

ABSTRACT
Chlamydia trachomatis is an obligate intracellular human pathogen that grows inside a membranous, cytosolic vacuole termed an inclusion. Septins are a group of 13 GTP-binding proteins that assemble into oligomeric complexes and that can form higher-order filaments. We report here that the septins SEPT2, -9, -11, and probably -7 form fibrillar structures around the chlamydial inclusion. Colocalization studies suggest that these septins combine with F actin into fibers that encase the inclusion. Targeting the expression of individual septins by RNA interference (RNAi) prevented the formation of septin fibers as well as the recruitment of actin to the inclusion. At the end of the developmental cycle of C. trachomatis, newly formed, infectious elementary bodies are released, and this release occurs at least in part through the organized extrusion of intact inclusions. RNAi against SEPT9 or against the combination of SEPT2/7/9 substantially reduced the number of extrusions from a culture of infected HeLa cells. The data suggest that a higher-order structure of four septins is involved in the recruitment or stabilization of the actin coat around the chlamydial inclusion and that this actin recruitment by septins is instrumental for the coordinated egress of C. trachomatis from human cells. The organization of F actin around parasite-containing vacuoles may be a broader response mechanism of mammalian cells to the infection by intracellular, vacuole-dwelling pathogens.
IMPORTANCE
Chlamydia trachomatis is a frequent bacterial pathogen throughout the world, causing mostly eye and genital infections. C. trachomatis can develop only inside host cells; it multiplies inside a membranous vacuole in the cytosol, termed an inclusion. The inclusion is covered by cytoskeletal “coats” or “cages,” whose organization and function are poorly understood. We here report that a relatively little-characterized group of proteins, septins, is required to organize actin fibers on the inclusion and probably through actin the release of the inclusion. Septins are a group of GTP-binding proteins that can organize into heteromeric complexes and then into large filaments. Septins have previously been found to be involved in the interaction of the cell with bacteria in the cytosol. Our observation that they also organize a reaction to bacteria living in vacuoles suggests that they have a function in the recognition of foreign compartments by a parasitized human cell.
INTRODUCTION
Chlamydiae are obligate intracellular bacteria and the causative agents of a number of frequent diseases in humans and in animals. The species Chlamydia trachomatis has a number of serovars that cause human sexually transmitted disease throughout the world, as well as eye infections, including trachoma, a blinding infection of the eye, which is endemic in 50 countries (1, 2). Chlamydia has a peculiar developmental cycle and occurs in two morphologically distinct forms. The infectious elementary body (EB) is taken up by the host cell (typically an epithelial cell), and the Chlamydia-containing vacuole is transformed into a specialized compartment termed an inclusion. Within the inclusion, EBs develop into reticulate bodies (RBs), which divide numerous times before redifferentiating into EBs toward the end of the developmental cycle. This developmental cycle takes about 2 days for C. trachomatis in vitro and terminates with the release of the bacteria (3, 4). Development and growth inside a cytosolic membranous vacuole are thus a hallmark of chlamydial infection, a characteristic it shares with a number of other bacterial pathogens, such as Legionella or Salmonella.
Cytoskeletal systems have numerous functions in a cell, for instance, conveying motility and shaping the cell or arranging organelles and vesicular transport. The cytoskeleton plays substantial roles in the interaction of a host cell with intracellular bacteria, and many bacteria have developed patterns of interaction with host cytoskeletal structures. Examples include the actin tail of cytosolic Listeria monocytogenes (5) or, for Chlamydia, the nucleation of actin at the site of entry through a dedicated bacterial protein (6).
During growth of the chlamydial inclusion, cytoskeletal elements are arranged around the vacuole. “Cages” or “coats” have been found for all three major cytoskeletal filaments, namely, intermediate filaments, microtubules, and actin (7, 8). It has been suggested that intermediate filaments build a supporting scaffold around the chlamydial inclusion that stabilizes it and prevents its collapse (8). Microtubules guide the trafficking of the inclusion to the microtubule-organizing center (9).
Actin polymerization is involved in the release of C. trachomatis: ultrastructural analysis indicates that C. trachomatis inclusions can be released without killing the host cell, and this process has been described as exocytosis-like (10). A more recent study identified two modes of release, either cell lysis or the active extrusion of the intact inclusion. Based on inhibitor studies, it has been suggested that extrusion requires actin polymerization (11). Extrusion was recently shown to be linked to the activity of members of the myosin phosphatase pathway, probably triggered by the interaction of the chlamydial protein CT228 with myosin phosphatase target subunit 1 (12).
Exploitation of the actin-myosin system therefore appears to be a strategy used by Chlamydia for organized exit from human cells, and a recent study identifies bacterial and host cell involvement in the formation of the actin coat, which begins after about 20 h after entry of Chlamydia into the host cell (13). The available data suggest that extrusion is an exit pathway that has evolved during coevolution of C. trachomatis and the host cell.
Septins are a group of filamentous GTPases already found in yeast and sometimes referred to as the fourth type of cytoskeletal structures (14). In humans, 13 septins exist, divided into four homology groups. Septins can form large structures, including rings and long filaments. Septin assembly has been found to be codetermined by cellular membranes (15) and by actin and microtubules. In particular, the relationship with actin appears to be bidirectional, with both components being involved in the assembly of the other (14). Septins have further been found to be recruited to intracellular cytosolic bacteria that are found freely in the cytosol (14). During chlamydial infections, the addition of a chemical that can inhibit septins reduced the number of inclusions with actin coating (13).
We identified the septin SEPT2 as a substrate of the chlamydial protease CPAF. We followed up on this observation to test for a potential role of septins in the development of the inclusion. Our data suggest that septins are involved in the formation of the actin coat around the chlamydial inclusion and in the extrusion of the inclusion. The results therefore indicate that the role of septins in the response to intracellular bacteria is not limited to the coating of (free) cytosolic bacteria but extends to bacterial inclusions, where they may be involved especially in the exocytosis of the bacteria.
RESULTS
Septins form a filamentous “cage” or “coat” around the inclusion.
We initially were alerted to the potential role of septins in infection while studying the role of the chlamydial protease CPAF. We had conducted an analysis of CPAF substrates and found that SEPT2 was cleaved in cells ectopically expressing CPAF (the cells are described in reference 16) (see Fig. S1A in the supplemental material). Cleavage sites of SEPT2 and further septins were identified by a technique that quantifies N termini of cellular proteins by mass spectrometry (terminal amine isotopic labeling of substrates [TAILS]) (17). SEPT2 can be cleaved by CPAF at positions 67 and 129, and a cleavage product of the same size as during ectopic expression of CPAF was observed during infection of HeLa cells with C. trachomatis (see Fig. S1B). Fragment size matched the size predicted by the position 67 cleavage, and expression of SEPT2 tagged on either side confirmed cleavage close to the N terminus (not shown).
Septins were unequivocally cleaved upon expression of active CPAF in uninfected cells, and published studies agree that CPAF is found in the cytosol of infected cells (the same is the case in a recently described expression of epitope-tagged CPAF [18]). However, it is likely that a substantial amount of CPAF is released only during detergent extraction of infected cells, and the observed cleavage of CPAF substrates is much smaller when this is experimentally prevented (19). Indeed, when cells were extracted with chaotropic salt, no SEPT2 cleavage was detectable by Western blotting (see Fig. S1 in the supplemental material). CPAF can be inhibited by the peptide WEHD-fmk (20), and WEHD-fmk fails to block recruitment of SEPT2 to the inclusion (see Fig. S1C). The cleavage of septins by CPAF therefore appears not to be relevant for the establishment of septin “cages,” but since CPAF is probably an important Chlamydia protease and its role is hotly debated at present (19), we provide additional data on this in Fig. S1 (also see the legend there; a more detailed discussion on CPAF is found in reference 21).
This finding prompted us to investigate the structure of septins during chlamydial infection. Immunostaining for SEPT2 showed that SEPT2-containing fibers encased the chlamydial inclusion, starting from about 16 h postinfection (p.i.) (Fig. 1). In most cells, fibers that ran across the inclusion were observed, often visible as a ring around the inclusion in epifluorescence microscopy. Confocal imaging and orthogonal views of the reconstructed image show fibers containing SEPT2 (see Fig. S2A and B in the supplemental material) and SEPT9 (see Fig. S2C) at the bottom of the inclusion, around it, and at its top. The structures are suggestive of larger-order structures since they are well known to be formed by septins.
FIG 1 .

SEPT2 forms filamentous structures around the chlamydial inclusion. HeLa cells were infected with C. trachomatis for 16, 20, 24, or 39 h. Immunostaining for SEPT2 (red) and chlamydial Hsp60 (cHsp60) (green) was performed. Arrows point to chlamydial inclusions. Scale bar, 10 µm. Images are representative of 2 individual experiments.
Large septin structures typically are formed by heteromeric complexes that contain three or four different septins (22), suggesting that the observed structures around the inclusion contained other septins in addition to SEPT2. Our proteomic study of isolated C. trachomatis inclusions by mass spectrometry (MS) had identified SEPT2, -7, -9, and -11 on the inclusion, with substantial enrichment of SEPT2 and -9 (see the legend to Fig. 2). We therefore also stained for these septins in infected cells. We found staining very similar to that for SEPT2 with antibodies against SEPT9 and also with antibodies against SEPT11. Staining for SEPT7 was inconclusive, most likely because the antibodies available to us did not give a very clear signal in immunofluorescence (data not shown). The data, however, clearly identify fibers containing SEPT2, -9, and -11 on the C. trachomatis inclusion. Since SEPT7 appears to be a core component of most septin structures (22, 23) and it was identified by MS on the inclusion, its involvement here appears very likely.
FIG 2 .

(A) Fibers containing SEPT2, -9, and -11 encase the chlamydial inclusion. HeLa cells infected for 48 h were fixed and stained for SEPT2 or SEPT9 or infected for 30 h, fixed, and stained for SEPT11. Chlamydial inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of at least 3 independent experiments. Proteomic analysis found the four septins SEPT2, -7, -9, and -11 on purified inclusions (see Materials and Methods for details; the following numbers of peptides were identified by mass spectrometry in infected/uninfected cells: SEPT2, 64/29; SEPT7, 6/11; SEPT9, 13/5; SEPT11, 7/11). (B) Infection increases the amount of SEPT9 in SEPT2-containing complexes. Uninfected HeLa cells or HeLa cells infected with C. trachomatis for 30 h were lysed, and proteins in the lysate supernatants (Input) were immunoprecipitated with anti-SEPT2 antibodies. Input, unbound, and IP fractions from the immunoprecipitations (IPs) were analyzed by SDS-PAGE, followed by immunoblotting with anti-SEPT2, -7, -9, or -11 or anti-GAPDH antibody. The smaller band for SEPT2 is consistent with cleavage by CPAF during sample preparation. A number of isoforms exist for SEPT9; one isoform especially appears to be recruited to septin fibers during infection.
Septin bundles also exist in uninfected cells, where they are involved, for instance, in the organization of F actin around the nucleus (24). When we immunoprecipitated SEPT2, we found a constitutive association with SEPT7, -9, and -11. The same septins were copurified from infected cells. However, a substantial increase in the amount of copurified SEPT9 from infected cells was observed (Fig. 2B). The formation of septin fibers during infection may therefore alter composition of existing septin structures, or the formation of larger septin structures from small subunits during infection may be linked to an increased presence of SEPT9.
SEPT2, -9, and -11 colocalize with actin on the inclusion.
To understand the contribution of septins to the cytoskeletal embedding of the inclusion, we costained infected cells for septins and microtubules or actin. There appeared to be no appreciable colocalization of SEPT2 and tubulin (see Fig. S2A in the supplemental material). However, simultaneous staining for SEPT2 and actin filaments (using phalloidin) produced a very similar fibrillar picture and clear colocalization of the two structures (Fig. 3). Because of cross-reactivity of the secondary antibodies, we were unable to stain simultaneously for two septin species. However, staining for SEPT9 or SEPT11 also indicated colocalization of these septins with F actin, similar to the case with SEPT2 (Fig. 3; in some images, the bundles containing F actin and septins around the nucleus described earlier [24] are also visible). These results are strongly suggestive of the formation of fibers that contain actin together with SEPT2, -9, and -11 (and possibly also SEPT7), encasing/coating the chlamydial inclusion.
FIG 3 .

SEPT2, -9, and -11 colocalize with F actin on the inclusion. HeLa cells were infected for 30 h, fixed, and costained with phalloidin-Alexa 546 to detect F actin and with specific antibodies against SEPT2, -9, or -11. Actin and septin filaments showing colocalization on the inclusion are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of 3 independent experiments.
Role of septins in development of the inclusion.
We then aimed at evaluating the role of septins in growth of the bacteria and in the development of the inclusion. We first used the described inhibitor of septins, forchlorfenuron (FCF). FCF is a urea-derived cytokinin commonly used in plant morphogenesis studies (25). In human cells, FCF has been shown to disrupt septin assembly and to have effects on mitosis and cell migration similar to those of septin-specific RNAi (26). When added to infected cultures at earlier time points (such as after 18 h), FCF reduced growth of the chlamydial inclusion substantially. When added at 24 h p.i., this effect was relatively small (see Fig. S3 in the supplemental material). However, FCF had only a small effect on the formation of the SEPT2 structures around the inclusion, calling its efficacy into question. Furthermore, since FCF appeared to have a growth-retarding effect on the inclusion but the inclusions were still encased by septin fibers, the effect of FCF on chlamydial growth may be unrelated to septins. FCF has been reported to reduce the number of actin-positive chlamydial inclusions when added at 44 h but not at 68 h for 4 h (13). Although it is difficult to exclude a nonspecific effect, this may mean that septin activity is at some stage necessary to stabilize F actin on the inclusion.
We then used RNAi to reduce the expression of individual septins or septin combinations. Knockdowns were efficient (tested by Western blotting) in the case of several small interfering RNAs (siRNAs) against SEPT9 (see Fig. S4A to C in the supplemental material) and of inducible short hairpin RNA (shRNA) against SEPT2 (shSEPT2) in a HeLa cell line (see Fig. S4C). We also made a HeLa cell carrying inducible shRNA against SEPT7, but the knockdown in that case was less efficient and more variable (see Fig. S4C). Triple knockdown of SEPT2, -7, and -9 by siRNA was also efficient (see Fig. S4D).
We observed no effect of the knockdown of SEPT2 or SEPT9 on the growth of C. trachomatis as measured by inclusion size (see Fig. S5A in the supplemental material). Likewise, the generation of infectious progeny was no different in cells where SEPT9 had been knocked down (see Fig. S5B). It has been found that vesicular cargo exits the trans-Golgi network along microtubule tracks that also contain SEPT2 fibers (27). Chlamydia intercepts post-Golgi vesicular transport, possibly linked to Golgi fragmentation during infection (28). We therefore tested colocalization of the Golgi apparatus and septins during infection. As described before, the Golgi apparatus was fragmented and arranged around the inclusion. Although in a rare minority of cells, Golgi fragments appeared arrayed along septin fibers, this was not the case in the great majority of cells (see Fig. S5C). There is therefore no evidence that septin fibers may be used by Chlamydia for vesicular transport to the inclusion.
We did observe, however, that the knockdown of SEPT2 also disrupted the fibrillar structure of SEPT9 on the inclusion (Fig. 4). This further supports the view that the three (or four) septins indeed form these structures together.
FIG 4 .

Knockdown of SEPT2 disrupts the fibrillar structure of SEPT9 on the inclusion. HeLa cells stably carrying an inducible shSEPT2 construct were induced with 100 ng/ml AHT to silence the expression of human SEPT2 for 48 h. At 24 h postinduction, cells were infected with C. trachomatis; they were fixed at 30 h postinfection and labeled with an antibody against SEPT9. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of 3 independent experiments.
The colocalization of septins with actin and the reported reciprocal interactions of septins and actin suggested the possibility of a functional interdependence of these two components. We therefore targeted septins by RNAi and stained for F actin during infection. Indeed, knockdown of either SEPT9 (Fig. 5A; see also Fig. S6A in the supplemental material) or SEPT2 (Fig. 5B) caused the reduction or loss of identifiable actin fibers on the inclusion. Conversely, when actin polymerization was inhibited by treatment with cytochalasin D, SEPT2 and -9 structures on the inclusion were also disrupted (Fig. 5C).
FIG 5 .

Targeting septins by RNAi disrupts F actin structures on the inclusion, while inhibiting F actin disrupts septin cages. (A and B) HeLa cells were transfected with siRNA targeting SEPT9 (A), or SEPT2-specific shRNA was induced (B). At 24 h posttransfection/-induction, cells were infected with C. trachomatis, and they were fixed at 30 h postinfection and stained with phalloidin-Alexa 546 to detect F actin and with Hoechst dye. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. (C) HeLa cells were infected for 30 h and stained for SEPT2 or SEPT9 and F actin as indicated. To some aliquots, cytochalasin D was added 30 min prior to fixation. DNA was stained with Hoechst dye. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. All images are representative of 3 independent experiments.
To visualize this likely role of septins in F actin recruitment more directly, we generated cells expressing Lifeact-green fluorescent protein (GFP) (29). Lifeact is a short peptide that is incorporated into F actin structures and, when fused to GFP, can be used to visualize the formation of F actin in live cells. Lifeact-GFP-expressing HeLa cells were infected with C. trachomatis and examined by live fluorescence microscopy for the period between 25 and 29 h postinfection. During this time, dynamic recruitment of F actin structures around the inclusion could be observed. However, knockdown of SEPT9 abolished detectable recruitment of F-actin-containing structures around the inclusion (see Movie S1 in the supplemental material; Fig. S6B gives still frames from these videos).
Not surprisingly, there was no alteration of the microtubule structures around the inclusion when septins were knocked down (see Fig. S6C in the supplemental material), and blockade of microtubule polymerization with nocodazole had no effect on septin cages (see Fig. S6D). Similarly, vimentin structures around the inclusions were unaffected by septin knockdowns (see Fig. S6E). These results argue for a function of septins in the organization or stabilization of the actin cage around the chlamydial inclusion.
Microdomains containing active src kinases have been observed around the chlamydial inclusion (30). However, there was no obviously correlating spatial arrangement of these microdomains (staining for active src) with septin fibers (time points up to 48 h were analyzed) (see Fig. S7 in the supplemental material), and knockdown of septins did not alter microdomain appearance (see Fig. S7). The inactive form of the myosin phosphatase subunit MYPT1 was found to be specifically associated with the microdomains (12). Knockdown of nonmuscle myosin IIB did not alter the SEPT2- and -9-containing fibrils on the inclusion (see Fig. S8A and B). The myosin inhibitor blebbistatin had a small effect on septin structures (see Fig. S8C); a role of myosin in the formation of septin fibrils is therefore possible. Knockdown of septins also had no effect on the structure of myosin IIB around the inclusion (see Fig. S9). Myosin IIB is involved in the extrusion of the chlamydial inclusion at the end of the developmental cycle (12), but how this role is exerted is not known. Actin is also implicated in extrusion, and we did find a role for septins in extrusion (see below). Since targeting of myosin IIB has no effect on septin arrangement, however, the formation of septin structures around the inclusion is likely upstream of myosin IIB action.
Septins are required for normal extrusion of the inclusion.
Extrusion of the chlamydial inclusion at the end of the developmental cycle is inhibited by pharmacological blockade of actin polymerization (11) and appears to be organized through the Chlamydia-initiated operation of the myosin system (12). Since we had found that septins are required for the normal organization of the actin cage around the inclusion, a contribution of septins to the extrusion of the inclusion appeared plausible. Furthermore, a role for SEPT5 in granule exocytosis has been described for platelets (31) and for neuronal cells (32), suggesting a broader role of septins in the organization of actin-dependent vesicular transport.
We tested for such a role in chlamydial extrusion by counting the extrusions released between 48 h and 66 h p.i. (11, 12). Using either siRNA directed against SEPT9 alone or the combinations of siRNA specific for SEPT2, -7, and -9, we found a 2- to 3-fold reduction in the numbers of extrusions when the septins were knocked down (Fig. 6A). Determination of infectious bacteria released by extrusion showed an about 4-fold reduction when SEPT9 was knocked down (Fig. 6B). Septins thus appear to be required for the normal organization of chlamydial exit by extrusion.
FIG 6 .
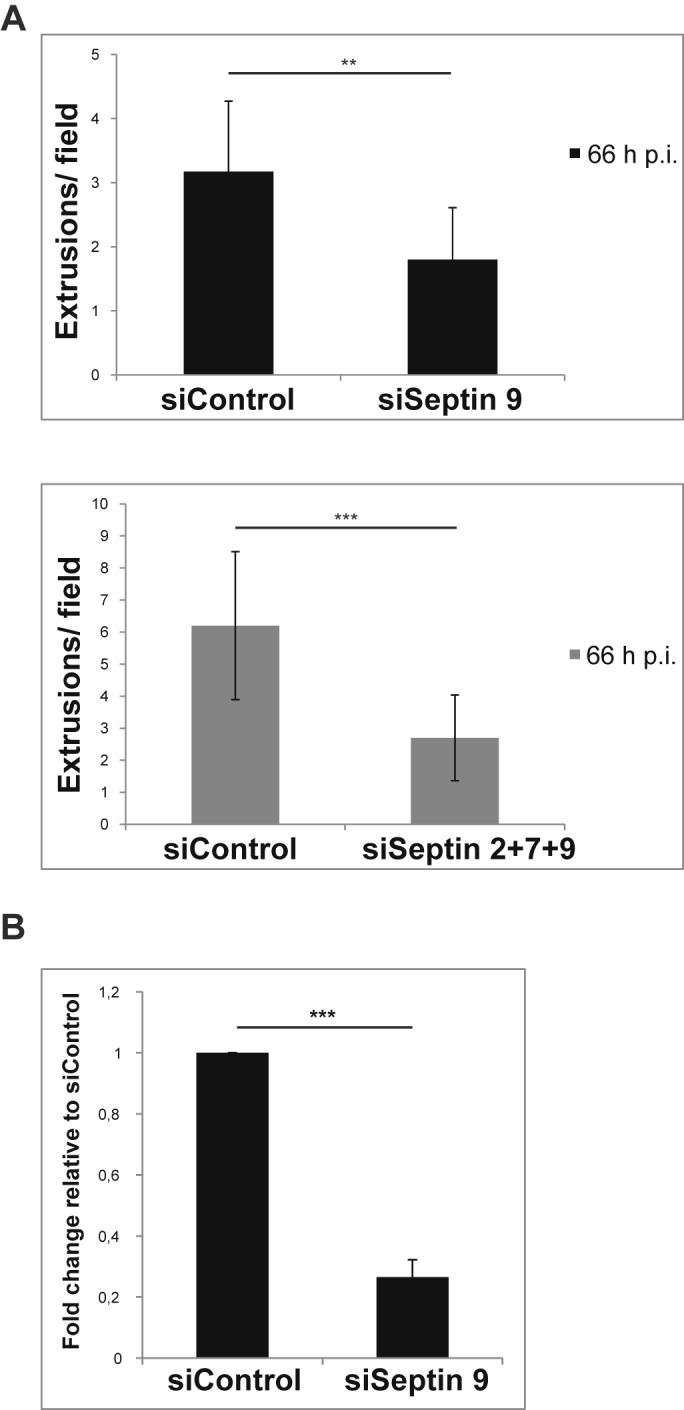
Septins are required for normal extrusion of the inclusion. (A) HeLa cells were transfected with siRNA directed against SEPT9 alone (top panel) or were transfected simultaneously with siRNAs targeting SEPT2, -7, and -9 (bottom panel), followed by infection with C. trachomatis about 4 h later. Extrusions released by host cells between 48 and 66 h p.i. were counted using fluorescence microscopy. Results were obtained from either 4 independent experiments (shown as means and SEM; P = 0.002) (siSeptin9, top panel) or 3 independent experiments (means and SEM; P = 0.0001) (siSeptin 2+7+9, bottom panel). (B) HeLa cells were transfected with siRNA targeting SEPT9, followed by infection with C. trachomatis about 4 h later. Culture supernatants containing extrusions released between 48 and 66 h p.i. were collected at 66 h p.i., extrusions were enriched by centrifugation and lysed, and bacteria were titrated on a fresh monolayer of HeLa cells. Twenty-five hours later, cells were fixed, and inclusions were counted using fluorescence microscopy. The IFU/ml of the siControl samples were set to 1. Fold change of results for siSEPT9 relative to those for siControl was calculated. Data represent means and SEM of 3 independent experiments; P = 0.0002.
DISCUSSION
We here report the arrangement of septins on the chlamydial inclusion. The staining patterns suggest an association of septins with F actin. Both the similarity of the individual septin patterns and the mutual effects of the loss of individual septins on others suggest that at least SEPT2, -9, and -11 form heteromeric fibers on the inclusion (clear localization results for SEPT7 could not be obtained with the reagents available but appear likely based on our proteomic studies and on the established involvement of SEPT7 in higher-order septin structures). The results further indicate that septins are necessary for the formation of F-actin fibers around the inclusion. Probably through this interaction, septins are also required for the normal release of chlamydial inclusions by extrusion.
Septins were cleaved subsequent to the activation of CPAF. How much of this cleavage also occurs during Chlamydia infection is difficult to say. Inhibitor studies showed that CPAF activity is not necessary for the recruitment of SEPT2 to the inclusion. Since the use of the CPAF inhibitor WEHD blocks chlamydial growth (20) (whether by inhibiting CPAF or by doing something else), we were unable to test for the possibility that CPAF-dependent cleavage of septins contributes to later effects, like extrusion. CPAF-dependent cleavage may aid the formation of the septin fibers, but it may also, by modulating the ability to oligomerize (see the legend to Fig. S1 in the supplemental material), be the attempt of Chlamydia to prevent the encaging by septins and actin (or it may have no effect at all).
The mammalian septins form four groups, based on structural similarity. Complexes containing 3 or 4 different septins can be isolated from mammalian cells, and the composition of the complexes appears to be cell type specific. Analyses of the crystal structure of the SEPT2-SEPT6-SEPT7 complex have identified a basic hexameric structure containing two molecules of each individual septin, which is then arranged into larger-order structures (33). Other complexes isolated from mammalian cells include SEPT7/9/11, -3/5/7, and -4/5/8 (reviewed in reference 22). More extensive studies using yeast two- or three-hybrid systems and studies of recombinant proteins suggested a complex of SEPT2/6/7/9 (22). Since SEPT11 shows binding and interaction preferences similar to those of SEPT6 (both are in the same group) (22), this complex is very similar to the septins we found arranged on the chlamydial inclusion and purified by immunoprecipitation (IP) with antibodies against SEPT2 (SEPT2/11/7/9). The costaining of all three, SEPT2, -9, and -11, with F actin supports this model, as does the interdependence of septin structures (targeting SEPT2 also disrupted SEPT9 fibers). All of these results are consistent with the model of one filamentous structure consisting of these four septins, together with F actin, arranged on the chlamydial inclusion.
The microscopic patterns of septins and of actin around the inclusion were the first suggestion that the two components are physically linked. Experimental downregulation of septins reduced the formation of the actin cage, as well as the dynamic recruitment of actin, and disruption of F actin also disrupted septin cages, arguing for a mutual dependency of these two components. The “cages” of vimentin and microtubules were not detectably affected by the loss of septins, and disrupting polymeric microtubular structures had no effect on septin cages. Vimentin structures have been proposed to be required for the stability of the inclusion (8). It has also been suggested that actin is necessary for the organization of vimentin (8). Since the loss of septins affected the actin structure around the inclusion but not the vimentin cage, this role of actin seems to be independent of its recruitment to the inclusion.
The molecular link in the interaction of septins and actin is not clear. It has been speculated that septins are involved in the regulation of actin nucleation through Arp2/3 or WASP (Wiskott-Aldrich syndrome protein) (14), but there seems to be no experimental evidence for this. Another possible link is through Rho GTPases. A Rho guanine nucleotide exchange factor has been found to interact with SEPT9b (34), and RhoA has been reported to be important for actin fiber formation around the chlamydial inclusion (8). SEPT2 has further been found to serve as a scaffold for myosin II (35). We found no obvious arrangement of myosin IIB with septins, and knockdown of either failed to affect the structures of the other, although inhibition of myosin with blebbistatin had a small effect on septin structures. Although molecular details remain to be worked out, these data are consistent with the organization of actin on the chlamydial inclusion through septins. Anillin is a contractile, actin-bundling protein that has also been found to bind to septins (for a review, see reference 36). We have stained for anillin in infected cells but found no association with the chlamydial inclusion (as already reported in an earlier study [37]). Anillin therefore does not seem to be involved in the F actin/septin bundles around the chlamydial inclusion.
Septins are therefore likely to recruit actin, but how are they recruited themselves? Very recent work shows that yeast plasma membrane and synthetic lipid bilayers can initiate the assembly of higher-order septin structures, probably from heterooctamers (15). How this is initiated is unknown, but it is conceivable that the chlamydial inclusion membrane can also have this function of triggering the assembly of septin complexes diffusing there from the cytosol. One possibility is that the known phospholipid interactions of septins play a role here. In particular, septins can interact with phosphatidylinositol-4,5-bisphosphate (PtdIns4,5P2, or PIP2 [38]) and –trisphosphate (PIP3), and PtdIns4,5P2-containing membranes can promote the formation of septin filaments (39). Although PIP4 rather than PIP2 or PIP3 has been found on the inclusion (40) (PIP2 has been reported to accumulate at the site of chlamydial entry [41]), this may be a possibility worth exploring.
We found that septins were required for normal extrusion of C. trachomatis. Based on inhibitor studies, it has been shown that extrusion required actin polymerization, WASP, myosin II, and RhoA (11). Although the depletion of septins reduced extrusion, this reduction was much less than that achieved by depletion of components of the myosin pathway (about 2- to 5-fold versus 20- to 50-fold) (12). Loss of septin expression prevents the accumulation of actin on the inclusion and extrusion; this suggests that either the actual actin coat on the inclusion mediates extrusion together with myosin or septin complexes elsewhere in the cell organize extrusion, most likely again through actin. Myosin may have a different function, however, such as the triggering of the process, which may depend on a local concentration of myosin; septins may have the role of arranging actin fibers on the inclusion, but actin polymerization may also be possible from other sites, without septins. Inhibition of actin function also blocked extrusion (11); since knockdown of septins disrupted the actin structures around the inclusion but only reduced extrusion (and did not block it completely), actin may also be able to act at other sites. Given the role of septins in the organization of the secretory pathway (31, 32), extrusion may be organized similarly to the exocytosis of secretory vesicles.
Septins thus have a function in organizing the actin coat of the inclusion. Is that a defense mechanism by the infected host cell, or are the bacteria coopting septins to facilitate intracellular replication? Septins have been investigated in a number of infections involving facultative intracellular bacteria (reviewed in reference 14). In the cytosolic bacteria tested (bacteria not surrounded by a membrane), such as Shigella, Listeria, and Mycobacterium, septins have been found to be recruited to the bacteria. Septins “cage” free bacteria and restrict their intracytosolic movement as well as cell-to-cell spreading; one possibility is even that septins support the delivery of the bacteria to the autophagy pathway. This structure appears different from what we describe here on the inclusion, not only in size but also in relationship to actin (septin and actin “collars” appear arranged next to each other on the bacteria and around actin tails) (42). In Shigella infection, septin depletion enhanced the number of bacteria with actin tails and spreading of the bacteria; septins therefore prevent the efficient formation of actin tails and restrict the movement of the bacteria (42). Septins have further been described to regulate entry of bacteria into the cell (14).
As far as we know, no role for septins in the growth or development microbes living in vacuoles has been reported so far. However, SEPT9 has been found on early phagosomes containing Listeria (43), and a proteomic screen of the Legionella-containing vacuole identified the same septins (SEPT2, -7, -9, and -11) that we have found on the Chlamydia inclusion (44). The encaging by septins and subsequently F actin may be a broader mechanism of dealing with pathogens that live in vacuoles inside eukaryotic host cells.
MATERIALS AND METHODS
Cell culture.
All cell lines were cultured at 37°C and with5% CO2. HeLa cells and 293-TRex-3xGyrB-CPAF cells (16) were maintained in Dulbecco modified Eagle’s minimal essential medium (DMEM) supplemented with 10% fetal calf serum (FCS) (tetracycline negative; PAA Laboratories) or 10% fetal bovine serum (FBS) (Gibco). For the 293-TRex-3xGyrB-CPAF cells, 5 µg/ml blasticidin (PAA Laboratories) and 350 µg/ml zeocin (Invivogen) were added. To induce and activate CPAF, 5 ng/ml anhydrotetracycline (AHT) (IBA Life Sciences) and 1 µM coumermycin (CM) (Sigma) were added (16). For some experiments, cytochalasin D (Sigma), nocodazole (Sigma), or blebbistatin (Calbiochem/ EMD Millipore) was added.
Infection with C. trachomatis.
Chlamydia trachomatis LGV2 was obtained from the American Type Culture Collection (ATCC) and stored in SPG medium (0.2 M sucrose, 8.6 mM Na2HPO4, 3.8 mM KH2PO4, and 5 mM glutamic acid, pH 7.4) at −80°C. One day prior to infection, cells were seeded in culture medium. Bacteria were added directly to the cells at a multiplicity of infection (MOI) of 3. To inhibit CPAF activity, 75 µM z-WEHD-fmk (R&D Systems) was added 9 h postinfection.
Antibodies and cell stains.
The following primary antibodies were used in this study: mouse anti-chlamydia Hsp60 (cHsp60; Enzo Life Sciences), mouse anti-α-tubulin (Sigma), rabbit antivimentin (Acris), mouse anti-phospho-Src Tyr 416 (pTyr-Src) (clone 9A6; Millipore) (12), rabbit anti-SEPT2 (11397-1-AP; Proteintech) or rabbit anti-SEPT2 (HPA018481; Sigma), rabbit anti-SEPT7 (H-120; Santa Cruz), mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Millipore), mouse anti-β-actin (Sigma), rabbit anti-GFP (Cell Signaling), rabbit anti-SEPT9 (made by immunization with SEPT9 peptide), rabbit anti-SEPT11 (Abcam), and mouse anti-nonmuscle myosin IIB (clone 3H2; Abcam). Secondary antibodies were donkey anti-rabbit Cy5, donkey anti-mouse Cy5, donkey anti-rabbit Alexa Fluor 647, donkey anti-rabbit fluorescein isothiocyanate (FITC), and goat anti-mouse DyLight 488 (all from Dianova) and goat anti-mouse Alexa Fluor 568 and goat anti-rabbit Alexa Fluor 488 (Invitrogen); and secondary antibodies conjugated to horseradish peroxidase (HRP) used for immunoblotting were goat anti-rabbit (A6667; Sigma) and goat anti-mouse (115-035-166; Jackson ImmunoResearch). Phalloidin Alexa Flour 546 (Life Technologies) was used to detect F actin, bisbenzimide Hoechst 33342 trihydrochloride (Sigma) was used to visualize host nuclei and bacterial DNA, and Sytox green nucleic acid stain (Life Technologies) was used to detect extruded chlamydial inclusions.
Immunofluorescence and time-lapse microscopy.
For immunofluorescence, cells were seeded in 24-well plates onto coverslips according to the assay, infected, and at the indicated time point fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) (PAA Laboratories or Gibco) for 15 min or in methanol for 10 min, permeabilized for 10 min in 0.2% Triton X-100 (Sigma) in PBS, and blocked in PBS–5% bovine serum albumin (BSA) (Sigma) in PBS for 30 min. Antibody solutions in blocking buffer were added, and the samples were incubated for 1 h or overnight at 4°C with primary antibody, washed with PBS, and incubated for 1 h in fluorescently labeled secondary antibody. The samples were stained with Hoechst for 10 min and mounted in Permafluor medium (Thermo Scientific). Epifluorescence images were acquired using a BZ-9000E microscope (Keyence) and further processed with the BZ-II Analyzer 1.42 software program (Keyence). Confocal microscopy images were collected using an Inverted Axiovert 200-M microscope (Zeiss) with plan-apochromat objectives, equipped with a Yokogawa CSU-X1 spinning-disc confocal head (Tokyo, Japan), an emission filter wheel, and a Coolsnap HQ II digital camera and 488-nm, 561-nm laser lines, driven by the Metamorph imaging software program, version 7.7.11.0 (Universal Imaging). All images were assembled using the program Adobe Illustrator CS4, version 14.0.0 (Adobe).
For time-lapse microscopy, HeLa cells stably expressing Lifeact-GFP were seeded in glass-bottom dishes (MatTek, Ashland, MA) and were transfected with indicated siRNAs or left untreated. At 24 h posttransfection, cells were infected with C. trachomatis. At 25 h p.i., the cells were washed once with PBS, given fresh medium, and subjected to time-lapse microscopy at 37°C in a chamber with a humidified atmosphere (6.5% CO2 and 9% O2) for 4 h. Images were acquired every 5 min with the confocal microscope described above and assembled into a video using the Metamorph software.
Immunoblotting.
For lysis in radioimmunoprecipitation assay (RIPA) buffer, cells were washed once with cold PBS, trypsinized, and pelleted (500 × g, 5 min, 4°C). After a wash step with PBS, cells were resuspended in cold serum-free DMEM, in some cases containing 150 µM clasto-lactacystin β-lactone (Sigma), for 10 min at 4°C and centrifuged (500 × g, 5 min, 4°C). Pellets were resuspended in RIPA buffer (Sigma) supplemented with protease inhibitor (Complete; Roche) and Benzonase (25 U; Novagen). After a 30-min incubation on ice, cell debris was removed (17,000 × g, 10 min, 4°C), and supernatants were transferred to a fresh test tube, supplemented with Laemmli buffer, and heated to 99°C for 5 min. For lysis in guanidinium hydrochloride (Gua-HCl) buffer, cell processing was done as described above, but the lysis buffer contained 4 M Gua-HCl (Sigma) and 100 mM HEPES (Roth) dissolved in water, pH 7.4. Unlike the case with RIPA buffer-extracted samples, the clarified cell lysates obtained at the final step were not immediately supplemented with Laemmli buffer due to incompatibility with the Gua-HCl buffer. Protein solutions were precipitated using acetone (1:4, protein solution-acetone) overnight at −20°C. Following precipitation, samples were centrifuged (15,000 × g, 15 min, 4°C) and pellets were air-dried to eliminate residual acetone. For SDS-PAGE, samples were resuspended in Laemmli buffer and heated to 95°C for 5 min. For lysis in urea, cell culture plates were placed on ice, and cells were washed once with ice-cold PBS. Per 6-well plate, 400 µl of 8 M urea solution in water supplemented with 325 U/ml benzonase, in some cases with 150 µM clasto-lactacystin β-lactone, was added directly to cell monolayers. After a 10-min incubation on ice, cells were detached from the plate using a cell scraper and centrifuged (17,000 × g, 10 min, 4°C), and the supernatant was transferred to a fresh test tube, supplemented with Laemmli buffer and heated to 95°C for 5 min. Equal volumes of whole-cell lysates were loaded onto 10% SDS gels and transferred to nitrocellulose membranes. Membranes were incubated with primary antibody for 2 to 3 h at room temperature (RT), washed, and incubated with the secondary antibody for 1 h at RT. Detection was performed using the SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) or ECL prime detection reagent (GE Healthcare).
IP.
Uninfected or infected HeLa cells were lysed in ice-cold immunoprecipitation (IP) lysis buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100, and 1× protease inhibitor mix [Roche]) for 30 min and cleared of cell debris by centrifugation at 17,000 × g for 10 min at 4°C. Protein concentrations were determined using the Bradford protein assay. Four hundred micrograms of proteins from each sample were immunoprecipitated with 0.2 µg of anti-SEPT2 antibody preincubated with 25 µl of protein G-agarose beads (Roche) in the presence of IP washing buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, and 1× protease inhibitor mix) for 4 to 5 h at 4°C with rotation. Twenty micrograms of proteins from each sample (input fraction) were additionally collected for subsequent SDS-PAGE. The IP was done overnight at 4°C with rotation. The samples were then centrifuged at 1,500 × g for 1 min at 4°C, and 1/20 of each sample was collected (unbound fraction). The IPs were washed 3 times with ice-cold IP lysis buffer. Elution was done with 35 µl of 3× Laemmli SDS buffer at 95°C for 5 min (IP fraction). Input, unbound, and IP fractions were analyzed by SDS-PAGE followed by Western blotting.
Proteomic studies of the inclusion.
Infected and uninfected cells were collected and washed, and intact inclusions were isolated from the infected cells as previously described (45). Purity of inclusions (4 biological replicates) was verified by Western blotting studies, immunofluorescence microscopy, and electron microscopy. Tryptic digests of the samples were subjected to liquid chromatography (EASYnLC; Proxeon, Odense, Denmark), coupled online to an LTQ-Orbitrap mass spectrometer (Thermo, Fisher, Bremen, Germany). A concatenated Fasta database of C. trachomatis, a human database, and the software program Scaffold 4.3.2 (Proteome Software Inc., Portland, OR) were used for identification.
siRNA transfections and septin inhibitor treatment.
siRNA targeting human SEPT9 (5′ GCACGAUAUUGAGGAGAAA 3′) (46) or human nonmuscle myosin IIB (5′ AAGGAUCGCUACUAUUCAGGA 3′) (47) was purchased from Eurofins MWG Operon. A nontargeting siRNA sequence for human and mouse (5′ GCGCAUUCCAGCUUACGUA 3′) (48) (Metabion) was used as a control. Stealth RNAi siRNAs were purchased from Life Technologies: three different siRNAs targeting human SEPT9, no. 1, HSS173895 (5′ AGGCGUACCGUGUGAAGCGCCUCAA 3′); no. 2, HSS173896 (5′ aGGCGCCUGCAUCACGGAACGAGAA 3′); and no. 3, HSS173897 (5′ GCCAUGAAGCAGGGCUUCGAGUUCA 3′); and Stealth RNAi siRNA GAPDH positive control (human) was used as a control. Transfections were performed using Lipofectamine RNAiMax (Invitrogen), and knockdown efficiency was validated by Western blotting. HeLa cells were seeded 1 day before transfection. siRNA was added to a final concentration of 20 nM to 125 µl Opti-MEM medium (Gibco), mixed with 1.25 µl Lipofectamine RNAiMax (Invitrogen) diluted in 125 µl Opti-MEM, and reaction mixtures were incubated for 20 min at RT. Cells were washed with PBS and given fresh culture medium, and the complexed siRNA was added to a final volume of 1.5 ml. Transfected cells were infected 24 h posttransfection.
Forchlorfenuron (FCF) (Sigma) was used as an inhibitor of septins. Fifty micromolar FCF dissolved in dimethyl sulfoxide (DMSO) was added at either 18 h p.i. or 24 h p.i., and the analysis was done at 30 h p.i.
Generation of stable cell lines.
To construct the stable YFP-Golgi HeLa cell line, a fusion protein of enhanced yellow fluorescent protein (EYFP) and the N-terminal 81 amino acids of human beta-1,4-galactosyltransferase (the membrane-anchoring signal peptide that targets the fusion protein to the transmedial region of the Golgi apparatus [49]) was cloned into the lentiviral vector pFCMV-TO-GW (a kind gift from John Silke, Melbourne, Australia). HeLa cells were lentivirally transduced and selected with 1.5 µg/ml puromycin (Invivogen).
For the generation of the stable Lifeact-GFP-expressing HeLa cell line, Lifeact-GFP (29) was cloned into the lentiviral vector pFCMV-TO-GW, described above. HeLa cells were lentivirally transduced and selected with 3 µg/ml puromycin.
shRNA SEPT2 (5′-AAGGTGAATATTGTGCCTGTC-3‘) (50) and shRNA SEPT7 (5′ GGCAGTATCCTTGGGGTGT 3′) (51) (Metabion) were stably transduced into HeLa cells constitutively expressing a tetracycline repressor (HeLa-TetR) using a lentiviral-based expression system (52). In brief, shRNAs were cloned into the pENTR-THT “entry” vector, followed by recombination into a lentiviral RNAi “destination” vector, pHR-Dest-SFFV-eGFP, using the Gateway cloning system. Lentiviruses were produced by transient transfection of 293T cells with the lentiviral destination vector together with the packaging vectors psPAX2 und pMD2G. Viruses were harvested from the supernatant and used for infection of HeLa-TetR cells in the presence of 5 µg/ml polybrene (Millipore). Cells transduced with lentiviruses coding for a nonmammalian shRNA (5′ GTATCATCTCTTGAATGAT 3′) were used as a control.
Enumeration of extrusions.
Extrusions were isolated and counted as described previously (13). HeLa cells were seeded in 6-well plates and transfected with siRNA directed against SEPT9 alone or transfected simultaneously with siRNAs targeting SEPT2, -7, and -9, followed by infection with C. trachomatis about 4 h later. At 48 h p.i., cells were washed with PBS, given fresh culture medium, and incubated at 37° C with 5% CO2. At 66 h p.i., culture supernatants containing extrusions were collected and cleared of cell debris by centrifugation (100 × g, 5 min, RT). Supernatants were further centrifuged (100 × g, 10 min, RT) to enrich extrusions. The resulting pellets were resuspended in 1 ml culture medium supplemented with Hoechst (1:15,000) and Sytox green (1:2,000). After 5 min of incubation at RT, labeled extruded inclusions free of host cell nuclei were plated as 10-µl drops onto 8-well imaging chambers (PAA Laboratories) and counted using epifluorescence microscopy.
For enumeration of infectious bacteria released by extrusions, supernatants were collected as described above, enriched for extrusions by centrifugation, and subsequently resuspended in 1 ml culture medium and further serially diluted and plated on HeLa cell monolayers in 24-well plates, following published protocols (13). After 25 h of infection, cells were fixed, stained with anti-chlamydial Hsp60 and Evans blue (Santa Cruz), and counted using fluorescence microscopy.
For enumeration of total infectious bacteria produced, both supernatant and cell lysates were collected at 66 h p.i., cells were lysed with glass beads and centrifuged, and supernatants were serially diluted, plated on HeLa cell monolayers, and processed for microscopy as described above.
Inclusion area measurement.
HeLa cells carrying the inducible shSEPT2 construct were induced with 100 ng/ml AHT. At 24 h postinduction, cells were infected with C. trachomatis. At 24 h or 48 h p.i., cells were fixed in methanol and stained with mouse anti-cHsp60 antibody followed by anti-mouse Cy5. HeLa cells transfected with siRNA against SEPT9 were infected 24 h later with C. trachomatis, and at 48 h p.i., cells were fixed in methanol and stained with mouse anti-cHsp60 antibody followed by anti-mouse DyLight 488. The inclusion area was measured by epifluorescence microscopy using the Dynamic Cell Count function of the BZ-II Analyzer software program.
Statistical analysis.
Data are presented as means ± standard errors of the means (SEM) from independent experiments. Significant differences between means were determined by using Student’s t test for paired samples.
Terminal-amine isotopic labeling of substrates.
293-TRex-3xGyrB-CPAF cells were induced for 8 h with AHT/CM to express active CPAF. Induced and uninduced cells were lysed in 3 M Gua-HCl–100 mM HEPES (pH 7.4) buffer, followed by acetone-methanol precipitation. Infected and noninfected HeLa cells were lysed in 4 M Gua-HCl–100 mM HEPES (pH 7.4) and further processed the same way. The TAILS procedure and mass spectrometry analysis were conducted as described previously (53).
SUPPLEMENTAL MATERIAL
Time-lapse confocal microscopy of Lifeact-GFP-HeLa cells infected with C. trachomatis. HeLa cells stably expressing Lifeact-GFP were either left untreated (A), transfected with nontargeting siRNA (B), or transfected with siRNA targeting SEPT9 (C). At 24 h posttransfection, cells were infected with C. trachomatis L2. At 25 h p.i., the cells were subjected to time-lapse microscopy for 4 h. Images were acquired every 5 min. Scale bar, 5 µm. The videos are representative of 2 independent experiments. Download
SEPT2 cleavage by CPAF and effect of CPAF inhibition changes. (A) CPAF was induced and activated by addition of AHT/CM (5 ng/ml, 1 µM) in T-REx-293-3xgyrB-CPAF cells (16) for 8 h. (B) HeLa cells were infected for 30 h with C. trachomatis and harvested. Prior to lysis, aliquots were treated with the CPAF inhibitor clasto-lactacystin (150 µM) for 10 min. Cells were lysed in RIPA buffer, 4 M guanidinium hydrochloride–100 mM HEPES (Gua-HCl), or 8 M urea in water. Immunoblot analysis of whole-cell lysates with antibodies against SEPT2, vimentin, and GAPDH is shown. Arrows indicate CPAF-generated protein fragments. *, unspecific band. Blots are representative of at least 3 independent experiments. These results demonstrate that SEPT2 is cleaved in a CPAF-dependent way, and the appearance of the same fragment during infection indicates that this cleavage occurs during infection. It has to be noted that there was substantially less SEPT2 cleavage when cells were extracted using buffers containing chaotropic salts (either 4 M guanidinium-hydrochloride [Gua-HCl] or 8 M urea). Under these conditions, SEPT2 cleavage was undetectable by Western blotting but the SEPT2 fragments were still identified by TAILS, which was done using 4 M Gua-HCl-buffer. It is impossible to know how much CPAF-dependent cleavage indeed occurs in vivo and how much during extraction. Further CPAF-dependent cleavage events were of SEPT6 (400), SEPT7 (21 and 403), and SEPT8 (331). The septin-structure observed is very likely the arrangement of octamers into higher order fibrils. Crystal structures of individual septins (33) show the complex arrangement of a number of alpha-helices that are stabilized into dimers by GTP. The precise consequences of cleavage are not known. It is possible that cleaving off a small portion will facilitate arrangement into oligomers or higher-order structures. It is also possible that it will destabilize the arrangement of any septin, destabilizing in turn the octamer and then the higher-order structures. The identified CPAF cleavage sites were indeed toward either the N or C terminus (position 67 in SEPT2, 400 in SEPT6 [of a total of about 430 amino acids, depending on isoform], or 403 [of a total of 418 amino acids] in SEPT7). This may be a trimming of the end, which could either enhance or reduce binding to other septins. (C) Infected HeLa cells were treated with the CPAF inhibitor WEHD-fmk (75 µM) at 9 h p.i. or were left untreated. At 48 h p.i., cells were fixed and doubly stained for SEPT2 (red) and chlamydial Hsp60 to detect bacteria (green). Scale bar, 10 µm. Images are representative of 2 independent experiments. Download
SEPT2 and SEPT9 filaments are distributed around the chlamydial inclusion. (A) Confocal microscopy image of Chlamydia-infected HeLa cells fixed at 24 h p.i. and costained for SEPT2 (green) and α-tubulin (red) show no significant colocalization of the two cytoskeletal structures. Images are representative of 2 independent experiments. (B and C) HeLa cells infected for 30 h were processed for immunofluorescence, stained with antiseptin antibodies, and analyzed by confocal microscopy. A cross-section of an inclusion showing the SEPT2 (B) or SEPT9 (C) structure is shown. The images as well as reconstructed xz and yz views show the septin cage around the inclusion. The intersection of the red lines indicates the position within the cell where the confocal image was taken. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. Download
Effect of FCF on chlamydial development and SEPT2 cage formation. Infected HeLa cells were treated or not with 50 µM FCF at either 18 h p.i. or 24 h p.i., fixed at 30 h p.i., and stained with antibodies against SEPT2. Note that addition of FCF at 18 h p.i. inhibits normal growth of the inclusion but does not seem to inhibit SEPT2 cage formation. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of 2 independent experiments. Download
Knockdown efficacy of septin-directed RNAi. Immunoblot analysis of whole-cell lysates of HeLa cells transfected with siRNA or expressing shRNA directed against septins. At each time point, cells were lysed in RIPA buffer, and proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and probed with antibodies against SEPT2, -7, or -9, GAPDH, or actin. (A) HeLa cells were transfected with siRNA directed against SEPT9. The blot is representative of 2 independent experiments. (B) HeLa cells were transfected with either three different siRNAs targeting SEPT9 (commercially available and different from the one used in panel A; “stealth” siRNAs; siSeptin 9 number 1, number 2, and number 3), with siRNA targeting GAPDH (siGAPDH) or with a negative control for 24, 48, and 72 h. (C) HeLa cell lines carrying a tetracycline-inducible construct consisting of an shRNA against SEPT2 or -7 (or a control shRNA) and a GFP expression cassette were induced using 100 ng/ml anhydrotetracyclin (AHT) for 48 h. In parallel, HeLa cells were transfected with siRNA against SEPT9 (same siRNA as in panel A). Cells were infected with C. trachomatis at 24 h postinduction/posttransfection, harvested at 30 h postinfection, and lysed in RIPA buffer. Blots were probed with antibodies against SEPT2, -7, or -9 or GFP to confirm shRNA expression. GAPDH served as a loading control. Actin levels were further assessed because of the effects of septin knockdowns on F-actin fibers. Blots are representative of 4 independent experiments. (D) HeLa cells were transfected simultaneously with siRNAs targeting SEPT2, -7, and -9 for 48 h or left untreated. At 24 h posttransfection, cells were infected or not, and at 30 h p.i., cells were lysed in RIPA buffer. Blots were probed with antibodies against SEPT2, -7, and -9. GAPDH was used as a loading control. Download
Intact growth of inclusion and generation of infectious bacteria and intact Golgi fragmentation upon knockdown of septins. (A) (Top panel) HeLa cells carrying the inducible shSEPT2-construct (see Fig. S4C) were induced with 100 ng/ml AHT for 72 h in total. At 24 h postinduction, cells were infected with C. trachomatis. At 24 h or 48 h p.i., cells were processed for immunofluorescence and the inclusion area was measured by epifluorescence microscopy. At least 1,000 inclusions from each condition were analyzed. Results are expressed as means/SEM from 3 independent experiments. (Bottom panel) HeLa cells were transfected with siRNA against SEPT9 (see Fig. S4A) and were infected 24 h later with C. trachomatis. At 48 h p.i., cells were fixed and processed as described above. At least 800 inclusions from each condition were analyzed. Results are expressed as means/SEM from 3 independent experiments. (B) HeLa cells were transfected with siRNA targeting SEPT9, followed by infection with C. trachomatis about 4 h later. Fresh medium was given to the cells at 48 h p.i., and at 66 h p.i., both supernatant and cell lysates were combined. Numbers of total infectious bacteria were determined by titration on a fresh monolayer of HeLa cells, which were fixed 25 h later. Inclusions were counted using fluorescence microscopy. The numbers of IFU/ml of the siControl samples were set to 1. Fold change for siSEPT9 relative to siControl was calculated. Data represent means/SEM from 3 independent experiments; P = 0.36. n.s., nonsignificant. (C) HeLa cells stably expressing a construct targeting EYFP to the Golgi apparatus were transfected with siRNA targeting SEPT9, with the combination of siRNAs directed against SEPT2, -7, and -9 (see Fig. S4A and C) or with a nontargeting siRNA as a control. At 24 h posttransfection, cells were infected with C. trachomatis. At 48 h p.i., cells were fixed and stained with anti-SEPT9 antibody (red). Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of 2 independent experiments. Download
Knockdown of SEPT9 causes reduction of actin fibers on the inclusion, but septin knockdown causes no obvious alterations to microtubules or vimentin structures around the inclusion. Disruption of the microtubule network has no effect on septin cages. In HeLa cells (A, C, D, and E) or Lifeact-GFP-HeLa cells (B), various septins were knocked down as indicated (A, SEPT9 using “stealth” siRNA; B, C, and E, SEPT9-specific siRNA [same cells as in Fig. S4A] or SEPT2-, -7-, and -9-specific siRNA [simultaneous transfection]; SEPT2 was knocked down by induction of shRNA [as in Fig. S4C]). Transfection of siRNA and induction of shRNA were done 24 h prior to infection. At 30 h p.i., cells were fixed and stained (A, C, D, and E), or at 25 h p.i., cells were subjected to live cell time-lapse microscopy (B) (see Movie S1). (A) Cells were stained with phalloidin-Alexa 546 to detect F actin and Hoechst dye. (B) Still images of the cells from Movie S1. (Top panel) Untreated cell; (middle panel) cell transfected with nontargeting siRNA; (bottom panel) cell transfected with siRNA targeting SEPT9. Arrows indicate the inclusions (yellow), actin recruitment to the inclusion (red), or reduced actin recruitment to the inclusion (white). Scale bar, 5 µm. (C) Cells were stained with antibodies against α-tubulin and Hoechst dye. (D) Some aliquots were pretreated with nocodazole (20 µM) for 30 min prior to fixation. Cells were costained for α-tubulin and for either SEPT2 or SEPT9. (E) Cells were stained with antibodies against vimentin and Hoechst dye. (A, C, D, and E) Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of 3 independent experiments. (B) Scale bar, 5 µm. Images are representative of 2 independent experiments. Download
Src microdomains are recruited to the inclusion independently of septins and show no obvious spatial alignment with SEPT9-fibers. HeLa cells were transfected either with siRNA targeting SEPT9, SEPT2, -7, and -9 simultaneously or with a nontargeting siRNA. At 24 h posttransfection, cells were infected with C. trachomatis, fixed at 30 h p.i. (A), or fixed at 48 h p.i. (B and C) and costained with anti-phospho-Tyr419-Src (pTyr-Src) and with either anti-SEPT9 (A and B) or phalloidin-Alexa 546 to detect F actin (C). Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of either 2 independent experiments (A) or 3 independent experiments (B and C). Very similar results were also obtained at 40 h p.i. (not shown). Download
Knockdown of myosin IIB causes no obvious alterations in septin structures around the inclusion, while blebbistatin has a minor effect. (A) Immunoblot analysis showing knockdown efficacy of nonmuscle myosin IIB-directed RNAi after transfection of HeLa cells for 48 h and 72 h. Cells were lysed in RIPA buffer, and proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and probed with antibodies against myosin IIB or GAPDH. (B) HeLa cells were transfected with siRNA directed against myosin IIB for 48 h. At 24 h posttransfection, cells were infected with C. trachomatis, fixed at 30 h postinfection, and labeled with antibodies against SEPT2 (top panel) or against SEPT9 (bottom panel) and Hoechst dye. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. (C) HeLa cells were infected with C. trachomatis and at 26 h p.i. were treated or not with the myosin II inhibitor blebbistatin (34 µM), fixed at 30 h p.i., and costained with antibodies against myosin IIB and either SEPT2 (top) or SEPT9 (bottom). DNA was stained with Hoechst dye. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. All microscopy images are representative of 2 independent experiments. Download
Myosin IIB is recruited to the inclusion independently of septins. HeLa cells were transfected with siRNA targeting either SEPT9, SEPT2, -7, and -9 simultaneously or with a nontargeting siRNA. At 24 h posttransfection, cells were infected with C. trachomatis, fixed at 30 h p.i., and labeled with an antibody against myosin IIB. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of 2 independent experiments. Download
ACKNOWLEDGMENTS
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG) (http://www.dfg.de) to G.H. (priority program SPP1580). O.S. is supported by the DFG (SCHI 871/2 and SCHI871/5-1), a starting grant of the European Research Council (Programme “Ideas” call identifier ERC-2011-StG 282111-ProteaSys), and the Excellence Initiative of the German Federal and State Governments (DFG) (EXC 294, BIOSS). The proteomic study of the inclusion was supported by a grant to T.R. from the German Ministry for Education and Research (BMBF) (http://www.bmbf.de/; Medizinische Infektionsgenomik BMBF 0315834 A).
Franz Jehle and Bettina Mayer are acknowledged for technical assistance.
Footnotes
Citation Volceanov L, Herbst K, Biniossek M, Schilling O, Haller D, Nölke T, Subbarayal P, Rudel T, Zieger B, Häcker G. 2014. Septins arrange F-actin-containing fibers on the Chlamydia trachomatis inclusion and are required for normal release of the inclusion by extrusion. mBio 5(5):e01802-14. doi:10.1128/mBio.01802-14.
REFERENCES
- 1. Burton MJ, Mabey DC. 2009. The global burden of trachoma: a review. PLoS Negl. Trop. Dis. 3:e460. 10.1371/journal.pntd.0000460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stamm WE, Batteiger BE. 2010. Chlamydia trachomatis (trachoma, perinatal infections, lymphogranuloma venereum, and other genital infections), p 2443–2461 In Mandell GE, Principles and practice of infectious diseases, vol 2 Churchill: Linvingstone Elsevier, Philadelphia, PA [Google Scholar]
- 3. Moulder JW. 1991. Interaction of chlamydiae and host cells in vitro. Microbiol. Rev. 55:143–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hackstadt T, Fischer ER, Scidmore MA, Rockey DD, Heinzen RA. 1997. Origins and functions of the chlamydial inclusion. Trends Microbiol. 5:288–293. 10.1016/S0966-842X(97)01061-5 [DOI] [PubMed] [Google Scholar]
- 5. Cossart P. 2011. Illuminating the landscape of host-pathogen interactions with the bacterium Listeria monocytogenes. Proc. Natl. Acad. Sci. U. S. A. 108:19484–19491. 10.1073/pnas.1112371108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jewett TJ, Fischer ER, Mead DJ, Hackstadt T. 2006. Chlamydial TARP is a bacterial nucleator of actin. Proc. Natl. Acad. Sci. U. S. A. 103:15599–15604. 10.1073/pnas.0603044103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Campbell S, Richmond SJ, Yates PS. 1989. The effect of Chlamydia trachomatis infection on the host cell cytoskeleton and membrane compartments. J. Gen. Microbiol. 135:2379–2386 [DOI] [PubMed] [Google Scholar]
- 8. Kumar Y, Valdivia RH. 2008. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe 4:159–169. 10.1016/j.chom.2008.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grieshaber SS, Grieshaber NA, Hackstadt T. 2003. Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J. Cell Sci. 116:3793–3802. 10.1242/jcs.00695 [DOI] [PubMed] [Google Scholar]
- 10. Todd WJ, Caldwell HD. 1985. The interaction of Chlamydia trachomatis with host cells: ultrastructural studies of the mechanism of release of a biovar II strain from HeLa 229 cells. J. Infect. Dis. 151:1037–1044. 10.1093/infdis/151.6.1037 [DOI] [PubMed] [Google Scholar]
- 11. Hybiske K, Stephens RS. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. U. S. A. 104:11430–11435. 10.1073/pnas.0703218104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lutter EI, Barger AC, Nair V, Hackstadt T. 2013. Chlamydia trachomatis inclusion membrane protein CT228 recruits elements of the myosin phosphatase pathway to regulate release mechanisms. Cell Rep. 3:1921–1931. 10.1016/j.celrep.2013.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chin E, Kirker K, Zuck M, James G, Hybiske K. 2012. Actin recruitment to the Chlamydia inclusion is spatiotemporally regulated by a mechanism that requires host and bacterial factors. PLoS One 7:e46949. 10.1371/journal.pone.0046949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mostowy S, Cossart P. 2012. Septins: the fourth component of the cytoskeleton. Nat. Rev. Mol. Cell. Biol. 13:183–194. 10.1038/nrm3284 [DOI] [PubMed] [Google Scholar]
- 15. Bridges AA, Zhang H, Mehta SB, Occhipinti P, Tani T, Gladfelter AS. 2014. Septin assemblies form by diffusion-driven annealing on membranes. Proc. Natl. Acad. Sci. U. S. A. 111:2146–2151. 10.1073/pnas.1314138111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paschen SA, Christian JG, Vier J, Schmidt F, Walch A, Ojcius DM, Häcker G. 2008. Cytopathicity of Chlamydia is largely reproduced by expression of a single chlamydial protease. J. Cell Biol. 182:117–127. 10.1083/jcb.200804023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kleifeld O, Doucet A, auf dem Keller U, Prudova A, Schilling O, Kainthan RK, Starr AE, Foster LJ, Kizhakkedathu JN, Overall CM. 2010. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol. 28:281–288. 10.1038/nbt.1611 [DOI] [PubMed] [Google Scholar]
- 18. Bauler LD, Hackstadt T. 2014. Expression and targeting of secreted proteins from Chlamydia trachomatis. J. Bacteriol. 196:1325–1334. 10.1128/JB.01290-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen AL, Johnson KA, Lee JK, Sütterlin C, Tan M. 2012. CPAF: a chlamydial protease in search of an authentic substrate. PLoS Pathog. 8:e1002842. 10.1371/journal.ppat.1002842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Christian JG, Heymann J, Paschen SA, Vier J, Schauenburg L, Rupp J, Meyer TF, Häcker G, Heuer D. 2011. Targeting of a chlamydial protease impedes intracellular bacterial growth. PLoS Pathog. 7:e1002283. 10.1371/journal.ppat.1002283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hacker G. 2014. The chlamydial protease CPAF: important or not, important for what? Microbes Infect. 16:367–370. 10.1016/j.micinf.2014.02.008 [DOI] [PubMed] [Google Scholar]
- 22. Sandrock K, Bartsch I, Bläser S, Busse A, Busse E, Zieger B. 2011. Characterization of human septin interactions. Biol. Chem. 392:751–761. 10.1515/BC.2011.081 [DOI] [PubMed] [Google Scholar]
- 23. Xie Y, Vessey JP, Konecna A, Dahm R, Macchi P, Kiebler MA. 2007. The GTP-binding protein Septin 7 is critical for dendrite branching and dendritic-spine morphology. Curr. Biol. 17:1746–1751. 10.1016/j.cub.2007.08.042 [DOI] [PubMed] [Google Scholar]
- 24. Kinoshita M, Field CM, Coughlin ML, Straight AF, Mitchison TJ. 2002. Self- and actin-templated assembly of Mammalian septins. Dev. Cell 3:791–802. 10.1016/S1534-5807(02)00366-0 [DOI] [PubMed] [Google Scholar]
- 25. Ricci A, Bertoletti C. 2009. Urea derivatives on the move: cytokinin-like activity and adventitious rooting enhancement depend on chemical structure. Plant Biol. (Stuttg.) 11:262–272. 10.1111/j.1438-8677.2008.00165.x [DOI] [PubMed] [Google Scholar]
- 26. Hu Q, Nelson WJ, Spiliotis ET. 2008. Forchlorfenuron alters mammalian septin assembly, organization, and dynamics. J. Biol. Chem. 283:29563–29571. 10.1074/jbc.M804962200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spiliotis ET, Hunt SJ, Hu Q, Kinoshita M, Nelson WJ. 2008. Epithelial polarity requires septin coupling of vesicle transport to polyglutamylated microtubules. J. Cell Biol. 180:295–303. 10.1083/jcb.200710039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heuer D, Relman Lipinski A, Machuy N, Karlas A, Wehrens A, Siedler F, Brinkmann V, Meyer TF. 2009. Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature 457:731–735. 10.1038/nature07578 [DOI] [PubMed] [Google Scholar]
- 29. Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. 2008. Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5:605–607. 10.1038/nmeth.1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mital J, Miller NJ, Fischer ER, Hackstadt T. 2010. Specific chlamydial inclusion membrane proteins associate with active Src family kinases in microdomains that interact with the host microtubule network. Cell. Microbiol. 12:1235–1249. 10.1111/j.1462-5822.2010.01465.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bartsch I, Sandrock K, Lanza F, Nurden P, Hainmann I, Pavlova A, Greinacher A, Tacke U, Barth M, Busse A, Oldenburg J, Bommer M, Strahm B, Superti-Furga A, Zieger B. 2011. Deletion of human GP1BB and SEPT5 is associated with Bernard-Soulier syndrome, platelet secretion defect, polymicrogyria, and developmental delay. Thromb. Haemost. 106:475–483. 10.1160/TH11-05-0305 [DOI] [PubMed] [Google Scholar]
- 32. Amin ND, Zheng YL, Kesavapany S, Kanungo J, Guszczynski T, Sihag RK, Rudrabhatla P, Albers W, Grant P, Pant HC. 2008. Cyclin-dependent kinase 5 phosphorylation of human septin SEPT5 (hCDCrel-1) modulates exocytosis. J. Neurosci. 28:3631–3643. 10.1523/JNEUROSCI.0453-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sirajuddin M, Farkasovsky M, Hauer F, Kühlmann D, Macara IG, Weyand M, Stark H, Wittinghofer A. 2007. Structural insight into filament formation by mammalian septins. Nature 449:311–315. 10.1038/nature06052 [DOI] [PubMed] [Google Scholar]
- 34. Nagata K, Inagaki M. 2005. Cytoskeletal modification of Rho guanine nucleotide exchange factor activity: identification of a Rho guanine nucleotide exchange factor as a binding partner for Sept9b, a mammalian septin. Oncogene 24:65–76. 10.1038/sj.onc.1208101 [DOI] [PubMed] [Google Scholar]
- 35. Joo E, Surka MC, Trimble WS. 2007. Mammalian SEPT2 is required for scaffolding nonmuscle myosin II and its kinases. Dev. Cell 13:677–690. 10.1016/j.devcel.2007.09.001 [DOI] [PubMed] [Google Scholar]
- 36. Finger FP. 2002. One ring to bind them. Septins and actin assembly. Dev. Cell 3:761–763. 10.1016/S1534-5807(02)00371-4 [DOI] [PubMed] [Google Scholar]
- 37. Sun HS, Wilde A, Harrison RE. 2011. Chlamydia trachomatis inclusions induce asymmetric cleavage furrow formation and ingression failure in host cells. Mol. Cell. Biol. 31:5011–5022. 10.1128/MCB.05734-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang J, Kong C, Xie H, McPherson PS, Grinstein S, Trimble WS. 1999. Phosphatidylinositol polyphosphate binding to the mammalian septin H5 is modulated by GTP. Curr. Biol. 9:1458–1467. 10.1016/S0960-9822(00)80115-3 [DOI] [PubMed] [Google Scholar]
- 39. Bertin A, McMurray MA, Thai L, Garcia G, III, Votin V, Grob P, Allyn T, Thorner J, Nogales E. 2010. Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. J. Mol. Biol. 404:711–731. 10.1016/j.jmb.2010.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moorhead AM, Jung JY, Smirnov A, Kaufer S, Scidmore MA. 2010. Multiple host proteins that function in phosphatidylinositol-4-phosphate metabolism are recruited to the chlamydial inclusion. Infect. Immun. 78:1990–2007. 10.1128/IAI.01340-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Balañá ME, Niedergang F, Subtil A, Alcover A, Chavrier P, Dautry-Varsat A. 2005. ARF6 GTPase controls bacterial invasion by actin remodelling. J. Cell Sci. 118:2201–2210. 10.1242/jcs.02351 [DOI] [PubMed] [Google Scholar]
- 42. Mostowy S, Bonazzi M, Hamon MA, Tham TN, Mallet A, Lelek M, Gouin E, Demangel C, Brosch R, Zimmer C, Sartori A, Kinoshita M, Lecuit M, Cossart P. 2010. Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe 8:433–444. 10.1016/j.chom.2010.10.009 [DOI] [PubMed] [Google Scholar]
- 43. Pizarro-Cerdá J, Jonquières R, Gouin E, Vandekerckhove J, Garin J, Cossart P. 2002. Distinct protein patterns associated with Listeria monocytogenes InlA- or InlB-phagosomes. Cell. Microbiol. 4:101–115. 10.1046/j.1462-5822.2002.00169.x [DOI] [PubMed] [Google Scholar]
- 44. Hoffmann C, Finsel I, Otto A, Pfaffinger G, Rothmeier E, Hecker M, Becher D, Hilbi H. 2014. Functional analysis of novel Rab GTPases identified in the proteome of purified Legionella-containing vacuoles from macrophages. Cell. Microbiol. 16:1034–1052. 10.1111/cmi.12256 [DOI] [PubMed] [Google Scholar]
- 45. Matsumoto A. 1981. Isolation and electron microscopic observations of intracytoplasmic inclusions containing Chlamydia psittaci. J. Bacteriol. 145:605–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Estey MP, Di Ciano-Oliveira C, Froese CD, Bejide MT, Trimble WS. 2010. Distinct roles of septins in cytokinesis: SEPT9 mediates midbody abscission. J. Cell Biol. 191:741–749. 10.1083/jcb.201006031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bao J, Jana SS, Adelstein RS. 2005. Vertebrate nonmuscle myosin II isoforms rescue small interfering RNA-induced defects in COS-7 cell cytokinesis. J. Biol. Chem. 280:19594–19599. 10.1074/jbc.M501573200 [DOI] [PubMed] [Google Scholar]
- 48. Zall H, Weber A, Besch R, Zantl N, Häcker G. 2010. Chemotherapeutic drugs sensitize human renal cell carcinoma cells to ABT-737 by a mechanism involving the Noxa-dependent inactivation of Mcl-1 or A1. Mol. Cancer 9:164. 10.1186/1476-4598-9-164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Watzele G, Berger EG. 1990. Near identity of HeLa cell galactosyltransferase with the human placental enzyme. Nucleic Acids Res. 18:7174. 10.1093/nar/18.23.7174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Spiliotis ET, Kinoshita M, Nelson WJ. 2005. A mitotic septin scaffold required for mammalian chromosome congression and segregation. Science 307:1781–1785. 10.1126/science.1106823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kremer BE, Haystead T, Macara IG. 2005. Mammalian septins regulate microtubule stability through interaction with the microtubule-binding protein MAP4. Mol. Biol. Cell 16:4648–4659. 10.1091/mbc.E05-03-0267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ploner C, Rainer J, Niederegger H, Eduardoff M, Villunger A, Geley S, Kofler R. 2008. The BCL2 rheostat in glucocorticoid-induced apoptosis of acute lymphoblastic leukemia. Leukemia 22:370–377. 10.1038/sj.leu.2405039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shahinian H, Loessner D, Biniossek ML, Kizhakkedathu JN, Clements JA, Magdolen V, Schilling O. 2014. Secretome and degradome profiling shows that Kallikrein-related peptidases 4, 5, 6, and 7 induce TGFbeta-1 signaling in ovarian cancer cells. Mol. Oncol. 8:68–82. 10.1016/j.molonc.2013.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse confocal microscopy of Lifeact-GFP-HeLa cells infected with C. trachomatis. HeLa cells stably expressing Lifeact-GFP were either left untreated (A), transfected with nontargeting siRNA (B), or transfected with siRNA targeting SEPT9 (C). At 24 h posttransfection, cells were infected with C. trachomatis L2. At 25 h p.i., the cells were subjected to time-lapse microscopy for 4 h. Images were acquired every 5 min. Scale bar, 5 µm. The videos are representative of 2 independent experiments. Download
SEPT2 cleavage by CPAF and effect of CPAF inhibition changes. (A) CPAF was induced and activated by addition of AHT/CM (5 ng/ml, 1 µM) in T-REx-293-3xgyrB-CPAF cells (16) for 8 h. (B) HeLa cells were infected for 30 h with C. trachomatis and harvested. Prior to lysis, aliquots were treated with the CPAF inhibitor clasto-lactacystin (150 µM) for 10 min. Cells were lysed in RIPA buffer, 4 M guanidinium hydrochloride–100 mM HEPES (Gua-HCl), or 8 M urea in water. Immunoblot analysis of whole-cell lysates with antibodies against SEPT2, vimentin, and GAPDH is shown. Arrows indicate CPAF-generated protein fragments. *, unspecific band. Blots are representative of at least 3 independent experiments. These results demonstrate that SEPT2 is cleaved in a CPAF-dependent way, and the appearance of the same fragment during infection indicates that this cleavage occurs during infection. It has to be noted that there was substantially less SEPT2 cleavage when cells were extracted using buffers containing chaotropic salts (either 4 M guanidinium-hydrochloride [Gua-HCl] or 8 M urea). Under these conditions, SEPT2 cleavage was undetectable by Western blotting but the SEPT2 fragments were still identified by TAILS, which was done using 4 M Gua-HCl-buffer. It is impossible to know how much CPAF-dependent cleavage indeed occurs in vivo and how much during extraction. Further CPAF-dependent cleavage events were of SEPT6 (400), SEPT7 (21 and 403), and SEPT8 (331). The septin-structure observed is very likely the arrangement of octamers into higher order fibrils. Crystal structures of individual septins (33) show the complex arrangement of a number of alpha-helices that are stabilized into dimers by GTP. The precise consequences of cleavage are not known. It is possible that cleaving off a small portion will facilitate arrangement into oligomers or higher-order structures. It is also possible that it will destabilize the arrangement of any septin, destabilizing in turn the octamer and then the higher-order structures. The identified CPAF cleavage sites were indeed toward either the N or C terminus (position 67 in SEPT2, 400 in SEPT6 [of a total of about 430 amino acids, depending on isoform], or 403 [of a total of 418 amino acids] in SEPT7). This may be a trimming of the end, which could either enhance or reduce binding to other septins. (C) Infected HeLa cells were treated with the CPAF inhibitor WEHD-fmk (75 µM) at 9 h p.i. or were left untreated. At 48 h p.i., cells were fixed and doubly stained for SEPT2 (red) and chlamydial Hsp60 to detect bacteria (green). Scale bar, 10 µm. Images are representative of 2 independent experiments. Download
SEPT2 and SEPT9 filaments are distributed around the chlamydial inclusion. (A) Confocal microscopy image of Chlamydia-infected HeLa cells fixed at 24 h p.i. and costained for SEPT2 (green) and α-tubulin (red) show no significant colocalization of the two cytoskeletal structures. Images are representative of 2 independent experiments. (B and C) HeLa cells infected for 30 h were processed for immunofluorescence, stained with antiseptin antibodies, and analyzed by confocal microscopy. A cross-section of an inclusion showing the SEPT2 (B) or SEPT9 (C) structure is shown. The images as well as reconstructed xz and yz views show the septin cage around the inclusion. The intersection of the red lines indicates the position within the cell where the confocal image was taken. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. Download
Effect of FCF on chlamydial development and SEPT2 cage formation. Infected HeLa cells were treated or not with 50 µM FCF at either 18 h p.i. or 24 h p.i., fixed at 30 h p.i., and stained with antibodies against SEPT2. Note that addition of FCF at 18 h p.i. inhibits normal growth of the inclusion but does not seem to inhibit SEPT2 cage formation. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of 2 independent experiments. Download
Knockdown efficacy of septin-directed RNAi. Immunoblot analysis of whole-cell lysates of HeLa cells transfected with siRNA or expressing shRNA directed against septins. At each time point, cells were lysed in RIPA buffer, and proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and probed with antibodies against SEPT2, -7, or -9, GAPDH, or actin. (A) HeLa cells were transfected with siRNA directed against SEPT9. The blot is representative of 2 independent experiments. (B) HeLa cells were transfected with either three different siRNAs targeting SEPT9 (commercially available and different from the one used in panel A; “stealth” siRNAs; siSeptin 9 number 1, number 2, and number 3), with siRNA targeting GAPDH (siGAPDH) or with a negative control for 24, 48, and 72 h. (C) HeLa cell lines carrying a tetracycline-inducible construct consisting of an shRNA against SEPT2 or -7 (or a control shRNA) and a GFP expression cassette were induced using 100 ng/ml anhydrotetracyclin (AHT) for 48 h. In parallel, HeLa cells were transfected with siRNA against SEPT9 (same siRNA as in panel A). Cells were infected with C. trachomatis at 24 h postinduction/posttransfection, harvested at 30 h postinfection, and lysed in RIPA buffer. Blots were probed with antibodies against SEPT2, -7, or -9 or GFP to confirm shRNA expression. GAPDH served as a loading control. Actin levels were further assessed because of the effects of septin knockdowns on F-actin fibers. Blots are representative of 4 independent experiments. (D) HeLa cells were transfected simultaneously with siRNAs targeting SEPT2, -7, and -9 for 48 h or left untreated. At 24 h posttransfection, cells were infected or not, and at 30 h p.i., cells were lysed in RIPA buffer. Blots were probed with antibodies against SEPT2, -7, and -9. GAPDH was used as a loading control. Download
Intact growth of inclusion and generation of infectious bacteria and intact Golgi fragmentation upon knockdown of septins. (A) (Top panel) HeLa cells carrying the inducible shSEPT2-construct (see Fig. S4C) were induced with 100 ng/ml AHT for 72 h in total. At 24 h postinduction, cells were infected with C. trachomatis. At 24 h or 48 h p.i., cells were processed for immunofluorescence and the inclusion area was measured by epifluorescence microscopy. At least 1,000 inclusions from each condition were analyzed. Results are expressed as means/SEM from 3 independent experiments. (Bottom panel) HeLa cells were transfected with siRNA against SEPT9 (see Fig. S4A) and were infected 24 h later with C. trachomatis. At 48 h p.i., cells were fixed and processed as described above. At least 800 inclusions from each condition were analyzed. Results are expressed as means/SEM from 3 independent experiments. (B) HeLa cells were transfected with siRNA targeting SEPT9, followed by infection with C. trachomatis about 4 h later. Fresh medium was given to the cells at 48 h p.i., and at 66 h p.i., both supernatant and cell lysates were combined. Numbers of total infectious bacteria were determined by titration on a fresh monolayer of HeLa cells, which were fixed 25 h later. Inclusions were counted using fluorescence microscopy. The numbers of IFU/ml of the siControl samples were set to 1. Fold change for siSEPT9 relative to siControl was calculated. Data represent means/SEM from 3 independent experiments; P = 0.36. n.s., nonsignificant. (C) HeLa cells stably expressing a construct targeting EYFP to the Golgi apparatus were transfected with siRNA targeting SEPT9, with the combination of siRNAs directed against SEPT2, -7, and -9 (see Fig. S4A and C) or with a nontargeting siRNA as a control. At 24 h posttransfection, cells were infected with C. trachomatis. At 48 h p.i., cells were fixed and stained with anti-SEPT9 antibody (red). Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of 2 independent experiments. Download
Knockdown of SEPT9 causes reduction of actin fibers on the inclusion, but septin knockdown causes no obvious alterations to microtubules or vimentin structures around the inclusion. Disruption of the microtubule network has no effect on septin cages. In HeLa cells (A, C, D, and E) or Lifeact-GFP-HeLa cells (B), various septins were knocked down as indicated (A, SEPT9 using “stealth” siRNA; B, C, and E, SEPT9-specific siRNA [same cells as in Fig. S4A] or SEPT2-, -7-, and -9-specific siRNA [simultaneous transfection]; SEPT2 was knocked down by induction of shRNA [as in Fig. S4C]). Transfection of siRNA and induction of shRNA were done 24 h prior to infection. At 30 h p.i., cells were fixed and stained (A, C, D, and E), or at 25 h p.i., cells were subjected to live cell time-lapse microscopy (B) (see Movie S1). (A) Cells were stained with phalloidin-Alexa 546 to detect F actin and Hoechst dye. (B) Still images of the cells from Movie S1. (Top panel) Untreated cell; (middle panel) cell transfected with nontargeting siRNA; (bottom panel) cell transfected with siRNA targeting SEPT9. Arrows indicate the inclusions (yellow), actin recruitment to the inclusion (red), or reduced actin recruitment to the inclusion (white). Scale bar, 5 µm. (C) Cells were stained with antibodies against α-tubulin and Hoechst dye. (D) Some aliquots were pretreated with nocodazole (20 µM) for 30 min prior to fixation. Cells were costained for α-tubulin and for either SEPT2 or SEPT9. (E) Cells were stained with antibodies against vimentin and Hoechst dye. (A, C, D, and E) Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of 3 independent experiments. (B) Scale bar, 5 µm. Images are representative of 2 independent experiments. Download
Src microdomains are recruited to the inclusion independently of septins and show no obvious spatial alignment with SEPT9-fibers. HeLa cells were transfected either with siRNA targeting SEPT9, SEPT2, -7, and -9 simultaneously or with a nontargeting siRNA. At 24 h posttransfection, cells were infected with C. trachomatis, fixed at 30 h p.i. (A), or fixed at 48 h p.i. (B and C) and costained with anti-phospho-Tyr419-Src (pTyr-Src) and with either anti-SEPT9 (A and B) or phalloidin-Alexa 546 to detect F actin (C). Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of either 2 independent experiments (A) or 3 independent experiments (B and C). Very similar results were also obtained at 40 h p.i. (not shown). Download
Knockdown of myosin IIB causes no obvious alterations in septin structures around the inclusion, while blebbistatin has a minor effect. (A) Immunoblot analysis showing knockdown efficacy of nonmuscle myosin IIB-directed RNAi after transfection of HeLa cells for 48 h and 72 h. Cells were lysed in RIPA buffer, and proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and probed with antibodies against myosin IIB or GAPDH. (B) HeLa cells were transfected with siRNA directed against myosin IIB for 48 h. At 24 h posttransfection, cells were infected with C. trachomatis, fixed at 30 h postinfection, and labeled with antibodies against SEPT2 (top panel) or against SEPT9 (bottom panel) and Hoechst dye. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. (C) HeLa cells were infected with C. trachomatis and at 26 h p.i. were treated or not with the myosin II inhibitor blebbistatin (34 µM), fixed at 30 h p.i., and costained with antibodies against myosin IIB and either SEPT2 (top) or SEPT9 (bottom). DNA was stained with Hoechst dye. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. All microscopy images are representative of 2 independent experiments. Download
Myosin IIB is recruited to the inclusion independently of septins. HeLa cells were transfected with siRNA targeting either SEPT9, SEPT2, -7, and -9 simultaneously or with a nontargeting siRNA. At 24 h posttransfection, cells were infected with C. trachomatis, fixed at 30 h p.i., and labeled with an antibody against myosin IIB. Inclusions are indicated by arrows. Cell nuclei are marked with asterisks. Scale bar, 10 µm. Images are representative of 2 independent experiments. Download