


Abstract
We have developed the first fully integrated microfluidic system for DNA sequencing-by-synthesis. Using this chip and fluorescence detection, we have reliably sequenced up to 4 consecutive bps. The described sequencer can be integrated with other microfluidic components on the same chip to produce true lab-on-a-chip technology. The surface chemistry that was designed to anchor the DNA to elastomeric microchannels is useful in a broad range of studies and applications.
INTRODUCTION
The Sanger method of DNA sequencing (1) and its subsequent capillary array automation (2) has revolutionized biology and has led to the sequencing of the consensus human genome (3,4). However, this state-of-the-art technology has inherent limitations, especially in cost and read length, which make it impractical in moving to the next logical step: massive comparative genomics studies (3), aggressive disease-gene discovery and ubiquitous personalized medicine (5,6). In terms of speed and cost, sequencing only five persons took over 9 months, while the total budget of the project was over 3 billion dollars (http://www.ornl.gov/sci/techresources/Human_Genome/project/whydoe.shtmlbudget).
The limitations of the electrophoretic approach have prompted researchers to work on alternative methods: mass spectrometry (7–10), base addition with deprotection steps (11), pyrosequencing (12–16), sequencing by hybridization (17), massively parallel sequencing with stepwise enzymatic cleavage and ligation (18), polymerase colonies (19,20), sequencing using nanopores (21–24) and massively parallel single-molecule sequencing (25). While some of these methods are promising, none has yet bested electrophoresis separation. However, they may find important niche applications in areas such as SNP determination, mini-sequencing and gene expression analysis.
Here we describe a sequencing-by-synthesis method that uses microfluidic plumbing (µSBS). Briefly, the scheme involves exposing a primed DNA template to a mixture of a known type of standard nucleotide, its fluorescently tagged analog, and DNA polymerase. If the tagged nucleotide is complementary to the template base next to the primer’s end, the polymerase extends the primer with it and fluorescence signal is detected after a washing step. Iteration with each type of nucleotide reveals the DNA sequence. We implemented this scheme in a PDMS (polydimethylsiloxane) microfluidic chip and devised a novel surface chemistry to anchor the DNA to the PDMS microchannel to prevent DNA loss during feeds and flushes. The average read length was 3 bp, which constitutes proof of principle for the system as well as demonstrating the enabling technology—a heterogeneous assay fully integrated in a microfluidic system, combining active plumbing, specific surface chemistry and parallelism. This approach to DNA sequencing has advantages in economy of material and integrability under the lab-on-a-chip paradigm, while the microfluidic and surface chemistry technologies by themselves are immediately applicable to many other systems involving DNA studies, pyrosequencing being one example.
MATERIALS AND METHODS
Set-up and detection system
In each experiment, the microfluidic chip was housed in a custom-built aluminum holder, which was itself placed in a machined attachment to the translation stage of an inverted Olympus IX50 microscope. 23-Gauge steel tubes from New England Small Tube Corp. (Litchfield, NH 03052) were plugged into the chip’s control channel ports. Their other ends were connected through tygon tubing (Cole-Parmer, Vernon Hills, IL) to Lee-valve arrays (Fluidigm Corp., South San Francisco, CA) operated by LabView software on a PC computer. The same types of steel tubes and tygon plumbing were used to supply reagents to the chip’s flow channel ports.
The microscope was equipped with a mercury lamp (HBO 103 W/2 Osram), an Olympus Plan 10× objective (NA 0.25), an Olympus PlanApo 60× objective (NA 1.4) and a cooled CCD camera (SBIG ST-7I, Santa Barbara Instrument Group). Fluorescence detection was conducted using the following filter sets: Alexa Fluor 555 (ex D470/40, 500 DCLP, em D535/50), TAMRA, Lissamine and Cy3 (ex D540/25, dichroic 565 DCLP, em D605/55), both from Chroma Technology Corp., Brattleboro, VT.
Reagents
Chip fabrication. HMDS (hexamethyldisilazane) is from ShinEtsuMicroSi, Phoenix, AZ. Photoresist 5740 is from MicroChem Corp., Newton, MA. TMCS (tetramethylchlorosilane) is from Aldrich. PDMS (polydimethylsiloxane) is Sylgard 184 from Dow Corning, K.R. Anderson, Santa Clara, CA.
Surface chemistry. DAPEG is diacrylated polyethylene glycol SR610 from Sartomer, Exton, PA. The Pt catalyst is hydrogen hexachloroplatinate from Aldrich (26,258-7). The used polyelectrolytes are polyethyleneimine (PEI) (Sigma P-3143) and polyacrylic acid (PAcr) (Aldrich 41,604-5). Streptavidin Plus comes from Prozyme (San Leandro, CA). Trisb is a buffer: Tris 10 mM (NaCl 10 mM) pH 8. TrisMg is a buffer: Tris 10 mM (NaCl 10 mM, MgCl2 100 mM) pH 8. The dUTP-Cy3 from Amersham Biosciences (Piscataway, NJ) is a dUTP nucleotide tagged with the Cy3 dye.
Sequencing. DNA1 is an 89mer biotinylated DNA template (Biotin-5′-tcatcag tcatcag tcatcag tcatcag tcatcag tcatcag tcatcag tcatcag tcatcag tcatcag tcatcACACGGAGGTTCTA-3′) annealed to a 14mer primer tagged with the Cy3 fluorescent dye (Cy3-5′-TAGAACCTCCGTGT-3′). DNA2 is a 99mer biotinylated DNA template (biotin-5′-tttgcttcttattc tttgcttcttattc tttgcttcttattc tttgcttcttattc tttgcttcttattc tttgcttcttattc ttacacggaggttcta) annealed to the same type of primer. All DNA is from Operon Co., Alameda, CA. The sequencing feeds contained: A (10 µM dATP-Lis, 2 µM dATP, polymerase), C (10 µM ddCTP-TAMRA, 0.2 µM dCTP, polymerase), G (10 µM ddGTP-TAMRA, 3.3 µM dGTP, polymerase), U (8 µM ddUTP-TAMRA, 28 nM dTTP, polymerase), all in 1× Sequenase reaction buffer with 15 mM DTT. All tagged nucleotides are from PerkinElmer (Boston, MA). All standard nucleotides are from Boehringer Mannheim (Germany). In all cases, the polymerase used is Sequenase Version 2.0 DNA Polymerase from USB Corp. (Cleveland, OH).
Microfluidic chip fabrication
PDMS microfluidic chips with integrated micromechanical valves were built using soft lithography as described previously (26) with the following modifications. Silicon wafers were exposed to HMDS vapors for 3 min. Photoresist 5740 was spun at 2500 rpm for 60 s on a Model WS-400A-6NPP/LITE spinner from Laurel Technologies Corp. The wafers were baked at 105°C for 90 s on a hotplate. UV exposure through black-and-white transparency masks was done at 180 mW/cm2 for 25 s on a mask aligner (Karl Suss America Inc., Waterbury, VT). The molds were then developed for 3 min in a solution of 5:1 = deionized water:2401 MicroChem developer. Flow layer molds were baked at 100°C for 30 min on a hotplate to melt the 5740 and round the flow channels. Molds were characterized on Alpha-Step 500 (KLA-Tencor, Mountain View, CA). Channel height was between 9 and 11 µm, while main flow channel width was between 95 and 105 µm. Control channel profile was oblong, while flow channel profile was parabolic. Except for the height measurements and the flow channel rounding, the mold fabrication was conducted in a class-10 000 clean room.
Molds were exposed to TMCS vapors for 3 min. PDMS in 5:1 and 20:1 ratios were mixed and degassed using HM-501 hybrid mixer and cups from Keyence Corp. (Long Beach, CA). Then 35 g of the 5:1 was poured onto the control mold in a plastic Petri dish wrapped with aluminum foil. Five grams of the 20:1 was spun over the flow mold at 2500 rpm for 60 s on Spincoater P6700 (Specialty Coating Systems, Indianapolis, IN). Both were baked in an 80°C oven for 30 min. The control layer was taken off its mold and cut into respective chip pieces. Control line ports were punched using a 20-gauge luer-stub adapter (Beckton-Dickinson, Franklin Lakes, NJ). Control layer pieces were washed with ethanol, blown dry and aligned on top of the flow layer under a stereoscope. The result was baked in an 80°C oven for 1 h. Chip pieces were then cut out and peeled off the flow layer mold. Flow line ports were punched with the same 20-gauge luer-stub adapter. Meanwhile, 5:1 Sylgard was spun at 5000 rpm for 60 s over RCA-cleaned 22 mm #1 coverslips (27). The coverslips were then baked in an 80°C oven for 30 min. Chip pieces were washed in ethanol and blown dry before binding to the PDMS layer on the coverslips. The now assembled chips underwent final bake in an 80°C oven for 2 h.
Surface chemistry
The flow channels of the PDMS chip are filled with a mixture of DAPEG and the Pt catalyst at the volumetric ratio of 200:1 = DAPEG:catalyst. Then, the chip is baked in an oven at 80°C for 30 min. The DAPEG mixture is flushed out of the microchannels with high purity water. Alternating layers of PEI and PAcr are built using 5 min feeds of 20 mg/ml solutions at pH 8, by analogy with previous work at the macro scale (28). Next, the surface is biotinylated using a kit from Pierce (28). This is followed by deposition of Streptavidin Plus at 1 mg/ml in Trisb and biotinylated DNA at 7 µM in TrisMg. This completed the standard procedure for surface chemistry build up.
RESULTS
In this sequencing-by-synthesis scheme, the template to be sequenced is annealed to a shorter primer and exposed to one type of nucleotide and its fluorescently tagged analog in the presence of DNA polymerase. If the tagged nucleotide is the complement of the template base next to the 3′ end of the primer, the DNA polymerase would extend the primer with that nucleotide. Fluorescence signal is then observed after a washing step, as the tag would then be attached to the DNA template. Since the type of tagged nucleotide is known in advance, the presence of signal reveals the template base. On the other hand, if the supplied tagged nucleotide is not the correct one, the primer is not extended and no signal is observed after the washing step. Next, another type of nucleotide is fed in the same way. Once one base is determined, the scheme proceeds in exactly the same fashion to read out the next base in the template, and so on. Thus, the promise of this scheme lies in the fact that so long as the polymerase can keep extending the primer in this fashion, there is no inherent ceiling in read length, as with gels. Background signal build up and complications from FRET and fluorescence quenching can be eliminated with occasional steps of fluorescence bleaching.
In general, the dye molecules attached to the nucleotides can hinder the polymerase activity due to steric effects, such that the extension yield drops below 10% after the first tagged base (25). Thus, the feeds contain a mixture of a nucleotide and its tagged analog instead of just the tagged analog. Under suitably chosen feed concentrations, only a fraction of the DNA population extends with tagged nucleotides, while most of the DNA builds up with a standard nucleotide and thus remains available for further extension without steric hindrance. Hence, the readout can proceed to further bases at the expense of a small portion of the DNA population per successful incorporation.
Using microfluidic technology (26) as plumbing for this scheme has a number of advantages. At the microscale, diffusion happens within seconds, which facilitates reagent exchange times. Speed is important since the sequential nature of the scheme demands that the duration of each step be kept as short as possible, so that the overall duration of a sequencing run is comparable with or better than the one of the currently available technology. Microfluidics also provides parallelism and promises significant reduction in costs by economy of scale of reagents.
The sequencing chip is shown in Figure 1. The derivatization tree supplies reagents that build up the surface chemistry in all sequencing chambers at the same time. This parallelism is crucial in applications where a large number of chambers must undergo multi-step in-situ derivatizations. Next, the sequencing tree provides nucleotides and polymerase feeds to individually addressable sequencing chambers. Five separate sequencing experiments can be run in the same device after a single parallel chemistry-build up procedure. Thus, the architecture solves a wider problem of combining parallel processing with individual addressability, which is relevant to array-based applications beyond DNA sequencing.
Figure 1.

Chip architecture. (A) The microchannels are filled with food dyes to accentuate features—blue (flow layer), red (control layer). The derivatization tree supplies reagents to form surface chemistry in all five sequencing chambers at once. Lane controls direct flow of sequencing reagents from the sequencing tree into any chamber of choice. (B) Valves are formed only where wide control segments cross over flow segments. Arrows indicate the flow direction during operation—derivatization (green) and sequencing (purple). This microfluidic architecture is applicable in any situation combining parallel derivatization of an array with individual addressability of its elements.
We had developed previously surface chemistry that uses polyelectrolyte multilayers (PEM) to anchor DNA to glass (28) and provide electrostatic shielding against non-specific tagged nucleotide attachment. However, in our microfluidic application, the long bake that seals the coverslip to PDMS also fouls the hydroxyl groups on the glass surface, which are crucial for the PEM deposition. Shorter bakes allow PEM deposition but produce unreliable sealing. We introduced an additional featureless PDMS layer as a floor for the flow channels. That produced reliable sealing but required adapting the DNA anchorage chemistry to PDMS surfaces. PDMS polymerizes as SiH groups react with vinyl groups in the presence of a Pt catalyst. This process leaves unreacted SiH groups on the surface, which can be used as functionalization targets for vinyl groups during surface chemistry build up. On the other hand, the polar nature of polyethylene glycol (PEG) made it a suitable substrate for PEM deposition. Thus, we selected diacrylated PEG (DAPEG) as the link between PDMS and the proven PEM surface chemistry. While UV-based PEG and biotin grafts have been shown before (29,30), our surface chemistry is the first demonstrated non-UV-based, robust, specific and tunable anchoring of DNA to PDMS where the DNA remains sterically available for enzymatic biochemical reactions and fluorescence background from non-specific binding is suppressed through surface chemistry.
We built negatively terminated PEM on top of DAPEG-treated microchannel surfaces in a PDMS chip like the one shown in Figure 1. Next, 1 µM dUTP-Cy3 in Trisb was fed through the microchannel for a few minutes. After flushing with Trisb, pictures were taken and the average counts per pixel in the microchannel were obtained. One layer of PEI was built up to make the surface positively charged. Then, the dUTP-Cy3 feed, the flushing and the detection were repeated. Since the tagged nucleotide is negatively charged, positively terminated surfaces attached 267 times more nucleotide than their negatively terminated counterparts, while control experiments showed PEM would not assemble onto PDMS in the absence of DAPEG. This shows the shielding quality of our PDMS-DAPEG-PEM surface chemistry.
Further experiments showed that increasing the number of PEM layers increases the surface charge density, which improves the shielding. Four alternating layers are sufficient to prevent most of the non-specific attachment, while best signal-to-noise is reached at 12 layers.
To test biotinylation, the DAPEG-PEM surface chemistry was built with eight PEM layers in a PDMS microfluidic chip of essentially the same design as Figure 1. Then the biotinylation mixture (28) was fed at 5 mM for 1, 2, 4, 8 and 16 min into lanes 1 through to 5, respectively, followed in each case by a 2 min flushing of MES 10 mM buffer. Next, pictures of all chambers were taken as background signals before the fluorophore feed, using the rhodamine fluorescence filter set. Streptavidin Alexa Fluor 555 from Molecular Probes was fed at 1 mg/ml in Trisb for 2.5 min from the derivatization tree into all chambers simultaneously by keeping the derivatization valve array closed. All unattached streptavidin was washed away with Tris buffer and pictures were taken again with the same filter set. The net signal was extracted and converted into streptavidin surface density using a simple bulk calibration by volume fluorescence signal from a known probe concentration (Fig. 2A). The same calibration method was used henceforth.
Figure 2.

Chip surface chemistry diagnostics. (A) The chambers of the same device were biotinylated over varying times. Tagged streptavidin saturated the surface sites and measured biotin density. The linear dependence with time is a good tool to tune the DNA surface density as desired. (B) Surface chemistry is stable with prolonged continuous flushing since the signal from anchored tagged DNA (squares) is consistent with the fluorescence bleaching prediction (circles). (C) Signals from tagged DNA anchored in different chambers of the same device (black symbols) are consistent with the bleaching prediction (empty diamonds). The consistency in behavior among chambers shows desired homogeneity of conditions over the array. (D) Same data as in (C) plotted with respect to real time, shows surface chemistry is stable over 14 h at room temperature.
Since all available biotin sites were saturated by streptavidin by the time of signal detection, the observed linear dependence maps the biotinylation progress. This linearity provides a tool for tuning the DNA surface density in the PDMS microchannel. The mapped range is the one of relevance to fluorescence studies as further increase in density might bring the probes together close enough for fluorescence quenching to become an issue.
To test surface chemistry stability with continuous flow, we anchored DNA1 in a device of the same layout as the one in Figure 1, using 16 PEM layers. Then we continuously flushed Trisb through one of the sequencing chambers, while simultaneously taking data every 5 min (Fig. 2B). The ‘squares’ curve shows the surface density of emitting fluorophores versus real time of continuous flushing. The ‘circles’ curve shows the expected signal, based on prior photobleaching experiments conducted without flushing. Since the measured signal is consistent with the prediction, virtually all loss of signal must be due to bleaching of the Cy3 tag on the DNA rather than loss of DNA off the surface due to anchorage failure. Hence, the surface chemistry is stable over at least 1.5 h of continuous flushing.
To test surface chemistry stability over time, we anchored DNA2 in a device of the same layout as the one in Figure 1, using 12 PEM layers. Next, from time to time, we flushed Trisb for 1 min through all chambers simultaneously and then took pictures of them using the optical system described above. The emitting fluorophores surface densities are plotted in Figure 2C and D, where each curve shows the results from a respective chamber. The consistency of these curves demonstrates the uniformity of conditions in the array, as well as the reproducibility of the results. The signal is consistent with the bleaching prediction (empty diamonds in Fig. 2C). Thus the falloff across exposures is not due to loss of material off the surface but due to fluorescence bleaching. The real-time pattern (Fig. 2D) shows the anchorage is stable over at least 14 h at room temperature.
To test sequencing reagents stability over time, we anchored DNA1 in a device of the same layout as the one in Figure 1, using 14 PEM layers. After fluorescence detection confirmed the successful attachment of DNA in one of the microchambers, the Cy3 tags there were bleached. Next, ddGTP-TAMRA (100 µM in 1× Sequenase reaction buffer with 5 mM DTT) was fed into that chamber only, followed by a Trisb flush and fluorescence detection. Then, another solution containing 0.5 U/µl polymerase, but otherwise identical to the first solution, was fed into the same chamber, followed by a Trisb flush and fluorescence detection. Later, the same procedure was repeated with the next chamber, and so on. The results are shown in Figure 3A. Comparing the polymerase and non-polymerase cases for each chamber shows that everywhere the polymerase successfully extended the primer with the tagged nucleotide, while the non-specific attachment of the dye was minuscule in comparison. As the whole experiment was done with a single load of reagents, comparing the results across chambers shows that the polymerase and nucleotide remained active over at least 2 h at room temperature with no visible loss in activity.
Figure 3.
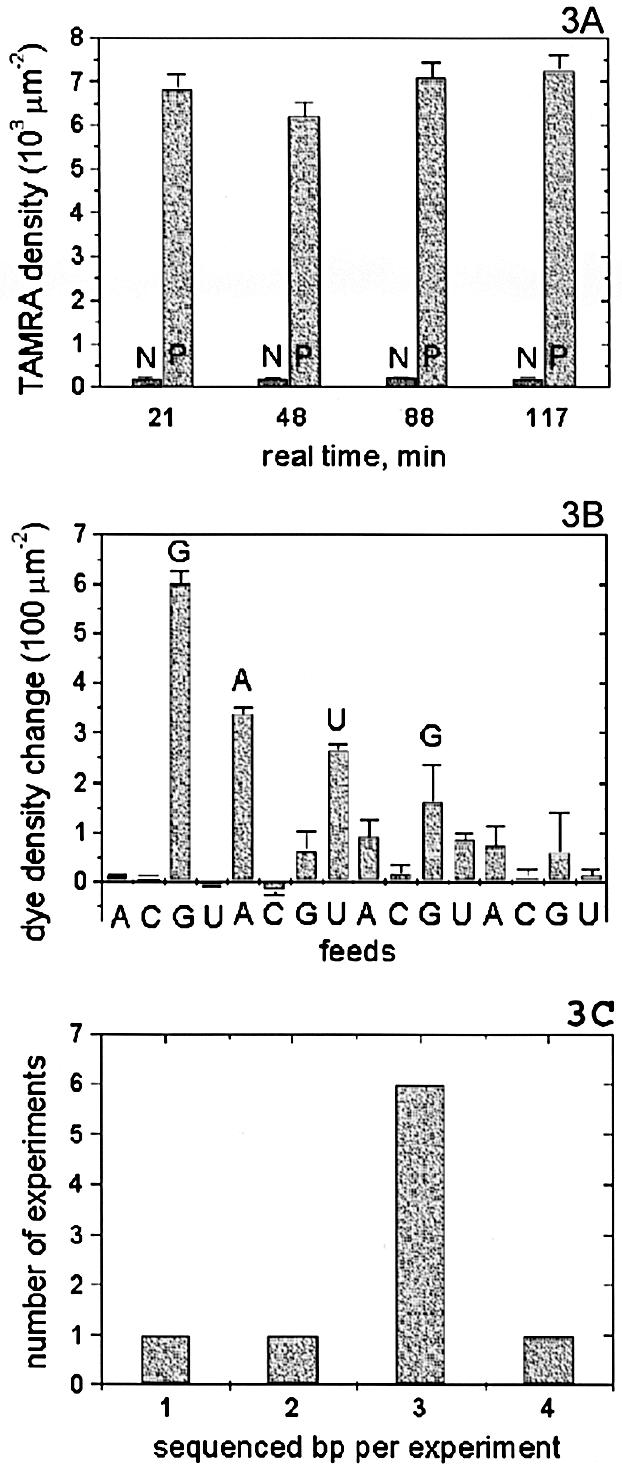
DNA sequencing. (A) The same sequencing reagents with polymerase (P) and without it (N) were fed into same-device chambers at different times. Tagged nucleotide incorporation was confirmed by the P-N difference in each case. P’s over time show sequencing reagents retain activity over at least 2 h. (B) A mixture of tagged and non-tagged nucleotides of the same type is fed into a single sequencing chamber. Incorporation is detected as increase in fluorophore surface density. The read GAUG corresponds exactly to the template sequence CTAC. (C) Statistics over nine sequencing experiments shows the most likely read length is 3 bp from a single chamber.
Taken together, these experiments showed that the device and reagents are stable and capable of performing nucleotide incorporation over many hours. Hence, we could proceed to sequencing.
DNA1 was anchored in a device identical to the one in Figure 1, using 16 PEM layers. After fluorescence detection confirmed the successful attachment of DNA, the Cy3 tags in one of the chambers were bleached. Next, a feed containing polymerase, a nucleotide, and its tagged analog, was followed by a Trisb flush and fluorescence detection. This process was iterated with different feeds in the same chamber, to collect the sequencing data. The net increase in the fluorescent signal after each feed was converted into a corresponding change in fluorophore surface density based on individual reagent calibrations. Next, the same experiment with the same sequence of feeds was repeated in another chamber of the same device, except for withholding the polymerase in all feeds. The similarly extracted data showed the level of non-specific attachment and was subtracted from the previous data to produce the final results for this experiment (Fig. 3B). The measured sequence, GAUG, exactly corresponds to the beginning of the known template sequence of CTACTG… Thus, we have shown successful sequencing of 4 consecutive bp in a single microfluidic chamber.
We pooled the data from nine such sequencing experiments with small variations in reagents, templates and feed schemes. The resulting histogram showed the most common read length to be 3 bp (Fig. 3C).
DISCUSSION
It is clear from the sequencing data that the signal-to-noise deteriorates so as to limit the read length in the chamber to 4 bp. We believe three factors conspire to produce this result.
First, while the surface chemistry shielding works very well, it is not perfect, so there is some dye-dependent attachment (∼50 dyes/µm2). Since it is non-specific, its magnitude varies enough to make it difficult to subtract as a DC-offset. Further improvements in the chemistry may eliminate this factor, but its overall impact is small next to the other two.
The second factor is that currently available polymerases have trouble incorporating even an unmodified nucleotide behind a tagged deoxynucleotide (typically 10% yield or less). Hence, at present, it makes no real difference if we use tagged deoxynucleotides or tagged terminators—in both cases, the primer does not extend any further. Every incorporation exponentially decreases the amount of DNA available for further incorporation, and so, the net signal drops with every sequenced base pair. The read length cut-off is where the net signal becomes comparable with the noise from other sources. Increasing DNA surface density would push the cut-off further along the base pair axis, but the surface density is limited on the upper end by quenching considerations and steric limitations. A better solution is to evolve polymerases whose incorporation yield is not affected by tagged nucleotides. Cleavable nucleotides (31,32) promise to accomplish the same in a different way.
The third factor is misincorporation—in the absence of competition from other types of bases in the solution, the polymerase can make a mistake and put in the wrong base. This results in loss of synchronicity, or dephasing, among the DNA strands in the sample, and thus the signal shrinks while the background increases. This can be addressed by evolving stricter polymerases and/or by use of kinetics decoy molecules to create competition among substrates.
When these techniques slow down the signal-to-noise falloff, read length will increase, while identification of repeats in the template sequence will become trivial through the multiplicity of the fluorescent signal.
Even at the current read length, µSBS is immediately applicable where the goal is to sequence only a few base pairs. We used 500 nl of reagents per feed—considerably less than is practical with conventional methods. The fundamental lower bound is the volume of the sequencing chamber, which is currently <500 pl; so, the potential for improvement is another three orders of magnitude. Although some fixed initial investment is always necessary to interface between the macro and micro world, it would ultimately be distributed over thousands of chambers on the chip (33).
The current readout length is also sufficient for single-nucleotide polymorphism (SNP) analysis. This field has received tremendous attention recently, with commercial platforms available from companies such as Pyrosequencing, ParAllele, Illumina, Orchid and Lynx, most of which are based around similar DNA polymerase extension reactions. The µSBS method is not yet as highly parallel as the industrial approaches, but in principle it can be extended to a similar scale. Microfluidic SNP analysis offers some advantages in fluid functionality, reagent consumption and integrability that may ultimately complement or improve the commercial platforms.
The scale of the µSBS devices couples with the detection system to require a very small amount of DNA for successful information extraction. The sequencing chamber contained <1 fmol of DNA, compared with 2 pmol in pyrosequencing (13), and at least tens of pmol in other schemes. The automated capillary method (2) reported a detection limit of 1 fmol per band, which introduces a factor of 600 for the total DNA amount necessary. The mass spectrometry method reported 5 fmol per fragment (8), or more (9), and so, suffers from the same factoring.
Such parsimonious requirements enable a further advantage of µSBS, namely, its integrability with a number of devices based on the same PDMS microfluidic technology but addressing different applications (34–36). The elimination of the need to switch back and forth between the macro and micro scales just for purposes of DNA sequencing, opens the way to a number of exciting applications, e.g. closed-loop evolution and analysis microfluidic systems.
The plumbing and surface chemistry described here are general and can serve as the basis platform for other sequencing schemes, such as pyrosequencing. Ronaghi et al. (13) used a four-enzyme system for incorporation, detection and waste elimination, and showed successful sequencing. However, the system has limitations (14), some of which are due to its solution-based format—each new extension reaction dilutes the sample and interferes with enzyme kinetics, while intermediate product accumulation limits read length. The system also loses accuracy in homopolymeric regions, because the primer extension takes longer, and the enzymatic nucleotide elimination activates too soon. The microfluidics and surface chemistry promise to eliminate all these problems. Anchoring of the template means intermediate products and nucleotides are removed fluidically without loss of DNA. This does away with enzymatic nucleotide elimination, and so, helps in homopolymeric regions. Each new feed is at the same optimal reagent concentration, which avoids dilution kinetics problems. In addition, the amount of DNA yielding the sequencing result in our system was 1 fmol versus the pyrosequencing amount of 2 (13) and 5 pmol (15). This parsimony would be important if the device is to be an integral part of a larger lab-on-chip system.
CONCLUSIONS
We have combined fluorescence, surface chemistry and microfluidic methods into a single PDMS device with broad applications to fluorescence studies of DNA. The system benefits from the standard microfluidic advantages of economy, speed and control, while our architecture combines parallel processing and individual addressability, and is thus significant to array applications. On the other hand, the novel surface chemistry described here ensures the DNA retention in the device and is the first demonstrated specific, robust and tunable grafting of DNA into PDMS microfluidic devices. This makes it an enabling technology for the related studies and applications, e.g. DNA pyrosequencing. Herein, we have proven the longevity and flow stability of the anchorage, as well as the longevity of our reagent preps. We have used the whole integrated system to demonstrate successful 3 bp DNA sequencing, which illustrates the developed technology and forms a major step towards microfluidic bulk-fluorescence DNA sequencing of longer read lengths, while the system remains fully integrable in PDMS lab-on-a-chip systems.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Markus Enzelberger and Marc Unger for fruitful discussions of chemistry. The authors also thank Todd Thorsen for technical help with the microfluidic chips fabrication and testing. Partial support for this work was provided by NIH grant HG01642, DARPA grant DAAD19-001-0392, and NIH training grant 5T32-GM07616.
REFERENCES
- 1.Sanger F., Nicklen,S. and Coulson,A.R. (1977) DNA sequencing with chain-terminating inhibitors. Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith L.M., Sanders,J.Z., Kaiser,R.J., Hughes,P., Dodd,C., Connell,C.R., Heiner,C., Kent,S.B.H. and Hood,L.E. (1986) Fluorescence detection in automated DNA sequence analysis. Nature, 321, 674–679. [DOI] [PubMed] [Google Scholar]
- 3.Lander E.S., Linton,L.M., Birren,B., Nusbaum,C., Zody,M.C., Baldwin,J., Devon,K., Dewar,K., Doyle,M., FitzHugh,W. et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 4.Venter J.C., Adams,M.D., Myers,E.W., Li,P.W., Mural,R.J., Sutton,G.G., Smith,H.O., Yandell,M., Evans,C.A., Holt,R.A. et al. (2001) The sequence of the human gemone. Science, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- 5.Trager R.S. (2002) Venter’s next goal: 1000 human genomes. Science, 298, 947. [DOI] [PubMed] [Google Scholar]
- 6.Pennisi E. (2002) Gene researchers hunt bargains, fixer-uppers. Science, 298, 735–736. [DOI] [PubMed] [Google Scholar]
- 7.Koster H., Tang,K., Fu,D.-J., Braun,A., van den Boom,D., Smith,C.L., Cotter,R.J. and Cantor,C.R. (1996) A strategy for rapid and efficient DNA sequencing by mass spectroscopy. Nat. Biotechnol., 14, 1123–1128. [DOI] [PubMed] [Google Scholar]
- 8.Fu D.-J., Tang,K., Braun,A., Reuter,D., Darnhofer-Demar,B., Little,D.P., O’Donnell,M.J., Cantor,C.R. and Koster,H. (1998) Sequencing exons 5 to 8 of the p53 gene by MALDI-TOF mass spectrometry. Nat. Biotechnol., 16, 381–384. [DOI] [PubMed] [Google Scholar]
- 9.Roskey M.T., Juhasz,P., Smirnov,I.P., Takach,E.J., Martin,S.A. and Haff,L.A., (1996) DNA sequencing by delayed extraction – matrix-assisted laser desorption/ionization time of flight mass spectrometry. Proc. Natl Acad. Sci. USA, 93, 4724–4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards J.R., Itagaki,Y. and Ju J. (2001) DNA sequencing using biotinylated dideoxynucleotides and mass spectroscopy. Nucleic Acids Res., 29, e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metzker L.M., Raghavachari,R., Richards,S., Jacutin,S.E., Civitello,A., Burgess,K. and Gibbs,R.A. (1994) Termination of DNA synthesis by novel 3′-modified-deoxyribonucleoside 5′-triphosphates. Nucleic Acids Res., 22, 4259–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hyman E.D. (1988) A new method of sequencing DNA. Anal. Biochem., 174, 423–436. [DOI] [PubMed] [Google Scholar]
- 13.Ronaghi M., Uhlen,M. and Nyren,P. (1998) A sequencing method based on real-time pyrophosphate. Science, 281, 363. [DOI] [PubMed] [Google Scholar]
- 14.Ronaghi M. (2001) Pyrosequencing sheds light on DNA sequencing. Genome Res., 11, 3–11. [DOI] [PubMed] [Google Scholar]
- 15.Pourmand N., Elahi,E., Davis,R.W. and Ronaghi,M. (2002). Multiplex pyrosequencing. Nucleic Acids Res., 30, e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ehn M., Ahmadian,A., Nilsson,P., Lundeberg,J. and Hober,S. (2002) Escherichia coli single-stranded DNA-binding protein, a molecular tool for improving sequence quality in pyrosequencing. Electrophoresis, 23, 3289–3299. [DOI] [PubMed] [Google Scholar]
- 17.Drmanac R., Labat,I., Brukner,I. and Crkvenjakov,R., (1989) Sequencing of megabase plus DNA by hybridization: theory of the method. Genomics, 4, 114–128. [DOI] [PubMed] [Google Scholar]
- 18.Jones D.H. (1997) An iterative and regenerative method for DNA sequencing. Biotechniques, 22, 938–946. [DOI] [PubMed] [Google Scholar]
- 19.Mitra R.D., Butty,V.L., Shendure,J., Williams,B.R., Housman,D.E. and Church,G.M. (2003) Digital genotyping and haplotyping with polymerase colonies. Proc. Natl Acad. Sci. USA, 100, 5926–5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitra R.D., Shendure,J., Olejnik,J., Edyta-Krzymanska-Olejnik and Church,G.M. (2003) Fluorescent in situ sequencing on polymerase colonies. Anal. Biochem., 320, 55–65. [DOI] [PubMed] [Google Scholar]
- 21.Akeson M., Branton,D., Kasianowicz,J.J., Brandin,E. and Deamer,D.W. (1999) Microsecond time-scale discrimination among polycytidylic acid, polyadenylic acid and polyuridylic acid as homopolymers or as segments within single RNA molecules. Biophys. J., 77, 3227–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deamer D.W. and Akeson,M. (2000) Nanopores and nucleic acids: prospects for ultrarapid sequencing. Trends Biotechnol., 18, 147–154. [DOI] [PubMed] [Google Scholar]
- 23.Howorka S., Cheley,S. and Bayley,H. (2001) Sequence-specific detection of individual DNA strands using engineered nanopores. Nat. Biotechnol., 19, 636–639. [DOI] [PubMed] [Google Scholar]
- 24.Nakane J.J., Akeson,M. and Marziali,A. (2003) Nanopore sensors for nucleic acid analysis. J. Phys. Condens. Matter, 15, R1365–1393. [Google Scholar]
- 25.Braslavsky I., Hebert,H., Kartalov,E. and Quake,S.R. (2003) Sequence information can be obtained from single DNA molecules. Proc. Natl Acad. Sci. USA, 100, 3960–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Unger M.A., Chou,H.-P., Thorsen,T., Scherer,A. and Quake,S.R. (2000) Monolithic microfabricated valves and pumps by multilayer soft lithography. Science, 288, 113–116. [DOI] [PubMed] [Google Scholar]
- 27.Unger M.A., Kartalov,E.P., Chiu,C.-S., Lester,H.A. and Quake,S.R. (1999) Single-molecule fluorescence observed with mercury lamp illumination. Biotechniques, 27, 1008–1014. [DOI] [PubMed] [Google Scholar]
- 28.Kartalov E.P., Unger,M.A. and Quake,S.R. (2003) Polyelectrolyte surface interface for single-molecule fluorescence studies of DNA polymerase. Biotechniques, 34, 505–510. [DOI] [PubMed] [Google Scholar]
- 29.Hu S.W., Ren,X.Q., Bachman,M., Sims,C.E., Li,G.P. and Allbritton,N. (2002) Surface modification of poly(dimethylsiloxane) microfluidic devices by ultraviolet polymer grafting. Anal. Chem., 74, 4117–4123. [DOI] [PubMed] [Google Scholar]
- 30.Shamansky L.M., Davis,C.B., Stuart,J.K. and Werner,G.K. (2001) Immobilization and detection of DNA on microfluidic chips. Talanta, 55, 909–918. [DOI] [PubMed] [Google Scholar]
- 31.Bai X., Li,Z., Jockusch,S., Turro,N.J. and Ju,J. (2003) Photocleavage of a 2-nitrobenzyl linker bridging a fluorophore to the 5′ end of DNA. Proc. Natl Acad. Sci. USA, 100, 401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z., Bai,X., Ruparel,H., Kim,S., Turro,N.J. and Ju,J. (2003) A photocleavable fluorescent nucleotide for DNA sequencing and analysis. Proc. Natl Acad. Sci. USA, 100, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thorsen T., Maerkl,S.J. and Quake,S.R. (2002) Microfluidic large scale integration. Science, 298, 580–584. [DOI] [PubMed] [Google Scholar]
- 34.Hansen C.L., Skordalakes,E., Berger,J.M. and Quake,S.R. (2002) A robust and scalable microfluidic metering method that allows protein crystal growth by free interface diffusion. Proc. Natl Acad. Sci. USA, 99, 16531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J., Enzelberger,M. and Quake,S. (2002) A nanoliter rotary device for polymerase chain reaction. Electrophoresis, 23, 1531–1536. [DOI] [PubMed] [Google Scholar]
- 36.Liu J., Hansen,C. and Quake,S.R., (2003) Solving the ‘world-to-chip’ interface problem with a microfluidic matrix. Anal. Chem., 75, 4718–4723. [DOI] [PubMed] [Google Scholar]