced with SD-N. For auxin treatment, IAA was added to a final concentration of 0.5 mM, and the same volume of solvent ethanol was added to control samples (Nishimura et al., 2009).
Immunoblotting
Yeast cell pellets (∼1.5–6.0 OD units) were suspended in 200 µl of 20% trichloroacetic acid, kept on ice for 15 min, and centrifuged at 15,000 g for 5 min. The pellets were washed with 1 ml of cold acetone, dried at room temperature, and suspended in (OD units × 50) µl of SDS sample buffer by mixing at 65°C for 10 min followed by cell disruption at room temperature using FastPrep-24 (MP Biomedicals) and 0.5-mm YZB zirconia beads (Yasui Kikai). These samples were boiled for 3 min and centrifuged at 15,000 g for 1 min; the supernatants were used for immunoblotting analysis (Nakatogawa and Ohsumi, 2012). To separate phosphorylated and unphosphorylated forms of nontagged Atg19, the Anderson gel system (10% acrylamide and bis-acrylamide at 77:1) was used (Anderson et al., 1973). Monoclonal antibodies against GFP (11814460001; Roche), AID (BioROIS), GST (B-14; Santa Cruz Biotechnology, Inc.), HA (3F10; Roche), and Myc (9E10; Santa Cruz Biotechnology, Inc.) sequences were used for detection of tagged proteins. The monoclonal antibody against Pgk1 was purchased from Invitrogen. The phosphate-binding reagent Biotinylated Phos-tag (Wako Pure Chemical Industries) was used to detect protein phosphorylation in combination with streptavidin-HRP (Invitrogen).
Microscopic image acquisition
Fluorescence microscopy was performed using an inverted fluorescence microscopy system (IX71; Olympus) equipped with a 150× objective lens (U Apochromat N 150×, NA 1.45; Olympus), a charge-coupled device camera (ImagEM Enhanced; Hamamatsu Photonics), blue (Sapphire 488–20; Coherent) and yellow lasers (85-YCA-010; Melles Griot), and band pass filters (FF495-Em02-25 and FF593-Em02-25; Semrock) for visualization of GFP and mCherry, respectively. Images were acquired using Aquacosmos software (Hamamatsu Photonics) and processed using MetaMorph 7.0r4 (Molecular Devices) and Photoshop CS6 (Adobe). The images were not manipulated other than contrast and brightness adjustments.
ALP assay
To quantify autophagic activity of yeast cells, an ALP assay was performed (Noda et al., 1995). In brief, yeast cells expressing a cytoplasm-localized mutant of vacuolar ALP (Pho8Δ60) were incubated under autophagy-inducing conditions, in which Pho8Δ60, as a part of the cytoplasm, was delivered into the vacuole via autophagy and activated by a vacuolar processing enzyme. The activity in cell lysates was measured using a fluorogenic substrate.
Immunoprecipitation
To examine the interaction between Atg19 and Atg11, yeast cells expressing Atg19-GFP and Atg11-3×HA were disrupted in buffer A (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 10% glycerol) containing 2 mM PMSF and 2× protease inhibitor cocktail (Complete, EDTA-free; Roche) using a Multi-beads Shocker (Yasui Kikai) and 0.5-mm YZB zirconia beads. Nonidet P-40 was added to the lysates to a final concentration of 0.01%, and the samples were rotated at 4°C for 30 min. The solubilized lysates were cleared by centrifugation at 15,000 g for 5 min, mixed with GFP-Trap_M beads (ChromoTek), and rotated at 4°C for 2 h. The beads were washed with buffer A, and the bound proteins were eluted by incubating the beads in SDS sample buffer at 65°C for 5 min. To examine the Atg36–Atg11 and Atg36–Pex3 interactions, cells expressing Atg36-GFP and either Atg11-3×HA or Pex3-9×Myc were spheroplasted by incubation in SD-N containing 1.2 M sorbitol, 0.1 M Tris-HCl, pH 8.0, and 0.2 mg/ml Zymolyase 100T (Seikagaku Biobusiness) at 30°C for 1 h, washed with buffer A containing 5 mM EDTA, 5 mM EGTA, 50 mM NaF, and 2 mM PMSF, and lysed in the same buffer additionally containing 2× protease inhibitor cocktail, 1× phosphatase inhibitor cocktail (PhosSTOP; Roche), 500 nM microcystin, and 0.5% (vol/vol) Triton X-100. The lysates were cleared by centrifugation and subjected to immunoprecipitation with GFP-Trap_M beads.
Phosphatase treatment
Cell lysates were prepared and incubated with GFP-Trap_M beads, as described in the previous paragraph. The beads were washed with buffer A and incubated with λ protein phosphatase (New England Biolabs, Inc.) at 30°C for 1 h under the recommended conditions in the presence or absence of 5× Halt phosphatase inhibitor cocktail (Thermo Fisher Scientific).
Protein purification
E. coli Rosetta cells carrying pGEX-6P-Atg19, -Atg11, -Atg32 (2–388), -Atg36, -Hrr25, and -Hrr25 K38A or the pGEX-6P-1 vector (for GST purification) were cultured in Luria broth medium (10 mg/ml tryptone, 5 mg/ml yeast extract, 10 mg/ml NaCl, and 1 mM NaOH) containing 50 µg/ml ampicillin and 50 µg/ml chloramphenicol at 37°C to an OD600 of ∼0.5, and protein expression was induced in the presence of 0.1 mM isopropyl β-D-thiogalactopyranoside at 16°C for 24 h. Cells were harvested and disrupted in buffer B (20 mM Hepes-KOH, pH 7.2, and 150 mM NaCl) containing 5 mM DTT, 0.5 mM EDTA, and 0.1 mM PMSF as described previously (Nakatogawa et al., 2007). The cleared lysates were rotated with glutathione–Sepharose 4B resin (GE Healthcare) at 4°C for 45 min. The resins were washed with buffer B, and bound proteins were eluted with 50 mM Tris-HCl, pH 8.0, containing 150 mM NaCl and 10 mM reduced glutathione. The GST-Hrr25–bound resins were treated with the PreScission Protease (GE Healthcare), which cleaves the recognition sequence between the GST and Hrr25 moieties, to elute Hrr25. The purified proteins were appropriately concentrated using Vivaspin columns (Sartorius) and stored at −80°C in solutions containing 25% glycerol.
In vitro kinase assay
0.3 µM GST, GST-Atg19, GST-Atg11, or GST-Atg36 was incubated at 30°C with 0.05 µM wild-type Hrr25 or Hrr25 K38A in 20 mM Hepes-KOH, pH 7.2, containing 150 mM NaCl, 1 mM MgCl2, 0.2 mM DTT, and 1 mM ATP.
Online supplemental material
Fig. S1 shows nonselective autophagy in Hrr25-depleted cells. Fig. S2 shows the localization of Atg proteins and Ape1 in Hrr25-depleted cells. Fig. S3 shows Atg11 localization in Sec23-depleted cells. Fig. S4 shows immunoprecipitation analysis of Hrr25. Fig. S5 shows selective autophagy of Ald6, ribosomes, and mitochondria in Hrr25-depleted cells. Table S1 lists yeast strains used in this study. Table S2 lists oligonucleotides used in this study. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201402128/DC1.
Supplementary Material
Acknowledgments
We thank the members of our laboratory for materials, discussions, and technical and secretarial support, Dr. Masato Kanemaki for suggestions on the AID system, and Dr. Daniel Klionsky, Dr. Koji Okamoto, and Dr. Nobuo Noda for providing plasmids. Materials for the AID system were provided by the National Bio-Resource Project of the Ministry of Education, Culture, Sports, Science and Technology of Japan.
This work was supported in part by the Funding Program for Next Generation World-Leading Researchers HO220017 (to H. Nakatogawa) and Grants-in-Aid for Scientific Research 25111003 (to H. Nakatogawa), 25711005 (to H. Nakatogawa), and 23000015 (to Y. Ohsumi) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- AID
- auxin-inducible degron
- ALP
- alkaline phosphatase
- Atg
- autophagy related
- CA
- casamino acids
- Cvt
- cytoplasm-to-vacuole targeting
- IAA
- indole-3-acetic acid
- mtDHFR
- mitochondrial matrix-targeted dihydrofolate reductase
- SD
- synthetic dextrose
- TAP
- tandem affinity purification
- YNB
- yeast nitrogen base
References
- Anderson, C.W., Baum P.R., and Gesteland R.F.. 1973. Processing of adenovirus 2-induced proteins. J. Virol. 12:241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki, Y., Kanki T., Hirota Y., Kurihara Y., Saigusa T., Uchiumi T., and Kang D.. 2011. Phosphorylation of Serine 114 on Atg32 mediates mitophagy. Mol. Biol. Cell. 22:3206–3217 10.1091/mbc.E11-02-0145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann, C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., and Boeke J.D.. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14:115–132 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2 [DOI] [PubMed] [Google Scholar]
- Breitkreutz, A., Choi H., Sharom J.R., Boucher L., Neduva V., Larsen B., Lin Z.Y., Breitkreutz B.J., Stark C., Liu G., et al. 2010. A global protein kinase and phosphatase interaction network in yeast. Science. 328:1043–1046 10.1126/science.1176495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMaggio, A.J., Lindberg R.A., Hunter T., and Hoekstra M.F.. 1992. The budding yeast HRR25 gene product is a casein kinase I isoform. Proc. Natl. Acad. Sci. USA. 89:7008–7012 10.1073/pnas.89.15.7008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic, V., and Levine B.. 2009. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 5:527–549 10.1016/j.chom.2009.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farré, J.C., Manjithaya R., Mathewson R.D., and Subramani S.. 2008. PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev. Cell. 14:365–376 10.1016/j.devcel.2007.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farré, J.C., Burkenroad A., Burnett S.F., and Subramani S.. 2013. Phosphorylation of mitophagy and pexophagy receptors coordinates their interaction with Atg8 and Atg11. EMBO Rep. 14:441–449 10.1038/embor.2013.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra, M.F., Liskay R.M., Ou A.C., DeMaggio A.J., Burbee D.G., and Heffron F.. 1991. HRR25, a putative protein kinase from budding yeast: association with repair of damaged DNA. Science. 253:1031–1034 10.1126/science.1887218 [DOI] [PubMed] [Google Scholar]
- Hutchins, M.U., and Klionsky D.J.. 2001. Vacuolar localization of oligomeric alpha-mannosidase requires the cytoplasm to vacuole targeting and autophagy pathway components in Saccharomyces cerevisiae. J. Biol. Chem. 276:20491–20498 10.1074/jbc.M101150200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara, N., Hamasaki M., Yokota S., Suzuki K., Kamada Y., Kihara A., Yoshimori T., Noda T., and Ohsumi Y.. 2001. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol. Biol. Cell. 12:3690–3702 10.1091/mbc.12.11.3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke, C., Magiera M.M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., and Knop M.. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 21:947–962 10.1002/yea.1142 [DOI] [PubMed] [Google Scholar]
- Johansen, T., and Lamark T.. 2011. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 7:279–296 10.4161/auto.7.3.14487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki, T., Wang K., Baba M., Bartholomew C.R., Lynch-Day M.A., Du Z., Geng J., Mao K., Yang Z., Yen W.L., and Klionsky D.J.. 2009a. A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol. Biol. Cell. 20:4730–4738 10.1091/mbc.E09-03-0225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki, T., Wang K., Cao Y., Baba M., and Klionsky D.J.. 2009b. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell. 17:98–109 10.1016/j.devcel.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki, T., Kurihara Y., Jin X., Goda T., Ono Y., Aihara M., Hirota Y., Saigusa T., Aoki Y., Uchiumi T., and Kang D.. 2013. Casein kinase 2 is essential for mitophagy. EMBO Rep. 14:788–794 10.1038/embor.2013.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J., Scott S.V., Oda M.N., and Klionsky D.J.. 1997. Transport of a large oligomeric protein by the cytoplasm to vacuole protein targeting pathway. J. Cell Biol. 137:609–618 10.1083/jcb.137.3.609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin, V., McEwan D.G., Novak I., and Dikic I.. 2009. A role for ubiquitin in selective autophagy. Mol. Cell. 34:259–269 10.1016/j.molcel.2009.04.026 [DOI] [PubMed] [Google Scholar]
- Kondo-Okamoto, N., Noda N.N., Suzuki S.W., Nakatogawa H., Takahashi I., Matsunami M., Hashimoto A., Inagaki F., Ohsumi Y., and Okamoto K.. 2012. Autophagy-related protein 32 acts as autophagic degron and directly initiates mitophagy. J. Biol. Chem. 287:10631–10638 10.1074/jbc.M111.299917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft, C., Deplazes A., Sohrmann M., and Peter M.. 2008. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 10:602–610 10.1038/ncb1723 [DOI] [PubMed] [Google Scholar]
- Li, S., Yang P., Tian E., and Zhang H.. 2013. Arginine methylation modulates autophagic degradation of PGL granules in C. elegans. Mol. Cell. 52:421–433 10.1016/j.molcel.2013.09.014 [DOI] [PubMed] [Google Scholar]
- Liu, L., Feng D., Chen G., Chen M., Zheng Q., Song P., Ma Q., Zhu C., Wang R., Qi W., et al. 2012. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14:177–185 10.1038/ncb2422 [DOI] [PubMed] [Google Scholar]
- Lord, C., Bhandari D., Menon S., Ghassemian M., Nycz D., Hay J., Ghosh P., and Ferro-Novick S.. 2011. Sequential interactions with Sec23 control the direction of vesicle traffic. Nature. 473:181–186 10.1038/nature09969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch-Day, M.A., and Klionsky D.J.. 2010. The Cvt pathway as a model for selective autophagy. FEBS Lett. 584:1359–1366 10.1016/j.febslet.2010.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto, G., Wada K., Okuno M., Kurosawa M., and Nukina N.. 2011. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell. 44:279–289 10.1016/j.molcel.2011.07.039 [DOI] [PubMed] [Google Scholar]
- Mizushima, N., and Komatsu M.. 2011. Autophagy: renovation of cells and tissues. Cell. 147:728–741 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- Mizushima, N., Yoshimori T., and Ohsumi Y.. 2011. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27:107–132 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- Motley, A.M., Nuttall J.M., and Hettema E.H.. 2012. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 31:2852–2868 10.1038/emboj.2012.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami, A., Kimura K., and Nakano A.. 1999. The inactive form of a yeast casein kinase I suppresses the secretory defect of the sec12 mutant. Implication of negative regulation by the Hrr25 kinase in the vesicle budding from the endoplasmic reticulum. J. Biol. Chem. 274:3804–3810 10.1074/jbc.274.6.3804 [DOI] [PubMed] [Google Scholar]
- Nakatogawa, H., and Ohsumi Y.. 2012. SDS-PAGE techniques to study ubiquitin-like conjugation systems in yeast autophagy. Methods Mol. Biol. 832:519–529 10.1007/978-1-61779-474-2_37 [DOI] [PubMed] [Google Scholar]
- Nakatogawa, H., Ichimura Y., and Ohsumi Y.. 2007. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 130:165–178 10.1016/j.cell.2007.05.021 [DOI] [PubMed] [Google Scholar]
- Nakatogawa, H., Suzuki K., Kamada Y., and Ohsumi Y.. 2009. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 10:458–467 10.1038/nrm2708 [DOI] [PubMed] [Google Scholar]
- Nakatogawa, H., Ishii J., Asai E., and Ohsumi Y.. 2012. Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy. 8:177–186 10.4161/auto.8.2.18373 [DOI] [PubMed] [Google Scholar]
- Nishimura, K., Fukagawa T., Takisawa H., Kakimoto T., and Kanemaki M.. 2009. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods. 6:917–922 10.1038/nmeth.1401 [DOI] [PubMed] [Google Scholar]
- Noda, N.N., Kumeta H., Nakatogawa H., Satoo K., Adachi W., Ishii J., Fujioka Y., Ohsumi Y., and Inagaki F.. 2008. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells. 13:1211–1218 10.1111/j.1365-2443.2008.01238.x [DOI] [PubMed] [Google Scholar]
- Noda, T., Matsuura A., Wada Y., and Ohsumi Y.. 1995. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 210:126–132 10.1006/bbrc.1995.1636 [DOI] [PubMed] [Google Scholar]
- Okamoto, K., Kondo-Okamoto N., and Ohsumi Y.. 2009. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell. 17:87–97 10.1016/j.devcel.2009.06.013 [DOI] [PubMed] [Google Scholar]
- Onodera, J., and Ohsumi Y.. 2004. Ald6p is a preferred target for autophagy in yeast, Saccharomyces cerevisiae. J. Biol. Chem. 279:16071–16076 10.1074/jbc.M312706200 [DOI] [PubMed] [Google Scholar]
- Petronczki, M., Matos J., Mori S., Gregan J., Bogdanova A., Schwickart M., Mechtler K., Shirahige K., Zachariae W., and Nasmyth K.. 2006. Monopolar attachment of sister kinetochores at meiosis I requires casein kinase 1. Cell. 126:1049–1064 10.1016/j.cell.2006.07.029 [DOI] [PubMed] [Google Scholar]
- Pfaffenwimmer, T., Reiter W., Brach T., Nogellova V., Papinski D., Schuschnig M., Abert C., Ammerer G., Martens S., and Kraft C.. 2014. Hrr25 kinase promotes selective autophagy by phosphorylating the cargo receptor Atg19. EMBO Rep. 15:862–870 10.15252/embr.201438932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer, T., Maco B., Petfalski E., Tollervey D., Böttcher B., Aebi U., and Hurt E.. 2006. Hrr25-dependent phosphorylation state regulates organization of the pre-40S subunit. Nature. 441:651–655 10.1038/nature04840 [DOI] [PubMed] [Google Scholar]
- Scott, S.V., Guan J., Hutchins M.U., Kim J., and Klionsky D.J.. 2001. Cvt19 is a receptor for the cytoplasm-to-vacuole targeting pathway. Mol. Cell. 7:1131–1141 10.1016/S1097-2765(01)00263-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani, T., and Klionsky D.J.. 2004. Cargo proteins facilitate the formation of transport vesicles in the cytoplasm to vacuole targeting pathway. J. Biol. Chem. 279:29889–29894 10.1074/jbc.M404399200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani, T., Huang W.P., Stromhaug P.E., and Klionsky D.J.. 2002. Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev. Cell. 3:825–837 10.1016/S1534-5807(02)00373-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski, R.S., and Hieter P.. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122:19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strømhaug, P.E., Reggiori F., Guan J., Wang C.W., and Klionsky D.J.. 2004. Atg21 is a phosphoinositide binding protein required for efficient lipidation and localization of Atg8 during uptake of aminopeptidase I by selective autophagy. Mol. Biol. Cell. 15:3553–3566 10.1091/mbc.E04-02-0147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, K.2013. Selective autophagy in budding yeast. Cell Death Differ. 20:43–48 10.1038/cdd.2012.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, K., Kirisako T., Kamada Y., Mizushima N., Noda T., and Ohsumi Y.. 2001. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 20:5971–5981 10.1093/emboj/20.21.5971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg, H., Shvets E., and Elazar Z.. 2011. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 80:125–156 10.1146/annurev-biochem-052709-094552 [DOI] [PubMed] [Google Scholar]
- Wild, P., Farhan H., McEwan D.G., Wagner S., Rogov V.V., Brady N.R., Richter B., Korac J., Waidmann O., Choudhary C., et al. 2011. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 333:228–233 10.1126/science.1205405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, Y., Ren J., Gao X., Jin C., Wen L., and Yao X.. 2008. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics. 7:1598–1608 10.1074/mcp.M700574-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., and Klionsky D.J.. 2010. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12:814–822 10.1038/ncb0910-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuga, M., Gomi K., Klionsky D.J., and Shintani T.. 2011. Aspartyl aminopeptidase is imported from the cytoplasm to the vacuole by selective autophagy in Saccharomyces cerevisiae. J. Biol. Chem. 286:13704–13713 10.1074/jbc.M110.173906 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.