


Abstract
Repair of DNA double-strand breaks (DSBs) in mammalian cells by nonhomologous end-joining (NHEJ) is initiated by the DNA–PK protein complex. Recent studies have shown inositol hexakisphosphate (InsP6) is a potent cofactor for DNA–PK activity in NHEJ. Specifically, InsP6 binds to the Ku component of DNA–PK, where it induces a conformational change and a corresponding increase in DNA end-joining activity. However, the effect of InsP6 on the dynamics of Ku, such as its mobility in the nucleus, is unknown. Importantly, these dynamics reflect the character of Ku’s interactions with other molecules. To address this question, the diffusion of Ku was measured by fluorescence photobleaching experiments using cells expressing green fluorescent protein (GFP)-labeled Ku. InsP6 was depleted by treating cells with calmodulin inhibitors, which included the compounds W7 and chlorpromazine. These treatments caused a 50% reduction in the mobile fraction of Ku–GFP, and this could be reversed by replenishing cells with InsP6. By expressing deletion mutants of Ku–GFP, it was determined that its W7-sensitive region occurred at the N-terminus of the dimerization domain of Ku70. These results therefore show that InsP6 enhances Ku mobility through a discrete region of Ku70, and modulation of InsP6 levels in cells represents a potential avenue for regulating NHEJ by affecting the dynamics of Ku and hence its interaction with other nuclear proteins.
INTRODUCTION
Generation of DNA double-strand breaks (DSBs) represents a potentially cataclysmic event for the viability of a cell. In mammalian cells, the principle mechanism of repair of DNA DSBs is by nonhomologous end-joining (NHEJ) (1). NHEJ is initiated by the protein complex DNA–PK, which is composed of Ku and a catalytic subunit (DNA–PKCS). Ku itself is a heterodimer composed of a 70 and 80 kDa subunit, termed Ku70 and Ku80, respectively. The sequence of events for the DNA end-joining reaction is that Ku first binds to the free ends of the DSB in a sequence- and structure-independent manner (2–5), and this is followed by recruitment of DNA–PKCS to the DNA–Ku complex (6–8). Assembly of DNA–PK at the DNA lesion leads to recruitment of additional DNA repair enzymes, including the XRCC4/DNA ligase IV complex (9,10). Related to its role in repair of DSBs, Ku functions in repair of DNA lesions that form as a result of ionizing radiation and chemical agents, as well as DSBs that occur during V(D)J recombination and immunoglobulin class switching in developing lymphocytes (11–16).
Structurally, the Ku heterodimer forms a ring, with DNA passing through the channel in the center of the complex (17). Recently, our studies have shown that Ku is highly mobile in cell nuclei, exhibiting a transient, high flux interaction with other molecules that include the nuclear matrix (18). These findings were obtained by fluorescence photobleaching experiments of cells expressing Ku molecules labeled with green fluorescent protein (GFP). Importantly, the dynamic nature characteristic of Ku is shared with other nuclear proteins that function in DNA and RNA synthesis and processing (19–22), thus suggesting this represents a paradigm for protein–nucleic acid interactions. Based on our finding regarding binding of Ku to nuclear matrix, we proposed a model where the nuclear matrix serves as a scaffold for assembly of NHEJ machinery, and Ku tethers the DNA ends to this assembly (18).
Recent studies have shown that inositol phosphates serve as a cofactor for DNA–PK in NHEJ activity (23–25). For example, inositol tetrakisphosphate (InsP4), inositol pentakisphosphate (InsP5) and inositol hexakisphosphate (InsP6) each are capable of enhancing end-joining activity by DNA–PK, with InsP6 being the most effective of these (23). The mechanism by which InsP6 serves as a cofactor for DNA–PK is unclear. However, it is known that InsP6 binds Ku, where it causes Ku to undergo a conformational change as detected by changes in its proteolytic sensitivity (24). One interpretation of these results is that InsP6 augments NHEJ by affecting interactions of Ku with other molecules through changes in its structure. As a protein’s mobility often reflects the nature of its intermolecular interactions, changes in the structure of Ku by InsP6 may also affect its mobility.
Physiologically, higher inositol phosphates are synthesized via an initial enzymatic step where phosphate is added to position 3 of Ins(1,4,5)-trisphosphate (InsP3) by InsP3 3-kinase to yield InsP4 (26). Interestingly, the C isozyme of InsP3 3-kinase is inhibited by Ca2+, and both the A and C forms are activated by calmodulin (27). Thus, treatment of cells with a calmodulin antagonist may effectively deplete cells of InsP6 and other higher inositol phosphates by inhibiting synthesis of the InsP4 precursor. Reduction in InsP6 pools by calmodulin inhibitors would therefore allow us to test the effect of lowering InsP6 levels on the mobility of Ku and its interactions with other molecules.
To test our hypothesis, HeLa cells were treated with the calmodulin antagonist N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide (W7). Chromatography showed W7 caused an 85% reduction in InsP6 levels in cells, and this correlated with a 50% decrease in the mobile fraction of Ku–GFP in the cell nucleus. Inhibition of Ku mobility by a separate calmodulin inhibitor, chlorpromazine (CPZ), was reversed by replenishing the cells with InsP6. By construction and expression of Ku70 deletion mutants, we mapped the site of W7 sensitivity to a 59 amino acid segment between residues 416 and 475, and this corresponds to the N-terminus of the dimerization domain of Ku70. These results suggest that regulation of InsP6 levels, such as through Ca2+ and calmodulin signaling, could regulate NHEJ by altering the structure and dynamics of Ku.
MATERIALS AND METHODS
Gene construction
Ku70–GFP and Ku70CD–GFP (residues 263–556) have been described previously (18). The Ku70 deletion mutants were generated by cloning PCR products into the mammalian expression vector pcDNA3.1 (Invitrogen, Carlsbad, CA) upstream of and in-frame to the coding sequence of enhanced GFP (eGFP; Clontech, Palo Alto, CA). A stop codon was located at the 3′ end of eGFP.
DNA encoding the respective constructs were amplified using the following oligonucleotides as primers for the coding strand in the PCR reaction: Ku70314–556, ACATCA TCTAGAATGAGCGATACCAAGAGGTCTCA; Ku70372–556, ACATCATCTAGAATGGAGTCGCTGGTGATTGG GAG; Ku70416–556, ACATCATCTAGAATGCAGGAAG AAGAGTTGGATGA; Ku70475–556, ACATCATCTAGA ATGAGTGACAGCTTTGAGAACCC; Ku70533–556, ACA TCATCTAGAATGGATTACAATCCTGAAGGGAA. The underlined residues indicate the start codon. The noncoding primer for each reaction was: TCTAGTGAATTCGTTG TTGTTGTTGTTCTTGGGCCTTTTGCTTCCAG. The residues in bold in the coding and noncoding primers indicate XbaI and EcoRI sites, respectively, that were used for subcloning the amplified products into the vector. Each of the noncoding primers contains a poly-glutamine insert between the C-terminus of the Ku subunit and the N-terminus of GFP. This was added in order to enhance folding of the separate protein domains. Glutamine was used for this purpose since it has the lowest propensity for forming secondary structures (28).
A separate construct containing Ku70 residues 263–416 followed by residues 475–556 was synthesized by separately amplifying the two gene segments with the following primers: ACATCATCTAGAATGCTCAACAAAGATATAGTGAT and TCATCCAACTCTTCTTCCTG were the coding and noncoding primers, respectively, for the gene segment encoding residues 263–416, and AGTGACAGCTTT GAGAACCC and TCTAGTGAATTCGTTGTTGTTGTTG TTCTTGGGCCTTTTGCTTCCAG for the coding and noncoding primers for amplifying the gene segment encoding residues 475–556. The residues in bold again indicate XbaI and EcoRI sites used for cloning into the expression vector. The separate gene segments were ligated at the blunt ends of the separate PCR products.
Cell culture and protein expression
HeLa and 293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with antibiotics and 10% fetal bovine serum. Cells were maintained at 37°C in the presence of 5% CO2. For fluorescence imaging experiments, 6.0 × 105 HeLa cells were seeded onto a coverslip in a 3.5 cm dish the day prior to the experiment. The cells were transfected using lipid carrier (Superfect, Qiagen, Valencia, CA) with 5 µg of plasmid DNA. For W7 treatment of cells, drug was diluted directly into culture medium from a 50 mM stock solution in 50% ethanol to a final concentration of 500 µM. CPZ and K-252a treatments consisted of a likewise addition of compound into culture media to a final concentration of 100 µM CPZ and 10 µM K-252a. Cells were incubated with the drugs for 2 h at 37°C prior to measurement. In experiments that included replenishment of InsP6 following treatment with CPZ, the samples were first washed following an initial 2 h incubation with CPZ, followed by addition of growth media containing 5 mM InsP6. Cells were maintained in InsP6-enriched media for 4 h prior to measurement.
Cell lysis, immunoprecipitation and immunoblotting
Transfected 293T cells were washed twice with phosphate-buffered saline (PBS) and lysed using 750 µl of a 10 mM NaCl, 3 mM MgCl2, 10 mM Tris (pH 7.4) (RSB) buffer containing 0.5% Triton X-100 (TX-100), 1.0 mM PMSF and 100 KU of aprotinin. After 10 min in lysis buffer, the samples were centrifuged for 5 min at 4000 r.p.m. using an Eppendorf desktop centrifuge (model 54150). The pellet containing intact nuclei was suspended in 500 ml of 150 mM NaCl, 1 mM EDTA, 20 mM Tris (pH 8.0), 0.1% TX-100, 10% glycerol, 1.0 mM PMSF and 100 KU of aprotinin, and the nuclei were lysed by sonication. The lysate was centrifuged, and the supernatant was removed and immunoprecipitated using a monoclonal antibody specific for GFP (Covance Research Products, Inc., Richmond, CA). Pansorbin (Calbiochem, San Diego, CA) was used as a solid phase for the immunoprecipitations. The immunoprecipitates were washed twice with 10 mM Tris (pH 8.0), 150 mM NaCl, 5 mM EDTA containing 0.10% TX-100, and eluted using SDS–PAGE sample buffer (29). The samples were separated by gel electrophoresis and detected by immunoblotting using a polyclonal antibody to Ku86 (Santa Cruz Biotech, Santa Cruz, CA). The membranes were developed using ECL (Amersham, Piscataway, NJ).
Inositol phosphate analysis
HeLa cells were grown in suspension in a mixture of 50% RPMI and 50% inositol-free RPMI supplemented with 10% dialyzed fetal calf serum. Cells were labeled for 4 days with 50 µCi/ml 3H-myo-inositol (Amersham). On the day of assay, aliquots (5 ml) were taken and the respective compounds were added as described above. Cells were then harvested by centrifugation, washed with HEPES-buffered saline, and quenched using 0.6 M perchloric acid. Neutralization and separation of inositol phosphates was performed as previously described (30).
Fluorescence microscopy
Images were collected using a Leica TCS laser scanning confocal microscope (William K. Warren Medical Research Institute, Oklahoma City, OK). GFP was excited at 488 nm and emission wavelengths 530–560 nm were collected for imaging.
Protein mobility was measured by fluorescence recovery after photobleaching (FRAP) as described previously (18). In brief, a region of the nucleus was photobleached using a brief (2.0 s) pulse of the 488 nm line of an Argon laser at 100% power. Recovery of fluorescence within the bleached region was monitored by collecting a frame every 1.7 s, and quantitated by measuring the fluorescence of the bleached region using IP Lab Spectrum software (Scanalytics, Vienna, VA). The mobile fraction of protein and time constant for its recovery was quantitated by fitting the recovery of the fluorescence to the function Ft / F0 = F∞ + F1 ∗ e(–t/τ), where F∞ represents the fraction of recovery at infinite time and indicates the mobile fraction of the molecule in the bleached region, or, inversely, its immobile fraction. τ is the time constant for recovery and it is inversely proportional to the diffusion coefficient. The recovery values were corrected for loss in signal during the experiment as described (18,19).
RESULTS
W7 depletes cells of InsP6, and inhibits Ku mobility
To determine the effect of calmodulin antagonists on the production of InsP6 and other species of inositol phosphates, HeLa cells were treated with 500 µM W7 for 1 h at 37°C prior to measurement of their inositol phosphates pools by strong anion exchange (SAX) chromatography. As shown in Figure 1, W7 caused depletion of multiple species of inositol phosphates, including an 85% reduction in the amount of InsP6 (fraction 61). This result shows W7 represents a useful tool for depleting cellular pools of InsP6 and other inositol phosphates.
Figure 1.

The calmodulin inhibitor W7 depletes cellular inositol phosphate pools. Lysates from 3H-myo-inositol-labeled HeLa cells were separated by SAX as described in Materials and Methods. Prior to lysis, the samples were treated with either 500 µM W7 for 1 h at 37°C, or left untreated. The peaks in the elution profile were identified using 3H-labeled standards.
To measure the effect of W7 on the mobility of Ku in the nucleus, FRAP measurements were performed using HeLa cells expressing a gene encoding Ku70 followed by GFP (Ku70–GFP). FRAP experiments provide a measure of protein mobility by determining the rate of protein diffusion into a photobleached region of the nucleus (31). Furthermore, we have shown previously that Ku70–GFP dimerizes with endogenous Ku80, and that the GFP-labeled molecules represent a faithful reporter of Ku (18). As shown in Figure 2, the FRAP experiments were performed using both W7-treated cells (Fig. 2A) and cells that received no treatment (Fig. 2B). In each example, only the nucleus of the cell is evident as the nuclear localization signal (NLS) of Ku70 efficiently targets protein to this compartment (32). Interestingly, Ku–GFP exhibited a reticulated pattern in W7-treated cells (arrowhead, Fig. 2A) that was lacking in cells that received no treatment. Furthermore, the mobility of Ku–GFP in W7-treated cells contrasted with that in untreated cells by exhibiting a lack of recovery of fluorescence following photobleaching. For example, in the experiment shown in Figure 2A and Supplementary Figure 1, essentially none of the fluorescence recovered after more than 30 s following photobleaching, whereas the untreated cell underwent full recovery in this time (Figure 2B and Supplementary Figure 2). Importantly, the recovery of GFP alone (i.e. not fused to Ku) was not effected by W7. For example, the W7-treated cell expressing GFP showed complete recovery following photobleaching (Figure 2C and Supplementary Figure 3), similar to that observed for the cell that received no treatment (Figure 2D and Supplementary Figure 4). As shown previously (18), GFP was localized to both the cytoplasm and nucleus. Hence, we have indicated the edge of the nucleus with black arrowheads.
Figure 2.

W7 specifically inhibits the mobility of Ku–GFP. (A–D) FRAP measurements of Ku70–GFP. (A and B) The cells were bleached using a 2 s pulse of laser illumination. The site of bleaching is indicated by the green squares. Each frame was collected at the indicated time post-bleaching. In (A), the sample was pre-treated with 500 µM W7 for 2 h prior to measurement. The white arrowhead in (A) indicates a Ku–GFP-enriched structure that appears reticulated throughout the nucleus. In (B), the sample was untreated before measurement. The white bars represent 5 µm. (C and D) The same as (A) and (B), except the cells expressed GFP. The black arrowheads indicate the boundary of the nucleus. (E) Recovery curves for the FRAP experiments in (A) and (B). The y-axis represents the normalized fluorescence intensity of the bleached region at each time point, and it was calculated as described (18). (F) The same as (E), except representing recovery curves for the experiments in (C) and (D). The mobile fraction from multiple FRAP measurements of Ku–GFP or GFP alone were averaged and plotted (G). Sample sizes were 17 for Ku–GFP in W7-treated cells, and 19 for the sample with Ku70–GFP but with no treatment. The sample sizes for the GFP experiments were 10 and 19 for the W7-treated and untreated samples, respectively.
The effect of W7 on the mobility of Ku70–GFP is further illustrated by the recovery curves in Figure 2E, which show that the bleached spot in the untreated cell fully recovered by 15 s, whereas recovery in the W7-treated cell was never >20%. Note in the untreated cell that the earliest point had a value of 70% recovery, and this is due to recovery that has occurred between the end of the photobleaching pulse and the acquisition of the first post-bleach frame (∼1.5 s). In the recovery curves, Ft / F0 at t = ∞ represents the mobile fraction of fluorophore. These results show that essentially the entire pool of Ku70–GFP was mobile in the untreated cell, but the mobile fraction was reduced to 20% in the cell that was treated with W7. Furthermore, the recovery curves for GFP alone shows its mobile fraction was not effected by treatment of the cells with W7, with both the untreated and W7-treated samples exhibiting essentially complete recovery (Fig. 2F).
FRAP measurements of Ku–GFP were repeated for cells that were either untreated or treated with W7, as well as with cells that expressed GFP. The results of these measurements are shown in Figure 2E. In summary, the average mobile fraction of Ku–GFP decreased by approximately one-half following treatment with W7, from a value of ∼0.9 for the untreated cells to <0.5 for the sample containing W7. In contrast, the mobile fraction of GFP was close to 1.0 in the treated cells, similar to that measured for the untreated sample. We conclude from these results that W7 specifically inhibited the mobility of Ku–GFP as reflected by a decrease in its mobile fraction, and this correlated with the depletion of cellular pools of InsP6 and other inositol phosphates by W7.
The reduction in Ku70–GFP mobility by calmodulin inhibitors is reversible
To determine if W7-mediated depletion of InsP6 and reduction in the mobile fraction of Ku70–GFP was specific to this compound alone, HeLa cells were treated with a separate calmodulin inhibitor, CPZ. SAX chromatography of cell lysates showed that CPZ caused a 69% reduction in the amount of InsP6 in the cells (Fig. 3A), and this correlated with a 50% reduction in the mobile fraction of Ku70–GFP (Fig. 3B–D). Interestingly, the calmodulin kinase II inhibitor K-252a, which did not affect InsP6 levels (data not shown), also did not alter the mobility of Ku70–GFP (Fig. 3). Thus, the reduction in the InsP6 content of cells and mobility of Ku70–GFP occurred with separate calmodulin inhibitors, but not when calmodulin kinase II-dependent pathways were inhibited.
Figure 3.

Loss of Ku70–GFP mobility by calmodulin inhibitors is reversed by addition of InsP6. (A) HeLa cells were treated with 100 µM CPZ for 2 h prior to cell lysis and measurement of inositol phosphates by SAX chromatography. CPZ caused a 69% reduction in the amount of InsP6 relative to the untreated sample. (B) FRAP measurements of HeLa cells expressing Ku70–GFP and treated with either 100 µM CPZ for 2 h, CPZ followed by 5 mM InsP6 for 4 h, or 10 µM K-252a for 2 h. Recovery curves for the FRAP experiments in (B) are shown in (C). The mobile fraction from multiple FRAP measurements of cells treated as in (B) were averaged and plotted (D). The sample sizes for (D) were 5, 8, 11 and 9 for the untreated sample, cells treated with CPZ alone, CPZ and then replenished with InsP6, and K-252a alone, respectively.
To determine if the change in the mobility of Ku70–GFP caused by calmodulin inhibitors was reversible, samples treated with CPZ were replenished with InsP6 as described (33). FRAP experiments showed the mobility of Ku70–GFP was rescued by replenishing cells with InsP6 (Fig. 3B–D). Importantly, maintaining the cells in media lacking InsP6 following treatment with CPZ failed to recover Ku70–GFP mobility (data not shown). These results therefore show that the reduction in the mobile fraction of Ku70–GFP by calmodulin inhibitors is reversible using InsP6.
Construction and expression of Ku70 deletion mutants
To identify the region of Ku70 conferring changes in the mobility of Ku–GFP by W7, five separate N-terminal deletion mutants were constructed, beginning with the central domain (CD, residues 263–556) that contains both the core region of Ku70 residues 255–450 (34) and its NLS residues from 539 to 556 (32) (Fig. 3). The constructs were engineered such that each began in a region that was void of significant secondary structure as defined by the crystal structure of the Ku–DNA complex (17). Furthermore, for detection by fluorescence microscopy, each was fused to GFP at its C-terminus. Confocal microscopy showed each of the constructs was efficiently expressed (Fig. 4). Deleting from the N-terminus of the CD protein maintained the NLS of Ku70, and Figure 4 shows each of the proteins was localized to the nucleus.
Figure 4.
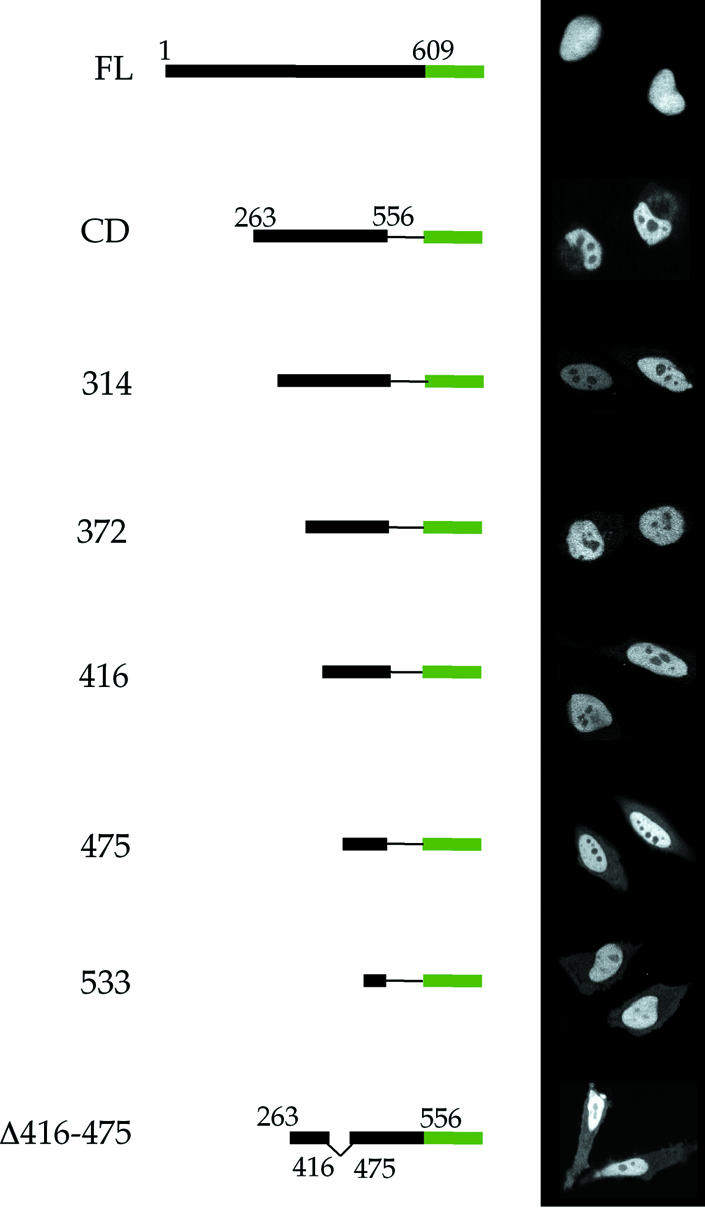
Targeting of Ku70–GFP deletion mutants to the nucleus. The central domain (CD) of Ku70 contains the core of full length (FL) Ku70 (residues 263–450), as well as the NLS (residues 535–550). Constructs 314, 372, 416, 475 and 533 consist of consecutive deletions of approximately 50 amino acids from the N-terminus of CD. Δ416–475 contains the central domain of Ku70 minus residues 416–475. Confocal images of transfected HeLa cells showed that all of the constructs were efficiently targeted to the nucleus (right).
Residues 416–475 of Ku70 mediate W7 sensitivity of Ku mobility
To measure the effect of W7 on the mobility of the Ku70 deletion constructs, FRAP measurements were performed using transfected HeLa cells that were either untreated or treated with W7 for 2 h prior to measurement. Between 10 and 15 measurements were made for each protein in each set of conditions. The results are illustrated in Figure 5A, and examples of FRAP experiments with each deletion construct are shown in Figure 5B. To summarize, CD, 314, 372, and 416 proteins each exhibited a significant decrease in their mobile fraction following treatment of the cells with W7, similar to that observed for full length (FL) Ku70–GFP. Conversely, deletion of the region between residues 416 and 475 resulted in proteins where the mobility was not altered by treatment with W7.
Figure 5.
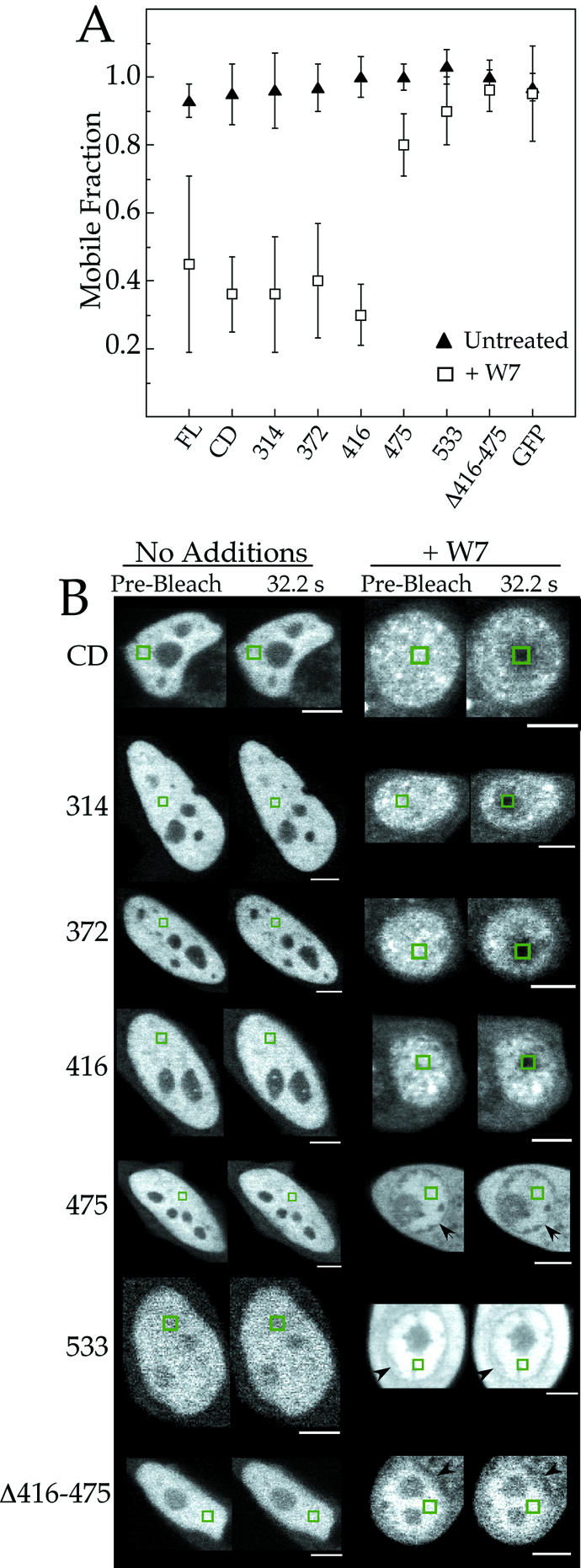
Mapping of the W7-sensitive region of Ku70 to residues 416–475. (A) The mobile fraction of Ku70, GFP and each of the Ku70 mutants (Fig. 3) was measured in W7- and untreated-HeLa cells. The values represent the average of 10–15 separate measurements. An example of each of the experiments in (A) is shown in (B). The black arrowheads in (B) point to the boundary of the nucleus in those conditions where the fluorescence labeling was not restricted to the nucleus. The white bars represent 5 µm.
To determine if deletion of the sequence between residues 416 and 475 was sufficient to disrupt the W7-mediated reduction in Ku mobility, an additional deletion mutant was constructed that encoded residues 263–416 of Ku70, followed by 475–556 (Δ416–475). The Δ416–475 mutant therefore represents an internal deletion of the Ku70CD, and confocal microscopy showed that it was efficiently expressed and targeted to the nucleus (Fig. 4). Furthermore, FRAP measurements showed W7 had no effect on the mobile fraction of the Δ416–475 mutant (Fig. 5). We conclude from these results that the W7-sensitive region of Ku70 occurs in the sequence between residues 416 and 475.
Interestingly, although a similar number of cells were measured for each sample in Figure 5, there was a significant difference in the standard deviation of full-length protein and that of the shorter constructs. One interpretation of this finding is that the large standard deviation of the full-length protein reflects heterogeneities in protein–protein and protein–DNA interactions, and these heterogeneities are lost by deleting portions of Ku70.
Another interesting observation is that the 475, 533, and Δ416–475 constructs were not restricted to the nucleus in cells treated with W7 (Fig. 5B). This is not due to disruption of the nuclear envelope since the other constructs remained concentrated in the nucleus in these conditions. Localization of 475, 533 and Δ416–475 in the cytoplasm may be related to changes in the permeability of the nuclear pore complex by W7, which could then effect the ability of these proteins to diffuse between the nucleus and cytoplasm.
Residues 416–475 of Ku70 contain its dimerization site
To correlate the W7- and CPZ-sensitive regions of Ku70 with their known structural and functional domains, the Ku70 deletion mutants and their sensitivity to W7 was mapped against the documented DNA-binding and dimerization domains of Ku70 (35) (Fig. 6A). These results show that deletion of the DNA-binding domain did not alter W7-mediated reductions in protein mobility. In contrast, deletion of the N-terminus of the Ku dimerization domain, beginning with construct 475 and including the Δ416–475 protein, abolished the changes in protein mobility by W7. These results suggest W7 mediates its effects on Ku70–GFP mobility through the Ku heterodimer in a manner unrelated to Ku binding to DNA.
Figure 6.

Mapping of W7 sensitivity to Ku70 functional and structural domains. (A) Loss of sensitivity of protein mobility to W7 corresponds to deletions in the Ku70 dimerization domain. (B) Crystal structure of the Ku70 (green)–Ku80 (blue) heterodimer complexed with DNA (gold). Residues 416–475 of Ku70 are labeled red. I is a view of the complex down the axis of the DNA, and II is turned ∼90° relative to I. (C) Measurement of protein co-immunoprecipitation with Ku80. Each of the Ku70–GFP deletion mutants were expressed and immunoprecipitated using monoclonal antibody to GFP. The samples were separated by protein electrophoresis, and the Ku80 that co-immunoprecipitated with expressed protein was detected by immunoblotting. Molecular weights (in thousands) are indicated on the right.
To further correlate W7 and CPZ sensitivity of Ku70–GFP with the structure of Ku, we mapped residues 416–467 of Ku70 on a ribbon diagram of the crystal structure of Ku (17) (Fig. 6B). Green and blue represent the Ku70 and Ku80 subunits, respectively, and the gold ribbon corresponds to the DNA that was co-crystallized with Ku. Residues 416–475 of Ku70 are labeled red. I and II represent two separate views of the Ku–DNA complex, each of which show that the 416–475 segment interfaces extensively with the Ku80 subunit, but is not proximal to the DNA. This is especially evident in II of Figure 5B, which shows that the 416–475 segment wraps about the surface of the β barrel structural domain of Ku80. These structural data further suggest that the sensitivity of Ku70–GFP mobility to calmodulin inhibitors is related to its heterodimerization. Consistent with this notion, Ku80 was effeciently co-immunoprecipitated with only the 314, 372, and 416 proteins (Fig. 6C).
DISCUSSION
In eukaryotic cells, DNA repair by NHEJ is initiated by the Ku heterodimer component of DNA–PK, which binds to the DNA free ends and recruits additional DNA repair machinery. Recent findings have shown that the inositol phosphates, especially InsP6, serve as a potent cofactor for DNA–PK, augmenting NHEJ activity via binding to Ku (23,24). In this study, we have shown that depletion of cellular pools of inositol phosphates by the calmodulin inhibitors W7 and CPZ caused a significant and specific decrease in the mobility of Ku–GFP, and this was reflected by a 50% or greater reduction in its mobile fraction. Importantly, replenishing cells with InsP6 following treatment with CPZ caused the mobile fraction of Ku70–GFP to return to a value similar to that measured in untreated control cells. This result is evidence that the loss of Ku70–GFP mobility by W7 and CPZ is due to their effect on InsP6 pools in cells. One interpretation of these results is that binding of InsP6 to Ku alters the physical structure of Ku in such a manner that the mobility of Ku is enhanced, and this is consistent with data showing InsP6-mediated changes in the conformation of Ku (23).
Based on our findings regarding the effect of W7 on cellular pools of inositol phosphates, including InsP6, and its effect on the mobility of Ku in the nucleus, we propose a model where InsP6 enhances Ku mobility by affecting the association of Ku with other molecules within this compartment (Fig. 7). In the absence of InsP6, Ku exists in a conformation that favors stable binding to these factors, resulting in a decreased pool of mobile protein. Binding of InsP6 causes a change in the conformation of Ku that favors the disassociation of Ku, thus yielding a larger mobile pool of Ku in the nucleus.
Figure 7.
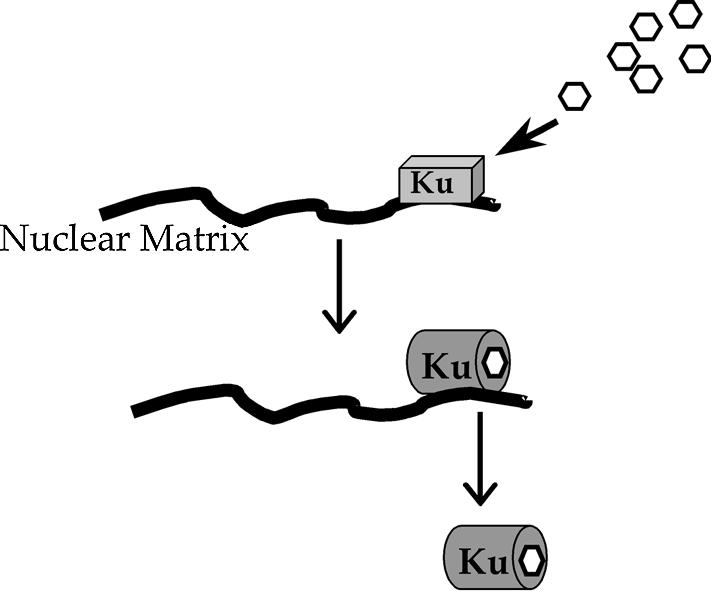
A model for W7-mediated changes in Ku mobility. Binding of InsP6 to Ku leads to a change in conformation that destabilizes association of Ku with protein factors, such as nuclear matrix. W7 depletes inositol phosphate levels, leading to a locking of Ku in the matrix-bound conformation, and a corresponding decrease in the mobile fraction in the nucleus.
One finding in this study is that deletion of the DNA-binding domain of Ku70 between residues 254 and 439 had no effect on W7-mediated loss of mobility. In contrast, deletion of the N-terminus of the Ku70 dimerization resulted in proteins whose mobility was no longer effected by W7. One conclusion from these results is that immobilization of Ku70–GFP occurs by binding of the Ku heterodimer to other factors in the nucleus in a manner that is independent of Ku binding to DNA. Importantly, the changes in the mobility of Ku by W7 and CPZ may be the outcome of multiple and cooperative interactions of Ku with other molecules that include separate regions of Ku70. Recent studies have shown that the C-terminus of Ku70 functions in suppressing apoptosis by interacting with Bax (36,37). Thus, discrete regions of Ku70 interact with other protein factors, and these may cooperate within the heterodimer in conferring immobilization by the calmodulin inhibitors.
We have previously shown Ku binds to the nuclear matrix through the Ku70 subunit (18). This result together with the abundance of nuclear matrix filaments suggest that the nuclear matrix is a good candidate for a ligand that could ‘tie-up’ Ku in its InsP6-low conformation. Interestingly, confocal images of cells treated with W7 showed Ku–GFP was condensed onto filamentous structures (Fig. 2A), similar to that observed for the matrix marker matrin 250 (18). A transient association of Ku–GFP with nuclear filaments in untreated cells has also been detected by fluorescence photobleaching experiments, and this correlates with association of Ku70–GFP with the nuclear matrix (18). These latter results therefore further suggest that the nuclear matrix is a principal site of stable binding of Ku in the absence of inositol phosphates. We are currently investigating the mechanisms by which Ku associates with the nuclear matrix, and the significance of this association on NHEJ.
Another property of fluorescence recovery following photobleaching is the rate of recovery, or the time constant. This value is proportional to the size of the molecule and the viscosity of its environment (38). Our measurements showed a 3–4-fold increase in the time constant for each of the Ku70–GFP constructs, as well as that of GFP alone. We therefore conclude that the decrease in the rate of recovery is nonspecific in nature, and may be related to changes in physical properties of the nucleus caused by W7, such as the viscosity of the compartment.
InsP6 is an abundant molecule in cells (39), and it is therefore likely that most of the Ku will be in the InsP6-bound state. This is consistent with our finding that essentially the entire pool of Ku–GFP in untreated cells is mobile. Given the role of InsP6 as a cofactor in DNA–PK activity (23–25) and the effect of its depletion on Ku mobility, it is possible that localized regulation of NHEJ is achieved by modifying the concentration of InsP6. For example, recent data has shown that inositol phosphate pools are not in equilibrium (40), suggesting that levels of separate inositol phosphate species can be discretely regulated. Thus, activation of enzymes that hydrolyze InsP6 to less substituted forms that do not function in modifying Ku structure could inhibit Ku mobility, and therefore the accessibility of DSBs to Ku and DNA repair. Interestingly, higher InsPs have been shown to function in regulating other important nuclear events, including RNA export, gene regulation, and regulation of recombination (41–45). Regulation of DSB repair by InsP6 through modification of the structural and dynamic properties of Ku would therefore be representative of regulation of other nuclear events by inositol phosphates.
In summary, we have shown that the calmodulin inhibitor W7 decreases cellular pools of inositol phosphates which serve as cofactors for DNA–PK, including InsP6. Furthermore, a reduction in InsP6 by W7 corresponded to a 50% or greater reduction in the mobile fraction of Ku–GFP. These data suggest that association of Ku with other molecules in the nucleus, such as nuclear matrix, is regulated by InsP6, and this could occur by InsP6-mediated changes in the structure of Ku. Inositol phosphates therefore represent one potential physiological avenue for regulation of DNA–PK.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank K. Rodgers for assistance with the molecular modeling, L. Hanakahi and J. D. Capra for helpful discussions. This work was supported by National Institutes of Health grant P50 RR015577, and S.T.S. is a Royal Society University Research fellow and is supported by The Royal Society and Tenovus Scotland.
REFERENCES
- 1.Smith G.C. and Jackson,S.P. (1999) The DNA-dependent protein kinase. Genes Dev., 13, 916–934. [DOI] [PubMed] [Google Scholar]
- 2.Mimori T. and Hardin,J.A. (1986) Mechanism of interaction between Ku protein and DNA. J. Biol. Chem., 261, 10375–10379. [PubMed] [Google Scholar]
- 3.Paillard S. and Strauss,F. (1991) Analysis of the mechanism of interaction of simian Ku protein with DNA. Nucleic Acids Res., 19, 5619–5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ono M., Tucker,P.W. and Capra,J.D. (1994) Production and characterization of recombinant human Ku antigen. Nucleic Acids Res., 22, 3918–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ono M., Tucker,P.W. and Capra,J.D. (1996) Ku is a general inhibitor of DNA-protein complex formation and transcription. Mol. Immunol., 33, 787–796. [DOI] [PubMed] [Google Scholar]
- 6.Dynan W.S. and Yoo,S. (1998) Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res., 26, 1551–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dvir A., Peterson,S.R., Knuth,M.W., Lu,H. and Dynan,W.S. (1992) Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc. Natl Acad. Sci. USA, 89, 11920–11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gottlieb T.M. and Jackson,S.P. (1993) The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell, 72, 131–142. [DOI] [PubMed] [Google Scholar]
- 9.Grawunder U., Wilm,M., Wu,X., Kulesza,P., Wilson,T.E., Mann,M. and Lieber,M.R. (1997) Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature, 388, 492–495. [DOI] [PubMed] [Google Scholar]
- 10.Ramsden D.A. and Gellert,M. (1998) Ku protein stimulates DNA end joining by mammalian DNA ligases: a direct role for Ku in repair of DNA double-strand breaks. EMBO J., 17, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu Y., Jin,S., Gao,Y., Weaver,D.T. and Alt,F.W. (1997) Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity and inability to support V(D)J recombination. Proc. Natl Acad. Sci. USA, 94, 8076–8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manis J.P., Gu,Y., Lansford,R., Sonoda,E., Ferrini,R., Davidson,L., Rajewsky,K. and Alt,F.W. (1998) Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J. Exp. Med., 187, 2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nussenzweig A., Chen,C., da Costa Soares,V., Sanchez,M., Sokol,K., Nussenzweig,M.C. and Li,G.C. (1996) Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature, 382, 551–555. [DOI] [PubMed] [Google Scholar]
- 14.Getts R.C. and Stamato,T.D. (1994) Absence of a Ku-like DNA end binding activity in the xrs double-strand DNA repair-deficient mutant. J. Biol. Chem., 269, 15981–15984. [PubMed] [Google Scholar]
- 15.Smider V., Rathmell,W.K., Lieber,M.R. and Chu,G. (1994) Restoration of X-ray resistance and V(D)J recombination in mutant cells by Ku cDNA. Science, 266, 288–291. [DOI] [PubMed] [Google Scholar]
- 16.Taccioli G.E., Gottlieb,T.M., Blunt,T., Priestley,A., Demengeot,J., Mizuta,R., Lehmann,A.R., Alt,F.W., Jackson,S.P. and Jeggo,P.A. (1994) Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science, 265, 1442–1445. [DOI] [PubMed] [Google Scholar]
- 17.Walker J.R., Corpina,R.A. and Goldberg,J. (2001) Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature, 412, 607–614. [DOI] [PubMed] [Google Scholar]
- 18.Rodgers W., Jordan,S.J. and Capra,J.D. (2002) Transient association of Ku with nuclear substrates characterized using fluorescence photobleaching. J. Immunol., 168, 2348–2355. [DOI] [PubMed] [Google Scholar]
- 19.Phair R.D. and Misteli,T. (2000) High mobility of proteins in the mammalian cell nucleus. Nature, 404, 604–609. [DOI] [PubMed] [Google Scholar]
- 20.McNally J.G., Muller,W.G., Walker,D., Wolford,R. and Hager,G.L. (2000) The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science, 287, 1262–1265. [DOI] [PubMed] [Google Scholar]
- 21.Chen D. and Huang,C. (2001) Nucleolar components involved in ribosome biogenesis cycle between the nucleolus and nucleoplasm in interphase cells. J. Cell Biol., 153, 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boisvert F.M., Kruhlak,M.J., Box,A.K., Hendzel,M.J. and Bazett-Jones,D.P. (2001) The transcription coactivator cbp is a dynamic component of the promyelocytic leukemia nuclear body. J. Cell Biol., 152, 1099–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanakahi L.A., Bartlet-Jones,M., Chappell,C., Pappin,D. and West,S.C. (2000) Binding of inositol phosphate to DNA–PK and stimulation of double-strand break repair. Cell, 102, 721–729. [DOI] [PubMed] [Google Scholar]
- 24.Hanakahi L.A. and West,S.C. (2002) Specific interaction of IP6 with human Ku70/80, the DNA-binding subunit of DNA–PK. EMBO J., 21, 2038–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma Y. and Lieber,M.R. (2002) Binding of inositol hexakisphosphate (IP6) to Ku but not to DNA–PKcs. J. Biol. Chem., 277, 10756–10759. [DOI] [PubMed] [Google Scholar]
- 26.Berridge M.J. and Irvine,R.F. (1989) Inositol phosphates and cell signalling. Nature, 341, 197–205. [DOI] [PubMed] [Google Scholar]
- 27.Dewaste V., Pouillon,V., Moreau,C., Shears,S., Takazawa,K. and Erneux,C. (2000) Cloning and expression of a cDNA encoding human inositol 1,4,5-trisphosphate 3-kinase C. Biochem. J., 352, 343–351. [PMC free article] [PubMed] [Google Scholar]
- 28.Minor D.L. Jr and Kim,P.S. (1994) Measurement of the beta-sheet-forming propensities of amino acids. Nature, 367, 660–663. [DOI] [PubMed] [Google Scholar]
- 29.Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 30.Safrany S.T. and Shears,S.B. (1998) Turnover of bis-diphosphoinositol tetrakisphosphate in a smooth muscle cell line is regulated by beta2-adrenergic receptors through a cAMP-mediated, A-kinase-independent mechanism. EMBO J., 17, 1710–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Axelrod D., Koppel,D.E., Schlessinger,J., Elson,E. and Webb,W.W. (1976) Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J., 16, 1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koike M., Ikuta,T., Miyasaka,T. and Shiomi,T. (1999) The nuclear localization signal of the human Ku70 is a variant bipartite type recognized by the two components of nuclear pore-targeting complex. Exp. Cell Res., 250, 401–413. [DOI] [PubMed] [Google Scholar]
- 33.Shamsuddin A.M. and Yang,G.Y. (1995) Inositol hexaphosphate inhibits growth and induces differentiation of PC-3 human prostate cancer cells. Carcinogenesis, 16, 1975–1979. [DOI] [PubMed] [Google Scholar]
- 34.Jin S. and Weaver,D.T. (1997) Double-strand break repair by Ku70 requires heterodimerization with Ku80 and DNA binding functions. EMBO J., 16, 6874–6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X. and Lieber,M.R. (1996) Protein-protein and protein–DNA interaction regions within the DNA end-binding protein Ku70–Ku86. Mol. Cell Biol., 16, 5186–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sawada M., Hayes,P., Matsuyama,S., Sun,W., Leskov,K. and Boothman,D.A. (2003) Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nature Cell Biol., 5, 352–357. [DOI] [PubMed] [Google Scholar]
- 37.Sawada M., Sun,W., Hayes,P., Leskov,K., Boothman,D.A. and Matsuyama,S. (2003) Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nature Cell Biol., 5, 320–329. [DOI] [PubMed] [Google Scholar]
- 38.Lippincott-Schwartz J., Snapp,E. and Kenworthy,A. (2001) Studying protein dynamics in living cells. Nature Rev. Mol. Cell Biol., 2, 444–456. [DOI] [PubMed] [Google Scholar]
- 39.Szwergold B.S., Graham,R.A. and Brown,T.R. (1987) Observation of inositol pentakis- and hexakis-phosphates in mammalian tissues by 31P NMR. Biochem. Biophys. Res. Commun., 149, 874–881. [DOI] [PubMed] [Google Scholar]
- 40.Orchiston E.A., Bennett,D., Leslie,N.R., Clarke,R.G., Winward,L., Downes,C.P. and Safrany,S.T. (2004) PTEN M-CBR3, a versatile and selective regulator of Ins(1,3,4,5,6)P5. Evidence for Ins(1,3,4,5,6)P5 as a proliferative signal. J. Biol. Chem., 279, 1116–1122. [DOI] [PubMed] [Google Scholar]
- 41.York J.D., Odom,A.R., Murphy,R., Ives,E.B. and Wente,S.R. (1999) A phospholipase C-dependent inositol polyphosphate kinase pathway required for efficient messenger RNA export. Science, 285, 96–100. [DOI] [PubMed] [Google Scholar]
- 42.Odom A.R., Stahlberg,A., Wente,S.R. and York,J.D. (2000) A role for nuclear inositol 1,4,5-trisphosphate kinase in transcriptional control. Science, 287, 2026–2029. [DOI] [PubMed] [Google Scholar]
- 43.Saiardi A., Caffrey,J.J., Snyder,S.H. and Shears,S.B. (2000) Inositol polyphosphate multikinase (ArgRIII) determines nuclear mRNA export in Saccharomyces cerevisiae. FEBS Lett., 468, 28–32. [DOI] [PubMed] [Google Scholar]
- 44.Messenguy F. and Dubois,E. (1993) Genetic evidence for a role for MCM1 in the regulation of arginine metabolism in Saccharomyces cerevisiae. Mol. Cell Biol., 13, 2586–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo H.R., Saiardi,A., Yu,H., Nagata,E., Ye,K. and Snyder,S.H. (2002) Inositol pyrophosphates are required for DNA hyperrecombination in protein kinase c1 mutant yeast. Biochemistry, 41, 2509–2515. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.