

Abstract
Study Objectives:
Although obstructive sleep apnea (OSA) causes cardiovascular morbidities through atherosclerosis induced by inflammation and endothelial dysfunction, OSA patients exhibit elevated plasma vascular endothelial growth factor (VEGF), which may represent an adaptive response to intermittent hypoxia. The aims of this study were to investigate whether in vitro endothelial wound healing and monocyte migration are affected by patient serum, and to determine the implication of circulating factors (VEGF and C-reactive protein).
Patients:
Serum was collected from healthy controls (HC), “healthy” OSA, and metabolic syndrome (MS) patients with or without OSA.
Measurements and Results:
Along with the presence of OSA and/or MS, both VEGF and hsCRP were significantly elevated in patient serum. Their specific role was tested with blocking antibodies on primary endothelial cells for wound healing assay and on human monocytes for migration assay. Endothelial wound healing was reduced with OSA compared to HC serum, and even more significantly using MS+OSA patient serum. Altered wound healing with OSA serum was unmasked when blocking VEGF and restored when blocking CRP. Monocyte migration was activated with OSA serum, and further enhanced by MS+OSA patient serum. Blocking CRP in serum inhibited this migration.
Conclusions:
Serum from OSA patient alters in vitro endothelial cell repair function and activates monocyte migration; this is further aggravated with the presence of metabolic syndrome. These effects are partly driven by VEGF and CRP, suggesting an unfavorable balance between the pro healing (VEGF) and pro injury (CRP) factors that may promote vascular injury in OSA with and without metabolic syndrome.
Citation:
Briançon-Marjollet A, Henri M, Pépin JL, Lemarié E, Lévy P, Tamisier R. Altered in vitro endothelial repair and monocyte migration in obstructive sleep apnea: implication of VEGF and CRP. SLEEP 2014;37(11):1825-1832.
Keywords: obstructive sleep apnea, metabolic syndrome, VEGF, hsCRP, endothelial repair
INTRODUCTION
Obstructive sleep apnea syndrome (OSA) is a highly prevalent sleep disorder that affects 5% to 20% of the population. It is a growing health concern worldwide, due to the associated increased risk of cardiovascular morbidity and mortality.1,2 The periodic upper airway collapse occurring during sleep in OSA patients induces chronic intermittent hypoxia (IH), which is thought to promote cardiovascular diseases through oxidative stress, sympathetic activation and systemic and vascular inflammation.3–5
Atherosclerosis results from a cascade of different processes that start with endothelium injuries to plaques constitution, mostly induced by shear stress.6,7 Owing to the repetitive surges in blood pressure at the end of apneas and hypopneas, shear stress is increased in OSA patients.8 Endothelial cell migration and proliferation are key mechanisms involved in endothelium healing, with migration supposed to repair small injuries. However, whereas endothelial cell migration is activated by laminar flow, it is impaired under disturbed flow conditions.9 Therefore, unfavorable balance between endothelium injuries and healing ability will promote atherosclerosis.10–12 In OSA patients, endothelium healing capacity indeed seems to be reduced.13,14 Jelic and colleagues suggested that this might be due to decreased endothelial progenitor cell levels,15 but little is known about endothelial cell migration in OSA patients. Moreover, atherosclerosis is characterized by monocyte migration and invasion of the sub-endothelial space. Abundant literature has proved that OSA induces vascular inflammation leading to leukocyte migration, structural and functional remodeling of the vessels, and finally to atherosclerosis.3–5
In this context, vascular endothelial growth factor (VEGF) and C-reactive protein (CRP) may appear as good candidates in promoting and impairing endothelial healing and monocyte migration in OSA patients. VEGF is well known as an inducer of endothelial proliferation and migration. OSA patients exhibit elevated circulating VEGF, and CPAP treatment restores VEGF levels back to normal values.16–20 VEGF may thus be a good candidate as a protector against vascular damage induced by chronic intermittent hypoxia and blood pressure surges in OSA. On the other hand, C-reactive protein, an inflammatory marker highly correlated with cardiovascular risk and prognosis, has been extensively studied in OSA. Recent data suggest that CRP also has functional consequences on endothelial cell migration21 and on monocyte activation.22 High sensitivity CRP (hsCRP) is often found elevated in OSA patients, especially if obesity, dyslipidemia, diabetes and cardiovascular diseases are present in association, suggesting that metabolic syndrome rather than OSA per se would induce hsCRP elevation.5,23,24 Obesity indeed represents a strong confounding factor of OSA that makes difficult to study the respective implications of OSA and obesity in cardiovascular alterations. OSA patients as well as animals exposed to chronic intermittent hypoxia develop insulin resistance, dyslipidemia, alteration of glucose metabolism and adipokines secretion.25–27
The objective of this study was to investigate how early mechanisms of atherosclerosis may be altered in OSA patients. To complete this goal we assessed the circulating levels of VEGF and hsCRP in 81 subjects depending of the OSA status associated or not with metabolic syndrome compared to healthy subjects, we evaluated the alterations of in vitro endothelial cell healing capacities and monocyte migration induced by patient serum, and finally we investigated the respective implication in these mechanisms of VEGF and CRP.
MATERIALS AND METHODS
Study Design
This study was a case-controlled study completed into HP2 lab in which 4 groups of patients and healthy controls have been evaluated. Ethical approval was obtained from our institutional review board (Comité de protection des personnes Sud-Est V) and all subjects gave written informed consent. This protocol conforms to the principles of Declaration of Helsinki.
Study Population and Assessment
A total of 81 patients and healthy subjects were recruited to complete 4 groups: (1) healthy controls (HC, n = 16); (2) OSA patients without metabolic syndrome (OSA, n = 31); (3) metabolic syndrome patients without OSA (MS, n = 13); (4) metabolic syndrome patients with OSA (MS+OSA, n = 21). Non-MS OSA patients and healthy control subjects were selected according to age and BMI, in order to achieve similar age and BMI between groups. Non-MS OSA patients were free of any history of cardiovascular disease, they were normotensive and had no antihypertensive medication. By contrast, patients in the MS and MS+OSA group fulfill the criteria of metabolic syndrome definition and by design had higher body mass index, lipid abnormalities, hypertension, and/or diabetes. All patients were non-smokers. All patients underwent clinical assessment and office blood pressure measurements; blood samples were taken fasten in the morning after an overnight polysomnography (see supplemental methods). OSA was defined as AHI > 15. Table 1 depicts baseline characteristics in the different groups.
Table 1.
Clinical data in the 4 groups of subjects.
Human sera were collected and stored at −80°C until use. For endothelial healing experiments, patient sera were pooled according to the presence of MS and/or OSA. For monocyte migration experiments, OSA patient sera were pooled into ter-tiles according to OSA severity: mild OSA = 15 < AHI < 25 (n = 11), moderate OSA = 25 < AHI < 45 (n = 10), severe OSA = AHI > 45 (n = 10).
VEGF and hsCRP measurements
Serum hsCRP level was measured using automated immunonephelometry (Behring Nephelometer II Analyzer, Dade Behring, Germany). Serum VEGF was measured in each patient using human VEGF DuoSet Elisa (R&D Systems). Each sample was assessed in duplicate.
Chemicals and Antibodies
rhVEGF, rhCRP, anti-hVEGF antibody, anti-hCD32a/Fcγ RIIA antibody antibodies were obtained from R&D Systems. Control isotype antibodies were from eBiosciences. Endotoxin level in rhCRP is less than 1.0 EU per 1 μg of protein by the LAL method.
Cell Culture
HMVEC (human microvascular endothelial cells, Lonza) were cultured in EGM2-MV according to supplier recommendations and used between passages 4 and 6. THP-1 monocytes were a kind gift from Fabienne Burger (Cardiology Division, University Hospital, Geneva) and maintained in RPMI medium with 10% FBS. For experiments, HMVEC were incubated in endothelial basal medium (EBM, Lonza) supplemented with 65% human sera, or rhVEGF (500 pg/mL), rhCRP (2 μg/mL), anti-hVEGF antibody (0.5 μg/mL), and/or anti-hCD32a (10 μg/ mL). THP1 were cultured in RPMI without FBS, supplemented with 65% human sera, or rhCRP (2 μg/mL) and/or anti-hCD32a (10 μg/mL).
Wound Healing Assay
HMVEC were seeded at 2.104 cells per well in 48-well dishes (Nunclon) to reach 100% confluence. After 24 h, a scratch was performed by crossing the wells from side to side with a sterile 10-100 μL cone. Detached cells were removed by changing the culture medium, and sera or drugs were added in the medium. Wound reparation was recorded with a dynamic microscope (AxioVert 200M, Zeiss). Pictures were taken every 30 min during 24 h under ×10 magnification (Metamorph). Two positions by well were recorded. The filled area was determined by subtracting the cell-free area at time t from the initial cell-free area with Image J software. Filled areas were expressed as percentages of control wells to normalise the values.
Monocyte Migration Assay
104 THP1 monocytes were pre-stimulated for 3 h in RPMI with 65% human serum or 20% SVF, with or without drugs; then migration was performed in Transwells with 8 μm pores (Corning) for 1 h, with stimulated cells on the upper chamber and 5nM MCP1 in the lower chamber. Transwells were removed and cells that migrated to the lower chamber were counted on 5 microscope fields under an inverted phase contrast microscope (Zeiss).
Statistical Analysis
Comparisons between groups were performed using oneway or two-way ANOVA, or Mann-Whitney test depending on whether the data were normally distributed. When significance was achieved, the appropriate test (Tukey or Dunn test) was used for post hoc analysis. Qualitative variables were tested using χ2 test. Correlations were assessed by using Spearman test. Results were considered significant when P values were < 0.05.
RESULTS
VEGF and hsCRP are Elevated in OSA Patients
Population characteristics are reported in Table 1. By design, BMI and office blood pressure were higher in MS patients. OSA patients with or without MS did not differ in terms of OSA severity.
Median serum VEGF concentration increased significantly according to the presence of OSA (367.61 vs 169.68 pg/mL, P = 0.044) and Metabolic Syndrome (474.52 vs 169.68 pg/mL, P = 0.023; Figure 1A). However, when OSA and MS condition were associated, we did not find further VEGF elevation. In subjects without MS, serum VEGF positively correlates with OSA severity (time spent with O2 saturation < 90%; Figure 1B, P = 0.046) and serum hsCRP (Figure 1C, P = 0.023). Median hsCRP serum level was also increased in OSA group (1.3 vs 0.6 mg/L, P < 0.05, Figure 1D). This seemed to be principally driven by the severity of OSA patients, since hsCRP was found elevated only in severe OSA patients (AHI > 45) compared to controls (1.7 vs 0.6 mg/L, P < 0.05) but not in mild or moderate OSA patients (see Figure S1, supplemental material). This was well illustrated in subjects without MS by the correlation between hsCRP and OSA severity expressed both by AHI and minimal oxygen saturation (P = 0.015, Figure 1E and P = 0.003, Figure 1F, respectively). Interestingly, these increases of hsCRP in OSA patients, especially in severe patients, remained significant after adjusting for BMI (P = 0.033 and P = 0.036, respectively).
Figure 1.

Serum VEGF and high sensitivity-CRP are elevated in OSA and MS patients. (A) Serum VEGF is elevated in 31 OSA patients and 13 MS patients compared to 16 non-OSA healthy controls, but not in 21 MS+OSA patients. * P < 0.05 vs controls, # P < 0.05 vs MS group, Two-way ANOVA. (B) Positive Spearman correlation between serum VEGF and time spent under 90% O2 saturation during sleep in OSA patients (P = 0.046) but not in healthy controls. (C) Positive Spearman correlation between serum VEGF and hsCRP in OSA patients and healthy controls (n = 47 subjects, P = 0.023). (D) Serum hsCRP is elevated in 31 OSA patients and 13 control MS patients compared to 16 non-OSA healthy controls, but not in 21 MS+OSA patients. * P < 0.05, Two-way ANOVA. (E) Positive Spearman correlation between hsCRP and severity of OSA measured by apnea-hypopnea index (AHI), n = 47 subjects, P = 0.015. (F) Negative Spearman correlation between hsCRP and minimal O2 saturation during sleep, both in healthy controls and OSA patients, n = 47 subjects, P = 0.003. In graphs B, C, E, and F, gray circles represent healthy controls (n = 16) and closed circles represent OSA patients (n = 31), both without MS.
As expected, the presence of metabolic syndrome highly impacted CRP levels but in these groups we did not find any further effect of OSA association (Figure 1D).
Serum VEGF and CRP Regulate Endothelial Wound Healing in vitro
To assay for functional consequences of human serum on endothelial cells, human microvascular endothelial cells (HMVEC) were submitted to endothelial wound healing assay in vitro. Reparation of endothelial wound was quantified along time, until confluence was obtained again, and normalized to control values (Figure 2A).
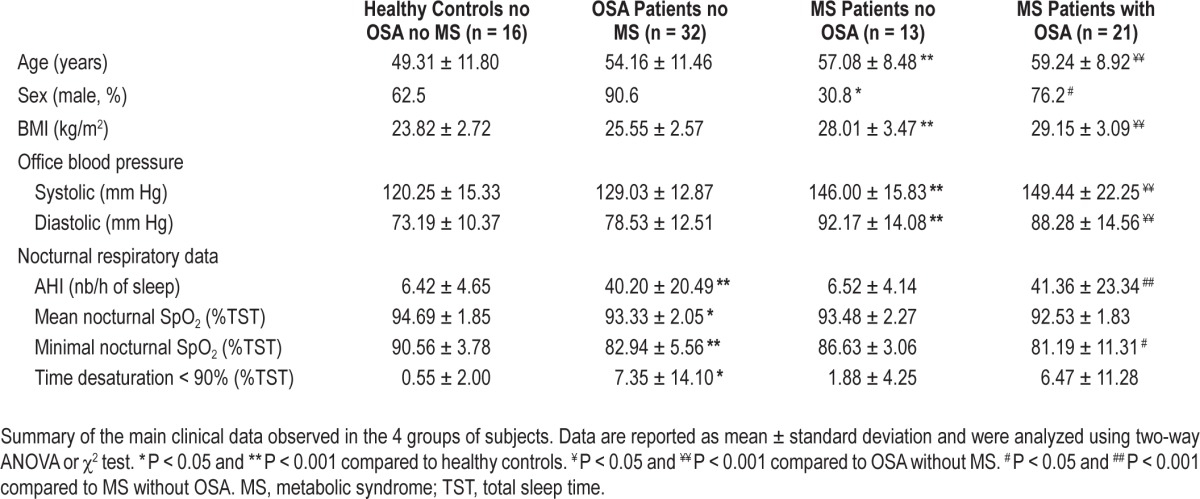
Figure 2.
Recombinant VEGF and CRP regulate endothelial repair in vitro. (A) Pictures in phase contrast microscopy (x10 magnification) showing examples of wounds at the beginning of experiment and after 7.5 h of healing by HMVEC (Bar = 50 μm). (B) VEGF (500 pg/mL) added in EBM medium activates wound healing compared to control EBM condition, and addition of anti-VEGF blocking antibody (0.5 μg/mL) or CRP (2 μg/mL) block this effect, whereas CRP alone has no significant effect. Addition of anti-CD32a (10 μg/mL) antibody reverses CRP action. One-Way ANOVA, ** P < 0.01 compared to EBM control, ## P < 0.01 compared to VEGF, $ P < 0.05 compared to VEGF+CRP. N = at least 5 independent experiments, error bars represent SEM.
First, rhVEGF (500 pg/mL) added in the EBM culture medium was able to enhance wound healing by 65%, and this effect was totally blocked by anti-VEGF blocking antibody at 0.5 μg/mL (Figure 2B). Moreover, CRP (2 μg/mL) had no effect per se but was able to inhibit strongly VEGF-induced wound healing. This effect was partially blocked by using an antibody raised against the CRP receptor CD32a (Figure 2B).
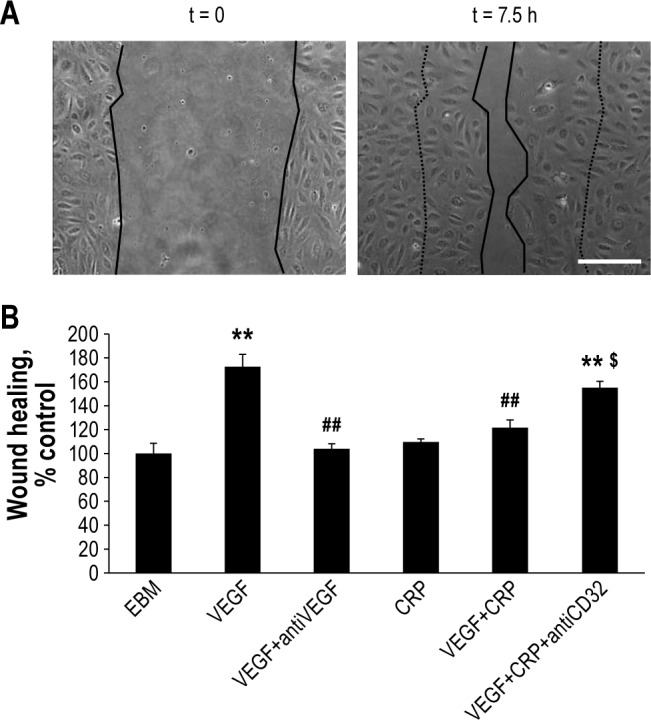
We subsequently incubated HMVEC with human sera. Serum from OSA patients without MS moderately but significantly inhibited repair capacity compared to healthy control serum (94% vs 100%, P < 0.001; Figure 3A and 3B). Addition of an anti-VEGF blocking antibody further impaired wound repair when HMVEC were incubated in presence of OSA patient serum (-10%, P = 0.016; Figure 3B), but not with healthy control serum, although the same tendency was observed (-5%, P = 0.065). Finally, addition of rhVEGF in OSA patient serum restored wound healing function to 128% of healthy controls serum (Figure S2, supplemental material). These data suggest that VEGF in OSA patient serum tends to increase wound healing, thereby counteracting impaired in vitro endothelial cell repair function.
Figure 3.
Serum VEGF activates endothelial repair in vitro. (A) Pictures in phase contrast microscopy (x10 magnification) showing examples of wounds after 7.5 h of healing by endothelial cells, in presence of pooled Healthy Controls serum or pooled OSA patients serum in EBM medium. Bar = 50 μm. (B) Serum from OSA patients diminish endothelial healing compared to healthy controls serum, and anti-VEGF blocking antibody (0.5 μg/mL) but not control antibody added in OSA patient serum inhibits endothelial healing. Two-Way ANOVA, * P < 0.001 compared to control, # P < 0.05 compared to OSA without antibody or control antibody. N = at least 6 independent experiments, error bars represent SEM.
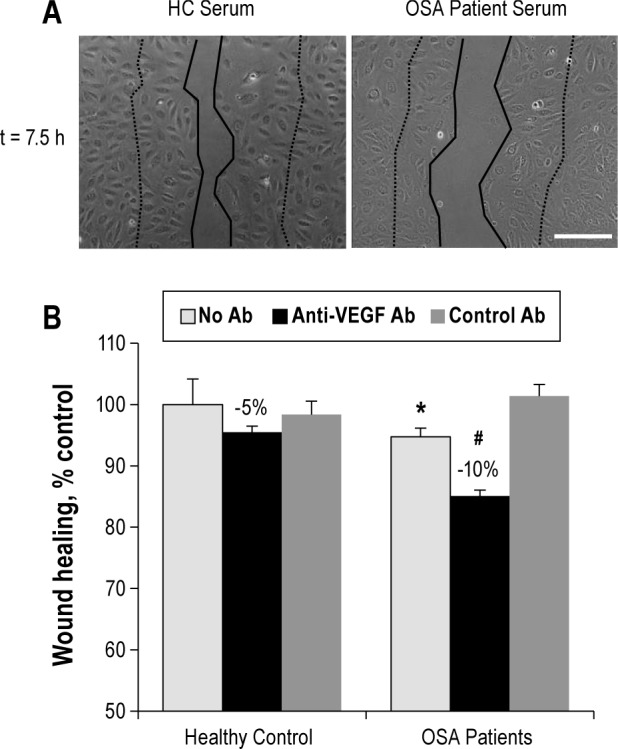
When 2 μg/mL CRP was added in serum of healthy controls, it was able to inhibit wound healing by 20% (Figure 4A). Moreover addition of an anti-CD32a antibody to block CRP in serum significantly enhanced endothelial wound healing, by 8% in control subjects and by 22% in OSA patient serum (Figure 4B). These results suggest that by contrast with VEGF, CRP was able to inhibit in vitro endothelial repair and could thus be involved in OSA-induced alteration of endothelial repair.
Figure 4.
CRP in OSA patient serum inhibits endothelial repair in vitro. (A) Recombinant CRP (2 μg/mL) added in pooled serum from healthy controls inhibits endothelial healing (Mann-Whitney rank sum test, P = 0.002). (B) Blocking anti-CD32a antibody added in pooled serum from control or OSA patients induces an increase in endothelial healing by 8% and 22%, respectively. Two-Way ANOVA, * P < 0.05 vs healthy controls, # P < 0.05 vs OSA patients without antibody. (C) MS combined to OSA reduces by 18% endothelial wound healing, expressed in percent of control subjects, whereas in non-MS subjects, this decrease is only 5%. Two-way ANOVA, * P < 0.05 vs healthy controls, # P < 0.01 vs MS group. N = at least 4 independent experiments, error bars represent SEM.
Finally, in order to determine the influence of metabolic syndrome on in vitro endothelial repair, HMVEC were incubated with serum from MS patients, with or without OSA. Interestingly, serum from MS without OSA enhanced in vitro repair compared to serum from healthy controls without MS (113% vs 100%, P < 0.05; Figure 4C). Moreover, MS+OSA serum induced a further reduced endothelial healing capacity compared to OSA without MS patients (-18% vs -5% compared to their respective non-OSA controls, P < 0.01). Thus OSA and MS may have deleterious synergistic effects on healing capacities of endothelial cells.
CRP in OSA Serum Activates Monocyte Migration in vitro
To achieve a better comprehension of early atherosclerosis mechanisms involved in OSA, we then tested whether human serum from OSA patients could also affect monocyte migration, and in particular if CRP contained in the serum could be responsible for the observed effects. First, we investigated the impact of exogenous CRP on THP-1 monocyte migration in vitro. Figure 5A shows that pre-incubation of the cells with 2 μg/mL of CRP induced a 32% increase in monocyte migration, and that this effect was blocked by the addition of 10 μg/mL anti-CD32a blocking antibody, but not by control antibody.
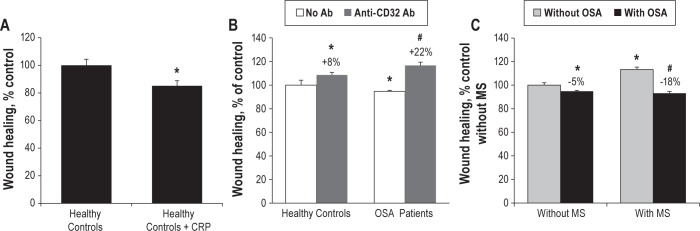
Figure 5.

CRP in OSA patient serum activates THP-1 monocyte migration in vitro. (A) Recombinant CRP (2 μg/mL) added in RPMI medium with 20% FBS activates THP1 migration through Transwells under MCP1 gradient (5 nM in lower chamber). Anti-CD32a blocking antibody (10 μg/mL) but not control antibody significantly blocks this effect. One-Way ANOVA, * P < 0.05 compared to RPMI control, # P < 0.05 compared to CRP. (B) Serum from moderate and severe but not from mild OSA patient activates monocyte migration compared to serum from healthy controls. Anti-CD32a blocking antibody (10 μg/mL) added in OSA patients serum inhibits monocyte migration. Two-Way ANOVA, * P < 0.05 compared to control without antibody, $ P < 0.05 compared to mild OSA patient, # P < 0.005 compared to respective condition without antibody. (C) Recombinant CRP (2 μg/mL) added in serum of healthy controls induces an increase in monocyte migration. Mann-Whitney test, P = 0,002. (D) OSA, MS and combination of OSA+MS induce monocyte migration, expressed in percent of control. Two-Way ANOVA, * P < 0.05 compared to healthy controls, # P < 0.05 compared to OSA only group, $ P < 0.05 compared to MS only group. N = at least 4 independent experiments, error bars represent SEM.
Then we tested monocyte migration in presence of human serums. Moderate (25 < AHI < 45) and severe (AHI > 45), but not mild (15 < AHI < 25) OSA patient serum, induced significant monocyte migration by 25% and 38%, respectively, compared to healthy controls (AHI < 15; Figure 5B). As expected, blocking CRP by anti-CD32a significantly inhibited monocyte migration in all OSA patient groups (-27% to -42% decrease, P < 0.005). Although the same tendency was observed in healthy controls, this did not reach significance (-16%, P = 0.057; Figure 5B). Moreover migration was enhanced by the addition of 2 μg/mL CRP to the healthy control serum, thereby reaching migration rate comparable to those obtained with OSA patient serum (Figure 5C).
Finally, we tested whether metabolic syndrome may influence monocyte migration. Figure 5D shows that MS serum itself has significant effect (+30%), and that MS combined to OSA serum further induces monocyte migration in apneic patients (+58%). Thus, OSA and MS may have synergistic effects on monocyte migration.
DISCUSSION
Our study addressed the question of whether circulating levels of VEGF and CRP may have functional involvement in early atherosclerosis of OSA patients. VEGF and CRP were elevated in OSA and metabolic syndrome patient serums, and these levels were sufficient enough to modulate endothelial cells and monocytes migration in vitro.
Increased circulating VEGF in OSA patients have been reported,16–20 although a study suggested an age-related relationship rather than a link with OSA severity.28 Herein, we observed a VEGF increase that was not age-related between patients and controls.
Although VEGF measurement is more accurate in plasma than in serum due to VEGF release from blood cells and platelets,17 we assayed VEGF in serum because endothelial cells had to be incubated into serum and not plasma. Therefore circulating VEGF measurements are a reliable assessment of VEGF concentration in contact with cells during the in vitro experiments.
Low grade inflammation as a mechanism of cardiovascular alteration has been largely studied in OSA patients, with CRP as a particular target. However, many discrepancies created controversy about the effect of OSA per se compared to obesity.5,23 We found a relationship between OSA severity and CRP concentration only in “healthy” OSA patients (i.e., without comorbidities; Figure 1D). The increase of CRP in these OSA patients remained significant after adjusting for BMI variations. However, this was not true for OSA with metabolic syndrome, supporting the hypothesis that cardiovascular disease and obesity associated with OSA also account for CRP elevation.
We subsequently wanted to evaluate the functional consequences of serum VEGF on endothelial cells in vitro. By using recombinant VEGF and VEGF blocking antibody, we showed that VEGF activates wound healing repair in HMVEC. However, despite higher serum VEGF concentration in “healthy” OSA than in control subjects (Figure 1A), wound healing decreased (Figure 3B). This suggests that other circulation factors are counteracting VEGF effects on in vitro endothelial repair. We therefore investigated the role of CRP on wound healing and observed that it inhibited VEGF-induced wound healing, despite having no effect when alone (Figure 2B). This suggests that the CRP effect on VEGF-induced cell migration is not due to any nonspecific toxicity effect. This is also consistent with previous studies showing inhibition of VEGF by CRP.21,29 We further found that anti-CD32a blocking antibodies activates wound healing repair, with a stronger impact in serum from OSA patient than in serum from healthy controls (Figure 4B), suggesting that this effect is related to higher CRP concentration observed in OSA patients (Figure 1D). Moreover, it should be noted that outside the field of OSA, these effects of recombinant VEGF and CRP in wound healings assays were already reported in the literature,21,29 although doses where higher (10-50 ng/mL for VEGF and 10-20 μg/mL for CRP). Herein, our data are original as addressing serum from the specific OSA population and therefore using much lower, physiological doses of VEGF and CRP (i.e., 500 pg/mL VEGF and 2 μg/mL CRP). By using the blocking antibody strategy, we were able to unravel the functional consequences of physiological variations of VEGF and CRP in patient serum.
In order to determine the respective implication of cell migration and proliferation in wound healing assays, we performed specific proliferation assays. No significant effect of patient serum, VEGF, or CRP was observed on HMVEC proliferation (Figure S3, supplemental material). Thus, wound healing regulation by these factors is likely mostly due to the regulation of cell migration.
Altogether, the data collected on endothelial cells suggested that VEGF and CRP may be two soluble factors with opposite effects on endothelial repair, compensating for each other. VEGF might be a promoting factor of endothelial repair that is antagonized by CRP. This hypothesis is supported by the fact that VEGF and CRP levels significantly correlate in non-MS subjects.
Endothelial damage is described as an early step of atherosclerosis, especially at sites prone to atherosclerosis lesion such as vessel bifurcations.11,12 Endothelial damage and dysfunction have also been reported in OSA patients, although studies focus on endothelial-dependent vasodilatation and endothelial activation13; and little has been described concerning endothelial repair capacity in OSA patients.15 Interestingly, only one in vitro study by Carreras and colleagues suggested that endothelial wound healing is increased by serum from rats submitted to recurrent obstructive apneas.30 The present work on human sera highlights the implication of soluble blood factors on altered endothelial repair capacity in OSA patients in vitro, therefore suggesting the lack of endothelial cell migration as a potential mechanism explaining endothelial lesions in OSA patients. Moreover, it points at the potential clinical interest of studying endothelial repair in vivo in OSA patients, particularly in conditions that require endothelial healing, such as stenting.31 Stenting indeed induces vascular lesions with endothelial denudation, and subsequent repair (including re-endothelialization) is needed, but a recent case report suggested that OSA might inhibit this repair, thereby inducing late in-stent thrombosis.32
Beside these serum consequences on endothelial cells migration in vitro, we wanted to test the impact of serum soluble factors, particularly CRP, on monocytes. We were able to show that CRP induced monocyte migration in vitro, which is consistent with previous data.33 Moreover, we performed monocyte proliferation assays and observed that serum or CRP had no significant effect on THP-1 monocyte proliferation (data not shown), suggesting that monocyte migration is specifically affected. We further showed that monocyte migration was induced by patient serum, especially by the serum of severe OSA patients (Figure 5C), which correlates with increasing CRP concentration in serum from these patients (Figure 1D). This is reinforced by the observation that when treated with anti-CD32a blocking antibody, there was no difference in migration induced by sera from different categories of patients (Figure 5B). Serum from MS+OSA patients further induced monocyte migration compared to OSA only patients (Figure 5C), suggesting that MS may promote monocyte migration. However this was observed in the absence of further CRP increase in MS+OSA serum compared to OSA or MS only serum. This suggests that CRP, although a key player, is not the only soluble factor involved in monocyte migration.
Several receptors for CRP have been described and studied.22,34 The work of Montecucco et al. suggests that among the 3 receptors CD32a, CD32b, and CD64, CD32a and CD64 could account for the larger part of CRP effects.22 However, in our experience anti-CD64 blocking antibody had no consistent effect on endothelial and monocyte migration (data not shown). We therefore focused on CD32a receptor as a mediator of CRP signaling.
Monocyte adhesion to endothelium and migration is involved in the development of atherosclerotic lesions, leading to vascular inflammation and remodeling.3–5 Thus, our results suggest that CRP in OSA patient serum could be implicated in monocyte activation and migration promoting early atherosclerosis.
There are some limitations to this study. We selected OSA patient with MS according to the international criteria for metabolic syndrome. However, this group may encompass several presentations and severities of MS that might be very heterogeneous, and this may have affected the results. It is also possible that the effects seen in vitro could be related to hypertensive vasculopathy rather than OSA or MS. This possibility is limited in OSA patients without MS who are normotensive, but plausible in MS patients. However since sera were pooled according to OSA severity and not blood pressure level, we cannot compare normotensive OSA patients to hypertensive OSA with MS in whom blood pressure was controlled by medication.
Altogether, our data suggest that serum from OSA patients contains factors altering endothelial repair and monocyte migration in vitro. Elevated VEGF may act as a beneficial factor leading to endothelial repair in OSA patients, counteracting the deleterious effects of CRP. CRP could further enhance monocyte migration, therefore adding to endothelial damage to facilitate early atherosclerotic lesions. Beside VEGF and CRP, other soluble factors might be taken into account to improve our understanding of these processes. Finally, metabolic syndrome aggravates both endothelial damage and monocyte migration, providing some new possible mechanistic explanations for the deleterious synergy between overweight and sleep apnea in OSA patients.
DISCLOSURE STATEMENT
This work was funded by INSERM, Grenoble University Joseph Fourier council grants. This work was also partly supported for an unrestricted grant from Agir@dom and a Projet Hospitalier de Recherche Clinique (PHRC 2006). The authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
The authors thank Guillaume Séchaud, Céline Beaumont and Florian Minot for their participation to this work and Sonia Dias-Domingos who performed data statistical analysis. We thank Fabienne Berger from Geneva University for making THP-1 monocytes available for experiments.
ABBREVIATIONS
- AHI
apnea-hypopnea index
- BMI
body mass index
- HC
healthy controls
- HMVEC
human microvascular endothelial cell
- hsCRP
high sensitivity C-reactive protein
- MS
metabolic syndrome
- OSA
obstructive sleep apnea
- VEGF
vascular endothelial growth factor
SUPPLEMENTAL MATERIAL
SUPPLEMENTAL MATERIALS AND METHODS
Sleep Studies
Diagnosis of OSA was obtained after full polysomnography. Continuous recordings were taken with electrode positions C3/ A2-C4/A1-Cz/01 of the international 10–20 Electrode Placement System, eye movements, chin electromyogram and ECG with modified V2 lead. Sleep was scored manually according to standard criteria.1 Airflow was measured with nasal pressure associated with the sum of oral and nasal thermistor signals. Respiratory efforts were monitored with abdominal and thoracic bands. An additional signal of respiratory effort (i.e., pulse transit time) was recorded concurrently. Oxygen saturation was measured using a digital pulse oximeter. An apnea was defined as a complete cessation of airflow for ≥ 10 sec and a hypopnea as a reduction ≥ 50% in the nasal pressure signal, or a decrease between 30% and 50% associated with either oxygen desaturation ≥ 3% or an EEG arousal (defined according to the Chicago report) and the later AASM statement, both lasting ≥ 10 s.2,3 Apneas were classified as obstructive, central, or mixed according to the presence or absence of respiratory efforts, on the pulse transit time,4 and the shape of the respiratory curve of nasal pressure (flow limited aspect or not).5 An AHI was calculated and defined as the number of apneas and hypopneas per hour of sleep (full polysomnography). OSA was defined as an AHI > 15.
Proliferation Assay
HMVEC were seeded in 96W plates at a density of 5,000 cells/well and incubated for 24h with or without rhVEGF (500 pg/mL), rhCRP (2 μg/mL) or 65% human sera in EBM medium. Then proliferation was assessed with the Cell Titer proliferation assay (Promega).
hsCRP elevation in OSA patients depends on OSA severity. Serum hsCRP is elevated in 31 OSA patients with AHI > 15 compared to 16 healthy controls, and among OSA patients is significantly elevated only in patients with severe OSA (AHI > 45, n = 10) but not in mild (15 < AHI < 25) or moderate (25 < AHI < 45) OSA patients. One-Way ANOVA, * P < 0.05.
VEGF added in OSA patient serum restores wound healing function. Recombinant VEGF (500 pg/mL) added in serum from OSA without MS patients significantly increased wound healing compared to healthy controls serum (n = 4 independent experiments, Student t-test P = 0.023).
Proliferation of HMVEC is not modified by VEGF, CRP or serum. (A) HMVEC proliferation is not modified by adding exogenous VEGF or CRP in EBM medium (One-way ANOVA, P = 0.385). (B) HMVEC proliferation is not significantly modified by incubation with serum from OSA patients compared to serum from healthy controls. (Student t-test, P = 0.059). n ≥ 3 independent experiments.
REFERENCES
- 1.Baguet JP, Barone-Rochette G, Tamisier R, Levy P, Pepin JL. Mechanisms of cardiac dysfunction in obstructive sleep apnea. Nat Rev Cardiol. 2010;9:679–88. doi: 10.1038/nrcardio.2012.141. [DOI] [PubMed] [Google Scholar]
- 2.Levy P, Pepin JL, Arnaud C, et al. Intermittent hypoxia and sleep-disordered breathing: current concepts and perspectives. Eur Respir J. 2008;32:1082–95. doi: 10.1183/09031936.00013308. [DOI] [PubMed] [Google Scholar]
- 3.Arnaud C, Dematteis M, Pepin JL, Baguet JP, Levy P. Obstructive sleep apnea, immuno-inflammation, and atherosclerosis. Semin Immunopathol. 2009;31:113–25. doi: 10.1007/s00281-009-0148-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levy P, Pepin JL, Arnaud C, Baguet JP, Dematteis M, Mach F. Obstructive sleep apnea and atherosclerosis. Prog Cardiovasc Dis. 2009;51:400–10. doi: 10.1016/j.pcad.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 5.McNicholas WT. Obstructive sleep apnea and inflammation. Prog Cardiovasc Dis. 2009;51:392–9. doi: 10.1016/j.pcad.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 6.VanderLaan PA, Reardon CA, Getz GS. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. 2004;24:12–22. doi: 10.1161/01.ATV.0000105054.43931.f0. [DOI] [PubMed] [Google Scholar]
- 7.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91:327–87. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dematteis M, Julien C, Guillermet C, et al. Intermittent hypoxia induces early functional cardiovascular remodeling in mice. Am J Respir Crit Care Med. 2008;177:227–35. doi: 10.1164/rccm.200702-238OC. [DOI] [PubMed] [Google Scholar]
- 9.Hsu PP, Li S, Li YS, et al. Effects of flow patterns on endothelial cell migration into a zone of mechanical denudation. Biochem Biophys Res Commun. 2001;285:751–9. doi: 10.1006/bbrc.2001.5221. [DOI] [PubMed] [Google Scholar]
- 10.Hagensen MK, Raarup MK, Mortensen MB, et al. Circulating endothelial progenitor cells do not contribute to regeneration of endothelium after murine arterial injury. Cardiovasc Res. 2012;93:223–31. doi: 10.1093/cvr/cvr278. [DOI] [PubMed] [Google Scholar]
- 11.Mullick AE, Soldau K, Kiosses WB, Bell TA, 3rd, Tobias PS, Curtiss LK. Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J Exp Med. 2008;205:373–83. doi: 10.1084/jem.20071096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Q. Disturbed flow-enhanced endothelial turnover in atherosclerosis. Trends Cardiovasc Med. 2009;19:191–5. doi: 10.1016/j.tcm.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 13.Atkeson A, Yeh SY, Malhotra A, Jelic S. Endothelial function in obstructive sleep apnea. Prog Cardiovasc Dis. 2009;51:351–62. doi: 10.1016/j.pcad.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng J, Zhang D, Chen B. Endothelial mechanisms of endothelial dysfunction in patients with obstructive sleep apnea. Sleep Breath. 2012;16:283–94. doi: 10.1007/s11325-011-0519-8. [DOI] [PubMed] [Google Scholar]
- 15.Jelic S, Padeletti M, Kawut SM, et al. Inflammation, oxidative stress, and repair capacity of the vascular endothelium in obstructive sleep apnea. Circulation. 2008;117:2270–8. doi: 10.1161/CIRCULATIONAHA.107.741512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imagawa S, Yamaguchi Y, Higuchi M, Neichi T, Hasegawa Y, Mukai HY, et al. Levels of vascular endothelial growth factor are elevated in patients with obstructive sleep apnea--hypopnea syndrome. Blood. 2001;98:1255–7. doi: 10.1182/blood.v98.4.1255. [DOI] [PubMed] [Google Scholar]
- 17.Lavie L, Kraiczi H, Hefetz A, et al. Plasma vascular endothelial growth factor in sleep apnea syndrome: effects of nasal continuous positive air pressure treatment. Am J Respir Crit Care Med. 2002;165:1624–8. doi: 10.1164/rccm.20110-040OC. [DOI] [PubMed] [Google Scholar]
- 18.Gozal D, Lipton AJ, Jones KL. Circulating vascular endothelial growth factor levels in patients with obstructive sleep apnea. Sleep. 2002;25:59–65. doi: 10.1093/sleep/25.1.59. [DOI] [PubMed] [Google Scholar]
- 19.Schulz R, Hummel C, Heinemann S, Seeger W, Grimminger F. Serum levels of vascular endothelial growth factor are elevated in patients with obstructive sleep apnea and severe nighttime hypoxia. Am J Respir Crit Care Med. 2002;165:67–70. doi: 10.1164/ajrccm.165.1.2101062. [DOI] [PubMed] [Google Scholar]
- 20.Teramoto S, Kume H, Yamamoto H, et al. Effects of oxygen administration on the circulating vascular endothelial growth factor (VEGF) levels in patients with obstructive sleep apnea syndrome. Intern Med. 2003;42:681–5. doi: 10.2169/internalmedicine.42.681. [DOI] [PubMed] [Google Scholar]
- 21.Schneeweis C, Grafe M, Bungenstock A, Spencer-Hansch C, Fleck E, Goetze S. Chronic CRP-exposure inhibits VEGF-induced endothelial cell migration. J Atheroscler Thromb. 2010;17:203–12. doi: 10.5551/jat.3004. [DOI] [PubMed] [Google Scholar]
- 22.Montecucco F, Steffens S, Burger F, Pelli G, Monaco C, Mach F. C-reactive protein (CRP) induces chemokine secretion via CD11b/ ICAM-1 interaction in human adherent monocytes. J Leukoc Biol. 2008;84:1109–19. doi: 10.1189/jlb.0208123. [DOI] [PubMed] [Google Scholar]
- 23.Lui MM, Sau-Man M. OSA and atherosclerosis. J Thorac Dis. 2012;4:164–72. doi: 10.3978/j.issn.2072-1439.2012.01.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taheri S, Austin D, Lin L, Nieto FJ, Young T, Mignot E. Correlates of serum C-reactive protein (CRP)--no association with sleep duration or sleep disordered breathing. Sleep. 2007;30:991–6. doi: 10.1093/sleep/30.8.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonsignore MR, McNicholas WT, Montserrat JM, Eckel J. Adipose tissue in obesity and obstructive sleep apnoea. Eur Respir J. 2012;39:746–67. doi: 10.1183/09031936.00047010. [DOI] [PubMed] [Google Scholar]
- 26.Jun J, Polotsky VY. Metabolic consequences of sleep-disordered breathing. ILAR J. 2009;50:289–306. doi: 10.1093/ilar.50.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pepin JL, Tamisier R, Levy P. Obstructive sleep apnoea and metabolic syndrome: put CPAP efficacy in a more realistic perspective. Thorax. 2012;67:1025–7. doi: 10.1136/thoraxjnl-2012-202807. [DOI] [PubMed] [Google Scholar]
- 28.Peled N, Shitrit D, Bendayan D, Peled E, Kramer MR. Association of elevated levels of vascular endothelial growth factor in obstructive sleep apnea syndrome with patient age rather than with obstructive sleep apnea syndrome severity. Respiration. 2007;74:50–5. doi: 10.1159/000095675. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz R, Osborne-Lawrence S, Hahner L, et al. C-reactive protein downregulates endothelial NO synthase and attenuates reendothelialization in vivo in mice. Circ Res. 2007;100:1452–9. doi: 10.1161/01.RES.0000267745.03488.47. [DOI] [PubMed] [Google Scholar]
- 30.Carreras A, Rojas M, Tsapikouni T, Montserrat JM, Navajas D, Farre R. Obstructive apneas induce early activation of mesenchymal stem cells and enhancement of endothelial wound healing. Respir Res. 2010;11:91. doi: 10.1186/1465-9921-11-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van der Heiden K, Gijsen FJ, Narracott A, et al. The effects of stenting on shear stress: relevance to endothelial injury and repair. Cardiovasc Res. 2013;99:269–75. doi: 10.1093/cvr/cvt090. [DOI] [PubMed] [Google Scholar]
- 32.Hrynkiewicz-Szymanska A, Szymanski FM, Filipiak KJ, et al. Can obstructive sleep apnea be a cause of in-stent thrombosis? Sleep Breath. 2011;15:607–9. doi: 10.1007/s11325-010-0361-4. [DOI] [PubMed] [Google Scholar]
- 33.Montecucco F, Burger F, Pelli G, et al. Statins inhibit C-reactive protein-induced chemokine secretion, ICAM-1 upregulation and chemotaxis in adherent human monocytes. Rheumatology (Oxford) 2009;48:233–42. doi: 10.1093/rheumatology/ken466. [DOI] [PubMed] [Google Scholar]
- 34.Devaraj S, Du Clos TW, Jialal I. Binding and internalization of C-reactive protein by Fcgamma receptors on human aortic endothelial cells mediates biological effects. Arterioscler Thromb Vasc Biol. 2005;25:1359–63. doi: 10.1161/01.ATV.0000168573.10844.ae. [DOI] [PubMed] [Google Scholar]
REFERENCES
- 1.Iber C, Ancoli-Israel S, Chesson A, Quan SF. The AASM manual for the scoring of sleep and associated events: rules, terminology and technical specifications. 1st ed. Westchester, IL: American Academy of Sleep Medicine; 2007. [Google Scholar]
- 2.Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research. The Report of an American Academy of Sleep Medicine Task Force. Sleep. 1999;22:667–89. [PubMed] [Google Scholar]
- 3.Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2012;8:597–619. doi: 10.5664/jcsm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Argod J, Pepin JL, Levy P. Differentiating obstructive and central sleep respiratory events through pulse transit time. Am J Respir Crit Care Med. 1998;158:1778–83. doi: 10.1164/ajrccm.158.6.9804157. [DOI] [PubMed] [Google Scholar]
- 5.Hosselet JJ, Norman RG, Ayappa I, Rapoport DM. Detection of flow limitation with a nasal cannula/pressure transducer system. Am J Respir Crit Care Med. 1998;157:1461–7. doi: 10.1164/ajrccm.157.5.9708008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
hsCRP elevation in OSA patients depends on OSA severity. Serum hsCRP is elevated in 31 OSA patients with AHI > 15 compared to 16 healthy controls, and among OSA patients is significantly elevated only in patients with severe OSA (AHI > 45, n = 10) but not in mild (15 < AHI < 25) or moderate (25 < AHI < 45) OSA patients. One-Way ANOVA, * P < 0.05.
VEGF added in OSA patient serum restores wound healing function. Recombinant VEGF (500 pg/mL) added in serum from OSA without MS patients significantly increased wound healing compared to healthy controls serum (n = 4 independent experiments, Student t-test P = 0.023).
Proliferation of HMVEC is not modified by VEGF, CRP or serum. (A) HMVEC proliferation is not modified by adding exogenous VEGF or CRP in EBM medium (One-way ANOVA, P = 0.385). (B) HMVEC proliferation is not significantly modified by incubation with serum from OSA patients compared to serum from healthy controls. (Student t-test, P = 0.059). n ≥ 3 independent experiments.