

Abstract
The acid- and volume-sensitive TASK2 K+ channel is strongly expressed in renal proximal tubules and papillary collecting ducts. This study was aimed at investigating the role of TASK2 in renal bicarbonate reabsorption by using the task2 –/– mouse as a model. After backcross to C57BL6, task2 –/– mice showed an increased perinatal mortality and, in adulthood, a reduced body weight and arterial blood pressure. Patch-clamp experiments on proximal tubular cells indicated that TASK2 was activated during 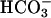 transport. In control inulin clearance measurements, task2 –/– mice showed normal NaCl and water excretion. During i.v. NaHCO3 perfusion, however, renal Na+ and water reabsorption capacity was reduced in –/– animals. In conscious task2 –/– mice, blood pH,
transport. In control inulin clearance measurements, task2 –/– mice showed normal NaCl and water excretion. During i.v. NaHCO3 perfusion, however, renal Na+ and water reabsorption capacity was reduced in –/– animals. In conscious task2 –/– mice, blood pH, 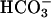 concentration, and systemic base excess were reduced but urinary pH and
concentration, and systemic base excess were reduced but urinary pH and  were increased. These data suggest that task2 –/– mice exhibit metabolic acidosis caused by renal loss of
were increased. These data suggest that task2 –/– mice exhibit metabolic acidosis caused by renal loss of 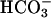 . Both in vitro and in vivo results demonstrate the specific coupling of TASK2 activity to
. Both in vitro and in vivo results demonstrate the specific coupling of TASK2 activity to 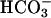 transport through external alkalinization. The consequences of the task2 gene inactivation in mice are reminiscent of the clinical manifestations seen in human proximal renal tubular acidosis syndrome.
transport through external alkalinization. The consequences of the task2 gene inactivation in mice are reminiscent of the clinical manifestations seen in human proximal renal tubular acidosis syndrome.
K+ channels are directly and indirectly involved in transport function in kidney. They stabilize the membrane voltage at hyperpolarized values, thereby increasing the driving force for secondary active rheogenic transport. In addition, they play an important role in K+ balance as a K+ secretory pathway (1). In proximal tubules, where mass transport of solutes and water takes place, luminal and basolateral K+ channels have been found. On the luminal side, Na+-coupled solute transport results in membrane depolarization that in turn activates voltage-activated K+ channels (2, 3). Other K+ channels are regulated by membrane stretch and cytosolic Ca2+ (4). The basolateral K+ conductance is supposed to be functionally coupled to Na+/K+ ATPase activity and regulated by cell volume and pH (5).
Among other solutes, proximal tubules reabsorb some 75% of filtered 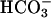 . Luminal Na+/H+ exchanger and H+ ATPase, carbonic anhydrases (type II and IV), and basolateral
. Luminal Na+/H+ exchanger and H+ ATPase, carbonic anhydrases (type II and IV), and basolateral 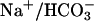 transporter are functionally coupled to transport
transporter are functionally coupled to transport 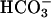 across the epithelium. Electrogenic
across the epithelium. Electrogenic 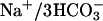 transport depolarizes the basolateral membrane. Thus, concomitant activation of basolateral K+ conductance is required for ongoing
transport depolarizes the basolateral membrane. Thus, concomitant activation of basolateral K+ conductance is required for ongoing 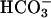 transport. Microperfusion studies indicated an increase of basolateral K+ conductance during
transport. Microperfusion studies indicated an increase of basolateral K+ conductance during 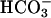 reabsorption and regulatory volume decrease (6), and picomolar concentrations of angiotensin II lead to parallel activation of basolateral NaHCO3 transport and K+ conductance (7). Therefore, defects in basolateral K+ channels may substantially diminish proximal tubular NaHCO3 transport.
reabsorption and regulatory volume decrease (6), and picomolar concentrations of angiotensin II lead to parallel activation of basolateral NaHCO3 transport and K+ conductance (7). Therefore, defects in basolateral K+ channels may substantially diminish proximal tubular NaHCO3 transport.
At the molecular level, several K+ channels have been identified in proximal tubules: e.g., KCNQ1/KCNE1 (2), KCNA10 (8), TREK-2b (a splice variant of TREK-2) (9), TWIK1 (10), Kir7.1 (11), Kir5.1, and Kir2.1 (12). A recent analysis of human kidney gene expression indicated the presence of Kir4.2 and TASK2 in proximal tubules (13). However, little is known about the role of these K+ channels in transport of proximal tubules and kidney function.
TASK2 is a K+ channel that belongs to the family of two P-domain channels characterized by four transmembrane domains and two pore-forming loops. The first mammalian member of this family was identified in 1996 and named TWIK1 (tandem of P-domains in a weak inwardly rectifying K+ channel) (14). Fourteen other family members are now identified and subclassified based on their sensitivity to fatty acids, stretch, or protons (15). TASK2 (TWIK-related acid-sensitive K+ channel 2) channels generate background K+ currents that are increased by external alkalinization in the physiological range of pH (16) and by cell swelling (17). Recently, TASK2 was shown to be involved in volume regulation of native renal proximal tubule cells (18). By radiation hybrid mapping, the human TASK2 gene (KCNK5) was localized on chromosome 6p21 (16). The present study was aimed at investigating the role of TASK2 K+ channels in kidney, and more specifically, in renal salt reabsorption. We found that this pH-sensitive K+ channel specifically senses the external basolateral pH increase resulting from 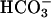 transport in primary cultured proximal tubular cells as well as in vivo. It serves as a molecular switch that adapts the K+ conductance to the
transport in primary cultured proximal tubular cells as well as in vivo. It serves as a molecular switch that adapts the K+ conductance to the 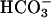 transport activity. Alteration of renal
transport activity. Alteration of renal 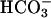 handling in task2 –/– mice are reminiscent of clinical manifestations seen in human proximal renal tubular acidosis establishing TASK2 as a candidate gene for the familial forms of this disease.
handling in task2 –/– mice are reminiscent of clinical manifestations seen in human proximal renal tubular acidosis establishing TASK2 as a candidate gene for the familial forms of this disease.
Materials and Methods
All animal experimentation was conducted in accord with the French and Swiss government animal welfare policies.
Primary Cell Cultures and Electrophysiological Studies. Proximal tubules were microdissected and dissociated cells were grown on collagen-coated dishes as described (18). Whole-cell currents were recorded after a 6- to 20-day culture. The compositions of solutions used for patch-clamp experiments are given in Table 1.
Table 1. Solutions for whole-cell experiments.
| Concentration, mmol/liter
|
|||||
|---|---|---|---|---|---|
| Solute | Bath 1 | Bath 2 | Pipette 1 | Pipette 2 | Pipette 3 |
| Na+ | 108 | - | 20 | - | - |
| K+ | 5 | 5 | 125 | 145 | 145 |
| Ca2+ | 0.3* | 1 | - | - | - |
| NMDG+ | - | 140 | - | - | - |
| Cl- | 20 | 142 | 25 | 20 | 20 |
| Gluconate | 95 | 5 | 100 | 100 | 125 |
| Glucose | 5 | 5 | - | - | - |
| HCO3- | - | - | 20 | 25 | - |
| Hepes† | 1, 30‡ | 1 | 10 | 10 | 10 |
| EGTA | - | - | 5 | 5 | 5 |
| Mg-ATP | - | - | 5 | 5 | 5 |
| Osmolality, mosmol/kg | 300 | 295 | 295 | 295 | 295 |
NMDG, N-methyl-d-glucamine.
Ca2+ concentration indicated corresponds to the free Ca2+ activity; the total Ca2+ concentration was 1 mM.
The pH was adjusted to 7.4 in the bath and 7.2 in the pipette solutions.
Solution osmolality was adjusted to 300 mosmol/kg with mannitol.
Task2 Knockout Mouse. The task2 knockout mouse was produced in 129/SV genetic background by exon trapping techniques (19, 20) and kindly provided by K. Mitchell, W. C. Skarnes, and coworkers (University of California, Berkeley). The vector pGTOTMpf containing LacZ and PLAP marker genes was inserted between exons 1 and 2 (18). Experiments were performed in task2 knockout (–/–) and littermate (+/+) animals after five to seven generations of backcross to the C57BL6 genetic background. Animals were kept on a standard diet and had free access to chow and water.
Dual-Energy X-Ray Absorptiometry Scan. Whole-body composition of anesthetized mice (ketamine 100 mg/kg of body weight i.p. plus xylazine 4 mg/kg of body weight i.p.) was analyzed by PIXImus dual-energy x-ray absorptiometry (General Electric). Total body analysis was acquired in 5 min, and the data were analyzed by using software provided by the manufacturer.
5-Bromo-4-chloro-3-indolyl β-d-Galactoside (X-Gal) Staining. Anesthetized mice were perfused via the left ventricle with 10 ml of heparinized 0.9% NaCl solution at 37°C and subsequently 50 ml of 3% paraformaldehyde solution at 37°C and pH 7.4. Cryosections of kidneys (10 and 20 μm) were stained with X-Gal for 24 h as described (18).
Clearance Studies. In anesthetized male mice (16–20 weeks old), a catheter was inserted into the left femoral vein for application of FITC-labeled inulin (Sigma). The right femoral artery was catheterized and used for blood sampling and measurement of arterial blood pressure (Harvard Apparatus). After injection of a 1% inulin bolus in 0.9% NaCl solution at 2 μl/g of body weight followed by continuous inulin infusion of 0.15 μl/g of body weight per min mice were allowed to stabilize for 30 min. During a 30-min control period, additional 0.9% NaCl at 0.045 μl/g of body weight per min was infused. Thereafter, 1 mol/liter NaHCO3 at 0.045 μl/min per g of body weight was infused during two periods of 30 min. Plasma inulin concentration was measured at the beginning and end of each period and averaged for calculation of inulin clearance. Ionic composition of urine and serum samples was determined by Dionex ion chromatography, and inulin concentration was measured with a spectrofluorometer (Shimadzu). The total blood volume taken during the experiment did not exceed 150 μl, and every blood sample was readily replaced by the same amount of 0.9% NaCl solution. Fractional excretions (e.g., Fe-Na+) were calculated as ratios of excreted amount and filtered amount of the respective substance.
Northern Blot. RNA was isolated from adult mice as described (21), and poly(A)+ mRNA was purified with the Oligotex mRNA kit (Qiagen, Valencia, CA). Two micrograms of each RNA sample was separated by electrophoresis on a 1% agarose gel and transferred onto HybondN nylon membranes (Amersham Pharmacia) to be hybridized with a 32P-labeled TASK2 probe (sequence 496-1700 from GenBank accession no. AF319542) in Express Hyb solution (Clontech) at 65°C. The washed blot was exposed to a screen for Fuji Film Bio-Imaging analyzer BAS-1500.
Blood Gas Analysis and Urine pH and HCO3– Measurements. Venous blood from 9-week-old conscious male mice was collected into heparin-treated capillary tubes by puncture of the retrobulbar plexus. Blood gas measurements were performed immediately on a Radiometer ABL 555 analyzer (Radiometer, Copenhagen). Spot urine was collected from 5-month-old male mice and allowed to equilibrate in a 5% CO2/95% air atmosphere for 1 h. pH and calculated 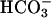 concentration were obtained on the ABL 555 analyzer.
concentration were obtained on the ABL 555 analyzer.
Statistics. Data are shown as mean values ± SEM from n observations. Paired as well as unpaired Student's t tests were used as appropriate. P < 0.05 was accepted to indicate statistical significance (*).
Results
General Appearance of task2 Knockout Mice. After breeding of heterozygous mice obtained from five to seven generations of backcross to C57BL6 genetic background, the percentage of task2 –/– mice in those litters was reduced: From a total of 305 mice at the age of weaning, 29 (9.5%) were task2 –/–, 79 (26%) were WT, and 197 (64.5%) were heterozygous. The low percentage of task2 –/– mice appears to be partially caused by an increased mortality during the neonatal period. Interestingly, this finding depended on the genetic background and was less pronounced at earlier stages of backcross from SV129 to C57BL6. Later on after weaning, task2 –/– mice thrived and were fertile but had a reduced body weight compared with WT mice (male mice: 30.8 ± 0.7 vs. task2 –/– 24.7 ± 0.7* g; female mice: 23.5 ± 0.6 vs. task2 –/– 19.8 ± 0.6* g, n = 9 each group). Bone mineral, fat, and lean contents were assessed by dual-energy x-ray absorptiometry scan. Bone mineral contents of WT and knockout mice were 0.36 ± 0.09 and 0.32 ± 0.01* g (n = 8 females each group); corresponding values for fat were 2.67 ± 0.21 and 2.03 ± 0.14* g; for lean 19.82 ± 1.03 and 16.61 ± 1.03* g. These data show that the body weight of task2 –/– mice is proportionally reduced for all three tested compartments.
Distribution of TASK2 in Mouse Tissues. Northern blot analysis revealed a strong expression of TASK2 in kidney and a lower expression in liver and trachea. TASK2 mRNA was not detected in brain, heart or skeletal muscle (Fig. 1A). The targeting vector used in the laboratory of W. Skarnes for the generation of task2 knockout mice contained a β-galactosidase gene, LacZ. In task2 –/– mice, β-galactosidase gene expression is controlled by the task2 promoter (22). The task2 promoter-driven X-Gal staining was observed in proximal tubules (all segments) and papillary collecting ducts (Fig. 1). Parallel experiments of WT tissues did not show such a staining.
Fig. 1.

Mouse TASK2 tissue distribution and localization in kidney. (A) Northern blot analysis of task2 expression in mouse adult tissues. Reprobing the same blot with a β-actin probe indicated the same poly(A)+ RNA content in each lane (not shown). The 4-kb task2 band was totally absent in blots made with RNA samples isolated from task2 –/– mice (data not shown). (B) TASK2 localization along the nephron. X-Gal staining was performed on a 20-μm-thick whole kidney cross section of a task2 +/– mouse. Blue staining was found in convoluted and straight proximal tubules and in papillary collecting ducts. The lower micrograph shows a cortical area at higher magnification (10-μm section).
Modulation of TASK2 Activity by 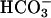 Transport in Cultured Renal Cells. We investigated the effects of
Transport in Cultured Renal Cells. We investigated the effects of 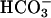 transport on TASK2 activity, using primary cultures of nephron segments microdissected from task2 +/+ and –/– mice. Whole-cell recordings were performed on confluent cells kept in a
transport on TASK2 activity, using primary cultures of nephron segments microdissected from task2 +/+ and –/– mice. Whole-cell recordings were performed on confluent cells kept in a 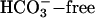 , weakly buffered bath solution (1 mM Hepes). The patch pipette was filled with a
, weakly buffered bath solution (1 mM Hepes). The patch pipette was filled with a 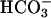 -containing solution allowing the diffusion of 25 mmol/liter
-containing solution allowing the diffusion of 25 mmol/liter 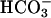 into the cytoplasm during whole-cell configuration. At the onset of experiments, 1 mM 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS), a blocker of
into the cytoplasm during whole-cell configuration. At the onset of experiments, 1 mM 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS), a blocker of 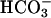 transport systems, was present in the bath solution. In these conditions, cells had a membrane voltage of –27.1 ± 11.4 mV (n = 8) and a whole-cell conductance of 2.1 ± 0.9 nS. After washout of DIDS, basolateral export of NaHCO3 was allowed to occur, presumably leading to alkalinization of the narrow basolateral space of the cell monolayer. In parallel, task2 +/+ cells hyperpolarized to –79.0 ± 6.7 mV and exhibited a large outward current resulting in an increase of whole-cell conductance to 20.4 ± 3.6 nS (Fig. 2A). When similar experiments were performed in a highly buffered bath solution (30 mM Hepes), no K+ outward current was elicited upon washout of DIDS (Fig. 2B). The reversal potential was –25.8 ± 7.5 mV, with a conductance of 3.7 ± 0.6 nS (n = 8) in the presence of DIDS. The values remained at –16 ± 5.8 mV and 4.8 ± 0.6 nS after DIDS removal. The likely mechanism of K+ channel activation is that
transport systems, was present in the bath solution. In these conditions, cells had a membrane voltage of –27.1 ± 11.4 mV (n = 8) and a whole-cell conductance of 2.1 ± 0.9 nS. After washout of DIDS, basolateral export of NaHCO3 was allowed to occur, presumably leading to alkalinization of the narrow basolateral space of the cell monolayer. In parallel, task2 +/+ cells hyperpolarized to –79.0 ± 6.7 mV and exhibited a large outward current resulting in an increase of whole-cell conductance to 20.4 ± 3.6 nS (Fig. 2A). When similar experiments were performed in a highly buffered bath solution (30 mM Hepes), no K+ outward current was elicited upon washout of DIDS (Fig. 2B). The reversal potential was –25.8 ± 7.5 mV, with a conductance of 3.7 ± 0.6 nS (n = 8) in the presence of DIDS. The values remained at –16 ± 5.8 mV and 4.8 ± 0.6 nS after DIDS removal. The likely mechanism of K+ channel activation is that 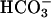 transport alkalinizes the extracellular fluid, which then activates pH-sensitive TASK2. Because the basolateral extracellular space of those cultured monolayers is very narrow, an alkalinization of the medium because of
transport alkalinizes the extracellular fluid, which then activates pH-sensitive TASK2. Because the basolateral extracellular space of those cultured monolayers is very narrow, an alkalinization of the medium because of 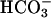 transport is likely to occur at low Hepes concentration as shown in Fig. 2 A. Such an alkalinization is diminished by the presence of 30 mM Hepes (Fig. 2B). As expected for a K+ conductance caused by TASK2 channels, the increase in outward current was prevented by 10 μM clofilium (Fig. 2D) and was not observed in task2 –/– cells (Fig. 2 C and D).
transport is likely to occur at low Hepes concentration as shown in Fig. 2 A. Such an alkalinization is diminished by the presence of 30 mM Hepes (Fig. 2B). As expected for a K+ conductance caused by TASK2 channels, the increase in outward current was prevented by 10 μM clofilium (Fig. 2D) and was not observed in task2 –/– cells (Fig. 2 C and D).
Fig. 2.

Whole-cell recordings of K+ currents on primary culture from proximal tubule cells from task2 +/+ and –/– mice. A large outward current was elicited upon DIDS washout in low buffered bath solution (A, 1 mM Hepes), which was absent in highly buffered external medium (B) and in task2 –/– cells (C). Solutions bath 1 and pipette 1 as described in Table 1. The membrane potential was held at –50 mV and stepped to test potential values between –100 and +120 mV in 20-mV increments. (D) Histograms of mean current values 200 ms after the onset of a pulse at +100 mV. Each value is the mean ± SEM of eight cells obtained from at least three distinct monolayers.
To test the requirement of Na+-coupled 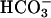 transport for TASK2 activation, Na+ was removed from the pipette, and N-methyl-d-glucamine chloride was used in the bath solution (Fig. 3 A and B). In these conditions,
transport for TASK2 activation, Na+ was removed from the pipette, and N-methyl-d-glucamine chloride was used in the bath solution (Fig. 3 A and B). In these conditions, 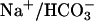 cotransport was abolished as reported (23), and the outward current was not activated. Subsequent perfusion of the monolayer with a Na+-containing, low-Cl– medium allowed the development of the clofilium-sensitive TASK2 current within 4 min. These results show that TASK2 outward current was Na+-dependent; in addition, a major contribution of Cl– influx to the outward current could be ruled out.
cotransport was abolished as reported (23), and the outward current was not activated. Subsequent perfusion of the monolayer with a Na+-containing, low-Cl– medium allowed the development of the clofilium-sensitive TASK2 current within 4 min. These results show that TASK2 outward current was Na+-dependent; in addition, a major contribution of Cl– influx to the outward current could be ruled out.
Fig. 3.

Electrolyte dependency of the K+ current. (A and B) Na+ and Cl– substitution experiments. (A) In the absence of Na+ ions (solutions bath 2 and pipette 2 in Table 1), no current was observed: reversal potential Erev =–10 ± 6.8 mV and conductance = 3.1 ± 0.8 nS. Subsequent perfusion of low-Cl– solution (Na-gluconate) allowed the development of the K+ conductance within 4 min: Erev =–76.4 ± 5.2 mV and conductance = 24.9 ± 4.9 nS, n = 10. (B) Histograms of mean current values 200 ms after the onset of a pulse at +100 mV. NMDG-Cl, N-methyl-d-glucamine chloride. Each value is the mean ± SEM of 10 cells obtained from at least three distinct monolayers. (C and D) Effect of the absence of cytosolic  .(C) When
.(C) When 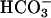 was omitted from the pipette solution (solutions bath 1 and pipette 3 in Table 1), no current was observed: conductance = 3.1 ± 0.7 nS and Erev =–28.3 ± 7.3 mV. Subsequent alkalization by changing external solution (bath 1 at pH 8 in Table 1) produced an increase in K+ conductance, 11.5 ± 0.8 nS and Erev =–79.8 ± 7.3 mV, n = 9. (D) Histograms of mean current values 200 ms after the onset of a pulse at +100 mV. Each value is the mean ± SEM of nine cells obtained from at least three distinct monolayers.
was omitted from the pipette solution (solutions bath 1 and pipette 3 in Table 1), no current was observed: conductance = 3.1 ± 0.7 nS and Erev =–28.3 ± 7.3 mV. Subsequent alkalization by changing external solution (bath 1 at pH 8 in Table 1) produced an increase in K+ conductance, 11.5 ± 0.8 nS and Erev =–79.8 ± 7.3 mV, n = 9. (D) Histograms of mean current values 200 ms after the onset of a pulse at +100 mV. Each value is the mean ± SEM of nine cells obtained from at least three distinct monolayers.
To further examine the need of 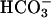 transport for activation of TASK2,
transport for activation of TASK2, 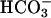 was omitted from the pipette. In this case, no TASK2 K+ current was observed upon DIDS washout (Fig. 3C). However, the
was omitted from the pipette. In this case, no TASK2 K+ current was observed upon DIDS washout (Fig. 3C). However, the 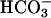 transport-induced alkalinization of the basolateral space could be mimicked by increasing the bath pH from 7.4 to 8.0, which elicited a clofilium-sensitive K+ current (Fig. 3 C and D).
transport-induced alkalinization of the basolateral space could be mimicked by increasing the bath pH from 7.4 to 8.0, which elicited a clofilium-sensitive K+ current (Fig. 3 C and D).
Kidney Function of task2 Knockout Mice. To test whether the findings obtained from primary cultured proximal tubular cells apply to proximal tubule function in vivo, we performed clearance measurements on anesthetized WT and task2 –/– mice. As a measure of glomerular filtration rate, we determined the inulin clearance. Under control conditions, task2 –/– mice showed a slightly (but not significantly) lower inulin clearance. Fractional excretions of Na+ and Cl– were slightly (but not significantly) higher in task2 –/– mice (Table 2). Mean arterial blood pressure was significantly lower in knockout mice (Table 3 and Fig. 4A). Under these conditions, arterial pH values and concentrations of plasma electrolytes of task2 –/– were not different from those of WT mice (Table 3).
Table 2. Urine parameters.
| Parameter | task2 +/+ | task2 -/- |
|---|---|---|
| Control | ||
| Vurine, ml/min/g | 0.069 ± 0.008 | 0.078 ± 0.014 |
| Inulin clearance, μl/min/g | 12.0 ± 1.1 | 9.3 ± 1.1 |
| Fe-Na+, % | 0.37 ± 0.09 | 0.94 ± 0.28 |
| Fe-K+, % | 27.3 ± 2.8 | 31.9 ± 4.0 |
| Fe-Cl-, % | 0.92 ± 0.16 | 1.67 ± 0.43 |
| Inulinurine/inulinplasma | 190 ± 25 | 185 ± 45 |
| Alkalosis | ||
| Vurine, μl/min/g | 0.067 ± 0.010 | 0.103 ± 0.015 |
| Inulin clearance, μl/min/g | 10.6 ± 1.0 | 11.1 ± 1.3 |
| Fe-Na+, % | 0.77 ± 0.18 | 1.56 ± 0.27* |
| Fe-K+, % | 30.8 ± 3.4 | 38.3 ± 4.0 |
| Fe-Cl-, % | 1.37 ± 0.29 | 2.20 ± 0.26* |
| Inulinurine/inulinplasma | 192 ± 31 | 112 ± 11* |
Results are mean ± SEM, n = 7-9 each. *, P < 0.05 indicates statistical significance.
Table 3. Plasma parameters and blood pressure.
| Parameter | task2 +/+ | task2 -/- |
|---|---|---|
| Control | ||
| Na+, mM | 145.3 ± 3.5 | 141.0 ± 2.8 |
| K+, mM | 4.0 ± 0.2 | 4.6 ± 0.3 |
| Cl-, mM | 116.9 ± 2.8 | 113.8 ± 5.4 |
| pH | 7.38 ± 0.02 | 7.36 ± 0.03 |
| Blood pressure, mm Hg | 76.0 ± 1.7 | 68.6 ± 2.8* |
| Alkalosis | ||
| Na+, mM | 150.2 ± 5.5 | 148.5 ± 4.7 |
| K+, mM | 3.9 ± 0.4 | 4.0 ± 0.2 |
| Cl-, mM | 116.7 ± 4.4 | 120.5 ± 7.5 |
| pH | 7.58 ± 0.03 | 7.61 ± 0.04 |
| Blood pressure, mm Hg | 75.1 ± 2.5 | 72.9 ± 1.8 |
Results are mean ± SEM, n = 7-9 each. *, P < 0.05 indicates statistical significance.
Fig. 4.

Effect of 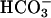 challenge on renal function. (A) Inulin clearance during alkalosis is shown in Bottom. After a 30-min control period, 1 mol/liter NaHCO3 at 0.045 μl/min per g of body weight was applied i.v. during two periods of 30 min (alk. 1 and alk. 2). n.s., not significant. Top shows the effect of NaHCO3 perfusion on arterial pH and Middle shows the effect on mean arterial blood pressure (art. femoralis). During perfusion with NaHCO3, the blood pressure of task2 –/– increased (n = 7–9 each). (B) Effect of alkalosis on Na+ and water excretion. During control, fractional Na+ excretion (Fe-Na+) was not different between WT +/+ and task2 –/– mice. After 60 min of alkalosis, Fe-Na+ was increased in task2 –/– mice. (C) Under these conditions, concentration of urine (ratio of urinary to plasmatic inulin concentrations) was decreased in task2 –/– but not in WT mice.
challenge on renal function. (A) Inulin clearance during alkalosis is shown in Bottom. After a 30-min control period, 1 mol/liter NaHCO3 at 0.045 μl/min per g of body weight was applied i.v. during two periods of 30 min (alk. 1 and alk. 2). n.s., not significant. Top shows the effect of NaHCO3 perfusion on arterial pH and Middle shows the effect on mean arterial blood pressure (art. femoralis). During perfusion with NaHCO3, the blood pressure of task2 –/– increased (n = 7–9 each). (B) Effect of alkalosis on Na+ and water excretion. During control, fractional Na+ excretion (Fe-Na+) was not different between WT +/+ and task2 –/– mice. After 60 min of alkalosis, Fe-Na+ was increased in task2 –/– mice. (C) Under these conditions, concentration of urine (ratio of urinary to plasmatic inulin concentrations) was decreased in task2 –/– but not in WT mice.
To examine kidney function under conditions with an increased proximal tubular 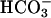 load, we performed inulin clearance experiments during alkalosis. After a 30-min control period, proximal tubular NaHCO3 reabsorption was challenged by i.v. perfusion of NaHCO3. The continuous NaHCO3 infusion induced an alkalinization of arterial blood pH from ≈7.4 to ≈7.6 (Table 3 and Fig. 4A). In parallel, mean arterial blood pressure of knockout mice increased to values similar to WT mice (Fig. 4A). Knockout mice excreted more diluted urine after NaHCO3 infusion as measured by the ratio of inulin concentrations in urine and plasma (inulinurine/inulinplasma, Table 2 and Fig. 4C). The low urine concentration of knockout mice was parallel to an increase in fractional excretion of Na+ and Cl– (Table 2 and Fig. 4B). No difference was observed in the fractional excretion of K+. These data are suggestive of an activation of TASK2 during proximal tubular NaHCO3 reabsorption in vivo. Knockout mice appear to have a reduced maximal transport capacity of proximal tubular cells for NaHCO3 resulting in an increased loss of Na+ and water.
load, we performed inulin clearance experiments during alkalosis. After a 30-min control period, proximal tubular NaHCO3 reabsorption was challenged by i.v. perfusion of NaHCO3. The continuous NaHCO3 infusion induced an alkalinization of arterial blood pH from ≈7.4 to ≈7.6 (Table 3 and Fig. 4A). In parallel, mean arterial blood pressure of knockout mice increased to values similar to WT mice (Fig. 4A). Knockout mice excreted more diluted urine after NaHCO3 infusion as measured by the ratio of inulin concentrations in urine and plasma (inulinurine/inulinplasma, Table 2 and Fig. 4C). The low urine concentration of knockout mice was parallel to an increase in fractional excretion of Na+ and Cl– (Table 2 and Fig. 4B). No difference was observed in the fractional excretion of K+. These data are suggestive of an activation of TASK2 during proximal tubular NaHCO3 reabsorption in vivo. Knockout mice appear to have a reduced maximal transport capacity of proximal tubular cells for NaHCO3 resulting in an increased loss of Na+ and water.
To test whether renal 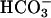 handling and acid–base metabolism are affected by the task2 gene disruption under control conditions, we measured blood gases (Table 4), pH, and
handling and acid–base metabolism are affected by the task2 gene disruption under control conditions, we measured blood gases (Table 4), pH, and 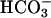 in spot urine. Retroorbital venous blood samples were taken from conscious mice to eliminate possible genotype-specific differences in the responsiveness toward anesthetics. task2 –/– mice had reduced venous blood pH and
in spot urine. Retroorbital venous blood samples were taken from conscious mice to eliminate possible genotype-specific differences in the responsiveness toward anesthetics. task2 –/– mice had reduced venous blood pH and 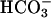 concentrations and more negative systemic base excess compared with WT mice. The urine pH and calculated
concentrations and more negative systemic base excess compared with WT mice. The urine pH and calculated 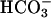 concentrations were higher in task2 –/– mice (pH = 6.77 ± 0.04*, and
concentrations were higher in task2 –/– mice (pH = 6.77 ± 0.04*, and 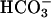 = 5.5 ± 1.0 mM*, n = 9), as compared with WT (pH = 6.51 ± 0.08, and
= 5.5 ± 1.0 mM*, n = 9), as compared with WT (pH = 6.51 ± 0.08, and  = 2.9 ± 0.7 mM, n = 9). These results are in agreement with a metabolic acidosis (via renal
= 2.9 ± 0.7 mM, n = 9). These results are in agreement with a metabolic acidosis (via renal  loss) of task2 –/– mice. Therefore, pH-sensitive TASK2 K+ channels appear to be important in renal acid–base metabolism.
loss) of task2 –/– mice. Therefore, pH-sensitive TASK2 K+ channels appear to be important in renal acid–base metabolism.
Table 4. Venous blood gas analysis of conscious male mice.
| Parameter | task2 +/+ | task2 -/- |
|---|---|---|
| pH | 7.33 ± 0.01 | 7.30 ± 0.01* |
| pCO2 | 47.5 ± 1.2 | 46.3 ± 1.3 |
| pO2 | 40.8 ± 0.7 | 39.7 ± 1.0 |
| HCO3- | 24.7 ± 0.8 | 21.9 ± 0.7* |
| SBEc | -0.48 ± 0.87 | -3.55 ± 0.78* |
Results are mean ± SEM, n = 21 and 22. SBEc, systemic base excess. *, P < 0.05 indicates statistical significance.
Discussion
Diversity of Renal K+ Channels. K+ channels have been shown to play an important role in vectorial transport in renal epithelia. By serial analysis of gene expression, as many as 28 different K+ channel genes have been recently shown to be expressed in human kidney (13). The precise function of only some of them is presently known: Mutations in ROMK in Bartter's syndrome have shed light on the essential role of this channel in transport in the thick ascending limb of Henle's loop (24–26). Alternatively, genetic manipulation of mice has allowed defining the role of KCNQ1/KCNE1 K+ channel complex in proximal tubule (2). In this study, we have investigated the renal phenotype of the task2 (kcnk5) knockout mouse (22). By using X-Gal staining as a convenient method to localize TASK2-expressing cells in kidneys, we found strong labeling in proximal tubule and in papillary collecting ducts. No labeling was observed in glomeruli, distal tubule, cortical collecting duct, and medulla (Fig. 1). These findings contrast with a previous study showing TASK2 mRNA expression in human distal tubules and cortical collecting ducts (16). This difference is probably not accountable for differences between mice and human, because TASK2 is one of 16 K+ channels that have been observed by serial analysis of gene expression in human proximal tubules (13).
Function of TASK2 in Primary Cultured Proximal Tubular Cells. Several different K+ conductances have been described by functional (patch-clamp and microperfusion) and immunohistochemical methods in proximal tubules. Luminal K+ channels are activated mainly during solute transport by depolarization of the luminal membrane and cytosolic Ca2+, and are probably inhibited by cGMP (2, 27–31). In the basolateral membrane, K+ channel activity is functionally coupled to Na+/K+ ATPase activity, thereby allowing K+ recycling and basolateral hyperpolarization. Moreover, basolateral K+ channels are activated by cell swelling and inhibited by intracellular ATP and low pH (4, 32, 33). In isolated perfused mouse and rabbit proximal tubules, regulatory cell volume decrease is dependent on K+ and 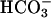 (6, 34). In the present report, we have demonstrated that a large K+ conductance was turned on in mouse cultured proximal tubular cells under conditions where
(6, 34). In the present report, we have demonstrated that a large K+ conductance was turned on in mouse cultured proximal tubular cells under conditions where 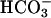 transport is activated. The activation of this conductance was almost absent in proximal tubular cells from task2 –/– mice, indicating that TASK2 underlies this conductance. The activation of TASK2 channels appears to be mediated by the rise in basolateral extracellular pH induced by
transport is activated. The activation of this conductance was almost absent in proximal tubular cells from task2 –/– mice, indicating that TASK2 underlies this conductance. The activation of TASK2 channels appears to be mediated by the rise in basolateral extracellular pH induced by 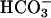 transport for four reasons: (i) the current was not observed in the presence of DIDS; (ii) it was observed when
transport for four reasons: (i) the current was not observed in the presence of DIDS; (ii) it was observed when 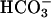 was present in the cytosol and basolateral
was present in the cytosol and basolateral 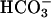 transport was allowed to take place (Fig. 2A); (iii) increased buffer capacity in the extracellular medium diminished the K+ current (Fig. 2B); and (iv) alkalinization of extracellular pH elicited a similar K+ conductance (Fig. 3C). Hence, TASK2 appears to be a basolateral K+ channel of proximal tubules physiologically activated during
transport was allowed to take place (Fig. 2A); (iii) increased buffer capacity in the extracellular medium diminished the K+ current (Fig. 2B); and (iv) alkalinization of extracellular pH elicited a similar K+ conductance (Fig. 3C). Hence, TASK2 appears to be a basolateral K+ channel of proximal tubules physiologically activated during 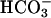 transport. The TASK2-induced hyperpolarization then provides the driving force for ongoing electrogenic
transport. The TASK2-induced hyperpolarization then provides the driving force for ongoing electrogenic  cotransport (35). Apart from TASK2, however, there are probably several other K+ channels in the basolateral membrane of proximal tubules that are mainly regulated by cytosolic pH or ATP, e.g., Kir7.1 (KCNJ13) (11), Kir4.2 (KCNJ15) (13, 36), Kir5.1 (KCNJ16) (13).
cotransport (35). Apart from TASK2, however, there are probably several other K+ channels in the basolateral membrane of proximal tubules that are mainly regulated by cytosolic pH or ATP, e.g., Kir7.1 (KCNJ13) (11), Kir4.2 (KCNJ15) (13, 36), Kir5.1 (KCNJ16) (13).
task2 –/– Mice Lose Na+, 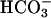 , and Water in Urine. Next, we tested whether TASK2 plays a role in renal
, and Water in Urine. Next, we tested whether TASK2 plays a role in renal 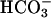 transport in vivo by characterizing the renal phenotype of task2 –/– mice by using inulin clearance measurements. Under control conditions, kidney function of task2 –/– mice was not significantly different from that of WT mice but they had the tendency to lose more Na+ in urine. Consistently, they displayed a reduced arterial blood pressure that could be caused by enhanced renal salt loss. To unmask defects in renal NaHCO3 handling, we performed a NaHCO3 challenge after the control period. As expected, arterial pH rose in both genotypes during continuous injection of NaHCO3, which should have resulted in maximal TASK2 activation in WT mice. After 1 h of NaHCO3 perfusion, task2 –/– exhibited an increased Na+ and water loss compared with WT mice. These results suggest that maximal reabsorption capacity for NaHCO3 is impaired in task2 –/– mice, leading to a pronounced loss of Na+ and water, which cannot, or at least not completely, be compensated by distal nephron segments. During NaHCO3 perfusion, arterial blood pressure and inulin clearance were selectively increased in task2 –/– mice. Probably, the lower blood pressure of task2 –/– mice during the control period was caused by contraction of the extracellular volume, which was too small to be directly detected by difference in Na+ excretion.
transport in vivo by characterizing the renal phenotype of task2 –/– mice by using inulin clearance measurements. Under control conditions, kidney function of task2 –/– mice was not significantly different from that of WT mice but they had the tendency to lose more Na+ in urine. Consistently, they displayed a reduced arterial blood pressure that could be caused by enhanced renal salt loss. To unmask defects in renal NaHCO3 handling, we performed a NaHCO3 challenge after the control period. As expected, arterial pH rose in both genotypes during continuous injection of NaHCO3, which should have resulted in maximal TASK2 activation in WT mice. After 1 h of NaHCO3 perfusion, task2 –/– exhibited an increased Na+ and water loss compared with WT mice. These results suggest that maximal reabsorption capacity for NaHCO3 is impaired in task2 –/– mice, leading to a pronounced loss of Na+ and water, which cannot, or at least not completely, be compensated by distal nephron segments. During NaHCO3 perfusion, arterial blood pressure and inulin clearance were selectively increased in task2 –/– mice. Probably, the lower blood pressure of task2 –/– mice during the control period was caused by contraction of the extracellular volume, which was too small to be directly detected by difference in Na+ excretion.
Some 75% of 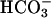 reabsorption is known to occur in proximal tubules, and the rest is reabsorbed in more distal nephron segments, such as thick ascending limb of Henle's loop, distal tubules, and collecting ducts (37). Because we had evidence for loss of Na+ and water even under control conditions, we tested whether acid–base metabolism was affected as well by analyzing urine bicarbonate excretion, blood pH, and gases on resting conscious animals. Consistent with a significant increase in urine pH and
reabsorption is known to occur in proximal tubules, and the rest is reabsorbed in more distal nephron segments, such as thick ascending limb of Henle's loop, distal tubules, and collecting ducts (37). Because we had evidence for loss of Na+ and water even under control conditions, we tested whether acid–base metabolism was affected as well by analyzing urine bicarbonate excretion, blood pH, and gases on resting conscious animals. Consistent with a significant increase in urine pH and  concentration, blood pH,
concentration, blood pH, 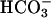 , and systemic base excess were significantly lower in task2–/– mice. Because blood CO2 was not different, all these parameters are pointing to a metabolic acidosis. The observed disturbance of acid–base metabolism in task2 –/– mice is caused probably by an increased proximal tubular loss of Na+,
, and systemic base excess were significantly lower in task2–/– mice. Because blood CO2 was not different, all these parameters are pointing to a metabolic acidosis. The observed disturbance of acid–base metabolism in task2 –/– mice is caused probably by an increased proximal tubular loss of Na+,  , and consecutively water, which cannot be fully compensated by distal nephron segments. At present, we cannot rule out a functional defect in TASK2-expressing papillary collecting ducts contributing to the renal
, and consecutively water, which cannot be fully compensated by distal nephron segments. At present, we cannot rule out a functional defect in TASK2-expressing papillary collecting ducts contributing to the renal 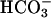 loss. However, the pivotal role of proximal tubules in renal
loss. However, the pivotal role of proximal tubules in renal 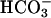 reabsorption and our study on primary cultured proximal tubular cells suggest that the renal phenotype of task2 –/– is caused mainly by defects in proximal tubular function. Interestingly, the acid–base status of task2 –/– mice is very similar to that observed in patients suffering from isolated proximal renal tubular acidosis (Online Mendelian Inheritance in Man no. 179830). So far, carbonic anhydrase II (38), luminal Na+/H+ exchanger (39), and basolateral
reabsorption and our study on primary cultured proximal tubular cells suggest that the renal phenotype of task2 –/– is caused mainly by defects in proximal tubular function. Interestingly, the acid–base status of task2 –/– mice is very similar to that observed in patients suffering from isolated proximal renal tubular acidosis (Online Mendelian Inheritance in Man no. 179830). So far, carbonic anhydrase II (38), luminal Na+/H+ exchanger (39), and basolateral 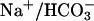 transporter (40) have been key players for
transporter (40) have been key players for 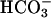 transport across proximal tubular epithelium. We propose TASK2 as another protein engaged in proximal tubular
transport across proximal tubular epithelium. We propose TASK2 as another protein engaged in proximal tubular 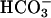 reabsorption: when activated by cell swelling and basolateral
reabsorption: when activated by cell swelling and basolateral 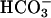 accumulation, TASK2 K+ channels hyperpolarize the cell membrane, thereby supporting ongoing electrogenic exit of Na+ and
accumulation, TASK2 K+ channels hyperpolarize the cell membrane, thereby supporting ongoing electrogenic exit of Na+ and 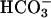 across the basolateral membrane (Fig. 5).
across the basolateral membrane (Fig. 5).
Fig. 5.

Model of putative TASK2 function in proximal tubule cells. Based on functional studies, TASK2 appears to be located in the basolateral membrane of proximal tubular cells. NaHCO3 reabsorption involves Na+/H+ exchange across the apical membrane. Na+ and 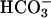 ions leave the cell by
ions leave the cell by 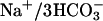 cotransporter thereby depolarizing the basolateral membrane. In the extracellular space, rise in
cotransporter thereby depolarizing the basolateral membrane. In the extracellular space, rise in 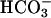 concentration causes an increase in pH that then activates basolateral TASK2 K+ channels. TASK2 activity recycles K+ accumulated by Na+/K+-ATPase and leads to repolarization of the membrane that is needed as a driving force for ongoing NaHCO3 export. CA, carbonic anhydrase; NHE, Na+/H+ exchanger.
concentration causes an increase in pH that then activates basolateral TASK2 K+ channels. TASK2 activity recycles K+ accumulated by Na+/K+-ATPase and leads to repolarization of the membrane that is needed as a driving force for ongoing NaHCO3 export. CA, carbonic anhydrase; NHE, Na+/H+ exchanger.
Involvement of several proteins in 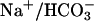 transport across proximal tubular epithelium is evocative of Na+ reabsorption in thick ascending limb of Henle's loop. In thick ascending limb, Na+ reabsorption requires luminal Na+2Cl–K+ cotransporter (NKCC2), luminal ROMK K+ channels, and basolateral Cl– channel (ClCKB and Barttin). Defects in each of these proteins are related to severe renal salt wasting (Bartter syndrome, Online Mendelian Inheritance in Man no. 241200). In proximal tubules, Na+/H+ exchanger, carbonic anhydrases,
transport across proximal tubular epithelium is evocative of Na+ reabsorption in thick ascending limb of Henle's loop. In thick ascending limb, Na+ reabsorption requires luminal Na+2Cl–K+ cotransporter (NKCC2), luminal ROMK K+ channels, and basolateral Cl– channel (ClCKB and Barttin). Defects in each of these proteins are related to severe renal salt wasting (Bartter syndrome, Online Mendelian Inheritance in Man no. 241200). In proximal tubules, Na+/H+ exchanger, carbonic anhydrases, 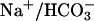 cotransporter, and TASK2 K+ channels appear to act in concert during NaHCO3 reabsorption.
cotransporter, and TASK2 K+ channels appear to act in concert during NaHCO3 reabsorption.
Acknowledgments
We thank Dr. K. Mitchell and Prof. Dr. W. Skarnes for generously providing the task2 –/– mice, M. M. Larroque for expert assistance, and Prof. Dr. G. Giebisch for reading the manuscript and fruitful discussions. This work was supported by the Centre National de la Recherche Scientifique and the Association Française Contre les Myopathies (J.B.), and by Forschungskcredit der Universtität Zürich, the Swiss National Science Foundation, and Deutsche Forschungsgemeinschaft Grant WA1274/4-1 (to R.W.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: DIDS, 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid; Fe, fractional excretion; X-Gal, 5-bromo-4-chloro-3-indolyl β-d-galactoside.
References
- 1.Giebisch, G., Hebert, S. C. & Wang, W. H. (2003) Pflügers Arch. 446, 289–297. [DOI] [PubMed] [Google Scholar]
- 2.Vallon, V., Grahammer, F., Richter, K., Bleich, M., Lang, F., Barhanin, J., Völkl, H. & Warth, R. (2001) J. Am. Soc. Nephrol. 12, 2003–2011. [DOI] [PubMed] [Google Scholar]
- 3.Lang, F., Messner, G. & Rehwald, W. (1986) Am. J. Physiol. 250, F953–F962. [DOI] [PubMed] [Google Scholar]
- 4.Giebisch, G. & Wang, W. (2000) Acta Physiol. Scand. 170, 153–173. [DOI] [PubMed] [Google Scholar]
- 5.Beck, J. S., Laprade, R. & Lapointe, J. Y. (1994) Am. J. Physiol. 266, F517–F527. [DOI] [PubMed] [Google Scholar]
- 6.Beck, J. S., Breton, S., Giebisch, G. & Laprade, R. (1992) Am. J. Physiol. 263, F453–F458. [DOI] [PubMed] [Google Scholar]
- 7.Coppola, S. & Frömter, E. (1994) Pflügers Arch. 427, 143–150. [DOI] [PubMed] [Google Scholar]
- 8.Yao, X., Tian, S., Chan, H. Y., Biemesderfer, D. & Desir, G. V. (2002) J. Am. Soc. Nephrol. 13, 2831–2839. [DOI] [PubMed] [Google Scholar]
- 9.Gu, W., Schlichthorl, G., Hirsch, J. R., Engels, H., Karschin, C., Karschin, A., Derst, C., Steinlein, O. K. & Daut, J. (2002) J. Physiol. (London) 539, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cluzeaud, F., Reyes, R., Escoubet, B., Fay, M., Lazdunski, M., Bonvalet, J. P., Lesage, F. & Farman, N. (1998) Am. J. Physiol. 275, C1602–C1609. [DOI] [PubMed] [Google Scholar]
- 11.Derst, C., Hirsch, J. R., Preisig-Muller, R., Wischmeyer, E., Karschin, A., Doring, F., Thomzig, A., Veh, R. W., Schlatter, E., Kummer, W., et al. (2001) Kidney Int. 59, 2197–2205. [DOI] [PubMed] [Google Scholar]
- 12.Derst, C., Karschin, C., Wischmeyer, E., Hirsch, J. R., Preisig-Muller, R., Rajan, S., Engel, H., Grzeschik, K., Daut, J. & Karschin, A. (2001) FEBS Lett. 491, 305–311. [DOI] [PubMed] [Google Scholar]
- 13.Chabardes-Garonne, D., Mejean, A., Aude, J. C., Cheval, L., Di Stefano, A., Gaillard, M. C., Imbert-Teboul, M., Wittner, M., Balian, C., Anthouard, V., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 13710–13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lesage, F., Guillemare, E., Fink, M., Duprat, F., Lazdunski, M., Romey, G. & Barhanin, J. (1996) EMBO J. 15, 1004–1011. [PMC free article] [PubMed] [Google Scholar]
- 15.Lesage, F. (2003) Neuropharmacology 44, 1–7. [DOI] [PubMed] [Google Scholar]
- 16.Reyes, R., Duprat, F., Lesage, F., Fink, M., Salinas, M., Farman, N. & Lazdunski, M. (1998) J. Biol. Chem. 273, 30863–30869. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann, E. K. & Hougaard, C. (2001) Comp. Biochem. Physiol. A Physiol. 130, 355–366. [DOI] [PubMed] [Google Scholar]
- 18.Barriere, H., Belfodil, R., Rubera, I., Tauc, M., Lesage, F., Poujeol, C., Guy, N., Barhanin, J. & Poujeol, P. (2003) J. Gen. Physiol. 122, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skarnes, W. C., Moss, J. E., Hurtley, S. M. & Beddington, R. S. P. (1995) Proc. Natl. Acad. Sci. USA 92, 6592–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerstin, K. M., Gong, D. H., Abdallah, M., Winegar, B. D., Eger, E. I. & Gray, A. T. (2003) Anesth. Analg. 96, 1345–1349. [DOI] [PubMed] [Google Scholar]
- 21.Chomczynski, P. & Sacchi, N. (1987) Anal. Biochem. 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell, K. J., Pinson, K. I., Kelly, O. G., Brennan, J., Zupicich, J., Scherz, P., Leighton, P. A., Goodrich, L. V., Lu, X., Avery, B. J., et al. (2001) Nat. Genet. 28, 241–249. [DOI] [PubMed] [Google Scholar]
- 23.Alpern, R. J. (1985) J. Gen. Physiol. 86, 613–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simon, D. B., Karet, F. E., Rodriguez-Soriano, J., Hamdan, J. H., DiPietro, A., Trachtman, H., Sanjad, S. A. & Lifton, R. P. (1996) Nat. Genet. 14, 152–156. [DOI] [PubMed] [Google Scholar]
- 25.Lorenz, J. N., Baird, N. R., Judd, L. M., Noonan, W. T., Andringa, A., Doetschman, T., Manning, P. A., Liu, L. H., Miller, M. L. & Shull, G. E. (2002) J. Biol. Chem. 277, 37871–37880. [DOI] [PubMed] [Google Scholar]
- 26.Lu, M., Wang, T., Yan, Q., Yang, X., Dong, K., Knepper, M. A., Wang, W., Giebisch, G., Shull, G. E. & Hebert, S. C. (2002) J. Biol. Chem. 277, 37881–37887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawahara, K., Hunter, M. & Giebisch, G. (1987) Am. J. Physiol. 253, F488–F494. [DOI] [PubMed] [Google Scholar]
- 28.Merot, J., Bidet, M., Le Maout, S., Tauc, M. & Poujeol, P. (1989) Biochim. Biophys. Acta 978, 134–144. [DOI] [PubMed] [Google Scholar]
- 29.Gögelein, H. & Greger, R. (1984) Pflügers Arch. 401, 424–426. [DOI] [PubMed] [Google Scholar]
- 30.Lang, F. & Rehwald, W. (1992) Physiol. Rev. 72, 1–32. [DOI] [PubMed] [Google Scholar]
- 31.Hirsch, J. R., Weber, G., Kleta, I. & Schlatter, E. (1999) J. Physiol. 519, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beck, J. S., Hurst, A. M., Lapointe, J. Y. & Laprade, R. (1993) Am. J. Physiol. 264, F496–F501. [DOI] [PubMed] [Google Scholar]
- 33.Noulin, J. F., Brochiero, E., Lapointe, J. Y. & Laprade, R. (1999) Am. J. Physiol. 277, F290–F297. [DOI] [PubMed] [Google Scholar]
- 34.Völkl, H. & Lang, F. (1988) Pflügers Arch. 412, 1–6. [DOI] [PubMed] [Google Scholar]
- 35.Yoshitomi, K., Burckhardt, B. C. & Frömter, E. (1985) Pflügers Arch. 405, 360–366. [DOI] [PubMed] [Google Scholar]
- 36.Lourdel, S., Paulais, M., Cluzeaud, F., Bens, M., Tanemoto, M., Kurachi, Y., Vandewalle, A. & Teulon, J. (2002) J. Physiol. 538, 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Capasso, G., Unwin, R., Rizzo, M., Pica, A. & Giebisch, G. (2002) J. Nephrol. 15, Suppl. 5, S88–S96. [PubMed] [Google Scholar]
- 38.Sly, W. S., Whyte, M. P., Sundaram, V., Tashian, R. E., Hewett-Emmett, D., Guibaud, P., Vainsel, M., Baluarte, H. J., Gruskin, A., Al Mosawi, M., et al. (1985) N. Engl. J. Med. 313, 139–145. [DOI] [PubMed] [Google Scholar]
- 39.Schultheis, P. J., Clarke, L. L., Meneton, P., Miller, M. L., Soleimani, M., Gawenis, L. R., Riddle, T. M., Duffy, J. J., Doetschman, T., Wang, T., et al. (1998) Nat. Genet. 19, 282–285. [DOI] [PubMed] [Google Scholar]
- 40.Igarashi, T., Inatomi, J., Sekine, T., Cha, S. H., Kanai, Y., Kunimi, M., Tsukamoto, K., Satoh, H., Shimadzu, M., Tozawa, F., et al. (1999) Nat. Genet. 23, 264–266. [DOI] [PubMed] [Google Scholar]