

ABSTRACT
Footrot is a contagious, debilitating disease of sheep, causing major economic losses in most sheep-producing countries. The causative agent is the Gram-negative anaerobe Dichelobacter nodosus. Depending on the virulence of the infective bacterial strain, clinical signs vary from a mild interdigital dermatitis (benign footrot) to severe underrunning of the horn of the hoof (virulent footrot). The aim of this study was to investigate the genetic relationship between D. nodosus strains of different phenotypic virulences and between isolates from different geographic regions. Genome sequencing was performed on 103 D. nodosus isolates from eight different countries. Comparison of these genome sequences revealed that they were highly conserved, with >95% sequence identity. However, single nucleotide polymorphism analysis of the 31,627 nucleotides that were found to differ in one or more of the 103 sequenced isolates divided them into two distinct clades. Remarkably, this division correlated with known virulent and benign phenotypes, as well as with the single amino acid difference between the AprV2 and AprB2 proteases, which are produced by virulent and benign strains, respectively. This division was irrespective of the geographic origin of the isolates. However, within one of these clades, isolates from different geographic regions generally belonged to separate clusters. In summary, we have shown that D. nodosus has a bimodal population structure that is globally conserved and provide evidence that virulent and benign isolates represent two distinct forms of D. nodosus strains. These data have the potential to improve the diagnosis and targeted control of this economically significant disease.
The Gram-negative anaerobic bacterium Dichelobacter nodosus is the causative agent of ovine footrot, a disease of major importance to the worldwide sheep industry. The known D. nodosus virulence factors are its type IV fimbriae and extracellular serine proteases. D. nodosus strains are designated virulent or benign based on the type of disease caused under optimal climatic conditions. These isolates have similar fimbriae but distinct extracellular proteases. To determine the relationship between virulent and benign isolates and the relationship of isolates from different geographical regions, a genomic study that involved the sequencing and subsequent analysis of 103 D. nodosus isolates was undertaken. The results showed that D. nodosus isolates are highly conserved at the genomic level but that they can be divided into two distinct clades that correlate with their disease phenotypes and with a single amino acid substitution in one of the extracellular proteases.
INTRODUCTION
Dichelobacter nodosus is a Gram-negative anaerobic bacterial pathogen that is the primary causative agent of footrot in sheep and other ruminants. In sheep, footrot is an economically significant debilitating disease that limits the mobility of the infected animal (1, 2). Footrot has been an ongoing problem in Australia and has reemerged in Europe (3, 4) with a recent outbreak being the first reported in Norway for many years.
The severity of ovine footrot can vary from benign disease, which presents as an interdigital dermatitis, to virulent disease, in which there is severe underrunning of the horn of the hoof. Progression of disease is dependent on climatic conditions, on host factors, and on the virulence of the infecting strain of D. nodosus. In the past, strains have been assigned a virulence phenotype based on the severity of disease, traditionally virulent or benign (5).
Major virulence factors are the type IV fimbriae and extracellular proteases produced by D. nodosus, with the fimbriae allowing the bacteria to move into the lesion while the proteases degrade the tissue (6–8). Diagnostic tests based on protease thermostability and elastase activity have been used for many years to distinguish benign and virulent strains (9–11). We now know that these tests detect a difference in activity between AprV2, one of the three extracellular serine protease produced by virulent D. nodosus strains, and its benign ortholog AprB2. This difference in function is attributable to a single amino acid change (Y92R) in the mature protease (8). Recently, a diagnostic quantitative real-time PCR test based on the nucleotide sequence encoding this difference was developed (12).
Determination of the genome sequence of the virulent D. nodosus isolate VCS1703A revealed that D. nodosus has a relatively small genome (1.39 Mb) that does not show any evidence of ongoing gene reduction, although approximately 20% of the genome appeared to be derived from lateral gene transfer (13). To better understand the genetic relationship between benign and virulent isolates and between isolates from different geographic regions, we have undertaken the genome sequencing of 103 D. nodosus isolates. A summary of the phenotypic data for many of these isolates is provided as part of the baseline meta-information. Our examination of the relationships between these genomes, inferred from conserved sections of the genome that are present in each of the strains, led to the observation of discernible geographic and disease phenotype correlations. In particular, a clear genetic division exists between isolates previously characterized as having virulent and benign disease phenotypes. This division correlates almost exactly with a classification based on the sequence difference at amino acid 92 between the mature AprV2 protease and its AprB2 ortholog (8, 14).
RESULTS AND DISCUSSION
Selection of strains.
To maximize the genetic diversity of the 103 sequenced strains, isolates with a range of disease phenotypes and geographic origins were selected. The collection comprised multiple isolates from Australia, Norway, Denmark, and Sweden and individual isolates from England, Nepal, Bhutan, and India (Table 1). The Australian collection included isolates acquired over a period of more than 40 years. In contrast, the Norwegian collection comprised isolates only dating back to 2008 (15), arising from an outbreak of virulent ovine footrot, a disease not observed in Norway for more than 60 years. This outbreak appears to have arisen from a single point of introduction of a virulent strain into one county in southern Norway, possibly after the importation of sheep from Denmark (16). It should be noted that this disease introduction was into a background where benign D. nodosus disease was endemic but previously undetected.
TABLE 1 .
Summary of D. nodosus strains
| Countrya | No. of isolates | AprV2/AprB2 typeb |
|
|---|---|---|---|
| Y | R | ||
| Australia (AU) | 38c | 29 | 8 |
| Bhutan (BT) | 1 | 0 | 1 |
| Denmark (DK) | 8 | 8 | 0 |
| India (IN) | 1 | 1 | 0 |
| Nepal (NP) | 1 | 1 | 0 |
| Norway (NO) | 36 | 19 | 17 |
| Sweden (SE) | 17 | 5 | 12 |
| United Kingdom (GB) | 1 | 1 | 0 |
| Total | 103 | 64 | 38 |
Two-letter country codes used in the figures and supplemental material appear in parentheses.
Amino acid encoded at residue 92 of the mature AprV2 protein. Note that one strain did not carry either gene.
The Australian isolate JIR1204 had no aprV2 or aprB2 ortholog; therefore, one Australian isolate had no AprV2/AprB2 type.
The genome sequence of D. nodosus is highly conserved.
A whole-genome shotgun sequencing approach was used to generate read sets of between 30- and 100-fold coverage for the genome of each isolate. These individual read sets were mapped to the D. nodosus VCS1703A genome sequence and also were assembled de novo using Velvet (17). A visual overview of the relationship of each of these genome sequences to that of VCS1703A was then produced using BLAST Ring Image Generator (BRIG) (18). Given the diversity of these isolates, the genomes of D. nodosus were remarkably conserved, as indicated by the areas of solid color in Fig. 1, with the 103 isolates showing >95% sequence identity. There were eight major regions of sequence variability, each of which will be discussed below.
FIG 1 .

BRIG diagram showing an overview of the genomic relationship between strain VCS1703A and the 103 sequenced strains of D. nodosus. Black arcs and labels at the edge of the circle indicate the eight nominally variable regions. Each sequenced isolate is represented as a colored ring, with a solid color representing greater than 95% sequence identity and a white region showing areas with less than 50% sequence identity to VCS1703A. A gradient of color is used to represent sequences in the range of 50 to 95% identity. The colors indicate the country of origin and the protease genotype. The major color groups are Denmark (gray), Sweden (light blue and dark blue), Norway (pink and red), and Australia (yellow and green). Gray, dark blue, red, and green indicate isolates with a virulent AprV2 (Y92) protease. Light blue, pink, and yellow indicate isolates with a benign AprB2 (R92) protease. The four innermost rings are the isolates from the United Kingdom, Nepal, Bhutan, and India, respectively.
D. nodosus genomes can be divided into two distinct clades.
Based on genetic relationships, these D. nodosus genomes can be divided into two major clades that correlate with the single amino acid difference (Y92R) between the mature AprV2 and AprB2 proteases and with the presumptive designation of an isolate as virulent or benign. The strength of the phylogenetic signal that underpins this division indicates a fundamental difference that spans the whole of the conserved regions of the D. nodosus genome.
The regions of the D. nodosus genome present in all of the sequenced isolates collectively defined the core conserved genome (Fig. 1), which was then analyzed as follows. First, we mapped reads from each of the isolates onto the VCS1703A genome sequence. Then, limiting the analysis to those regions of the VCS1703A genome where mapped reads were obtained from all 103 isolates, we identified those bases that varied in one or more of the isolates. We observed 31,627 core single nucleotide polymorphisms (SNPs); these differences were distributed across the entire core regions of the D. nodosus genome. The Neighbor-Net algorithm as implemented in SplitsTree (19) was used to infer the relationship between strains based on these core SNPs; a network diagram providing an overview of this relationship is shown (Fig. 2). The network clearly showed that the isolates group into two distinct clades, as indicated by a vertical orange line in Fig. 2. This grouping was strongly supported by statistical analysis, as indicated by the presence of these clades in 92% of a set of 500 bootstrap replicate trees tested. Our analysis indicated that the differences attributable to this phylogenetic signal were numerous and were distributed across the whole of the core conserved regions of the D. nodosus genome.
FIG 2 .

Network diagram showing the relationship between the genome sequences of 103 D. nodosus isolates. The data set used for the inference of the relationship comprised 31,627 bases from each isolate and represented conserved positions in the VCS1703A reference genome sequence; an orthologous sequence was present in each of the sequenced isolates, and the sequence in one or more of the isolates differed from that found in the reference genome sequence. The relationship shown was inferred using the parsimony-based method as implemented in SplitsTree (19). The tree is annotated with a vertical orange line, indicating a division of taxa into clade I and clade II. The taxon labels are further annotated by colors, with yellow indicating Australia, red indicating Norway, lilac indicating Sweden, and green indicating Denmark. Individual isolates from Bhutan (pink), Nepal (blue), India (orange), and the United Kingdom (gray) are also shown. The taxon labels have the following information: the name of the isolate, the serogroup, and the AprV2 type.
Clade I comprised isolates that encode a tyrosine residue at position 92 in the mature AprV2 protease, which we have previously shown to be essential for virulence (8). Clade II comprised isolates that encode an arginine residue at position 92 in the equivalent AprB2 protease of benign isolates. The two exceptions to this association were the RBG-17 isolate from Bhutan, which is located in clade I but encodes an AprB2-type protease, and the Australian strain JIR1204, which has no aprV2 or aprB2 gene because the entire DNO_1166 to DNO_1168 region is missing from this isolate.
For many of the 103 D. nodosus isolates, there was insufficient information to make a definitive decision as to whether the strain was virulent or benign. However, where it had previously been possible to make such a virulence designation, clade I comprised isolates that have a presumptive virulent disease phenotype and clade II comprised isolates that have a benign disease phenotype. Two isolates from Sweden did not conform to this pattern (see Fig. S1 in the supplemental material); each isolate was of a clade II (AprB2) type that was classified as having a virulent disease phenotype. However, it should be noted that there is considerable variation in the manners in which different laboratories designate isolates as virulent or benign. Since there is no uniformly accepted standard, these designations must be regarded as provisional and subject to further investigation and interpretation.
Network diagram inferred using the same data set as for Fig. 2. Taxon labels show the presumptive disease phenotype of the isolate. Isolates for which no presumptive disease phenotype was available are highlighted in gray. The two Swedish isolates that have a presumptive disease phenotype that is not consistent with an association between clade I (virulent isolates) and clade II (benign isolates) are highlighted in pink. The Bhutan isolate is highlighted in blue and was found to be unique among the isolates because of its aprB2 genotype and clade I grouping. Download Figure S1, PDF file, 0.2 MB (197.7KB, pdf) .
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
It is well established that the only consistent difference between the mature AprV2 and AprB2 proteases is the Y92R substitution (8, 14). This substitution alters the antigenicity of the protein and the structure of an exosite that determines the accessibility of the substrate binding pocket (8). AprV2/AprB2 SNPs contributed only 28 of 31,627 points in the data set used to infer the network diagram (Fig. 2; see Table S1 in the supplemental material). Remarkably, only the SNP responsible for the Y92R difference had an almost 100% correlation with the division between clade I and clade II. We therefore conclude that the AprV2/AprB2 polymorphism is central to the definition of a virulent isolate of D. nodosus.
Summary of SNPs present in the intact AprV2/B2 genes found in the 103 isolates in the study. Table S1, PDF file, 0.3 MB. (303.1KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
The AprV2/AprB2 type is the mechanistic basis for the traditional elastase and gelatin gel tests that are used for the differential diagnosis of D. nodosus (5, 8, 11). Recent studies by other workers have provided evidence that the AprV2/AprB2 status of a D. nodosus isolate correlates with the disease status of the individual sheep or the flock from which it was obtained (20), although these studies were not validated by pen or field virulence trials. Subsequently, a diagnostic competitive real-time PCR test based on the Y92R sequence difference was developed (12). Our SNP analysis is in agreement with the conclusions of that study and provides clear evidence that presumptive virulent and benign isolates of this bacterium represent two distinct clades, a finding that has significant implications for attempts to eradicate this important disease.
The aprV2 gene is not the only gene contributing to the phylogenetic signal dividing clade I and clade II. The widespread distribution of differences across the core conserved genome is predominantly in the form of single nucleotide differences. The implication is that the difference in disease phenotypes represented by clade I (virulent) and clade II (benign) is either encoded through loss of function (differences resulting in premature stop codons) or changes to function (as illustrated by AprV2 and AprB2). We note that almost no pseudogenes were observed in the analysis of the VCS1703A genome sequence (13) and that loss of function in such a small genome is not likely to be sustainable and does not contribute to the difference in phenotype between D. nodosus clade I and clade II isolates.
The occurrence of two distinct clades (Fig. 2) indicated that intermediate genomic states between clade I and clade II occur very rarely, if at all. Despite indications that recombination had occurred between benign and virulent isolates, there was no apparent impact on the phylogenetic signal that separated the clade I and clade II isolates. Detailed analysis of the SNP distributions in each gene led to the identification of 12 genes where the sequence differences correlated with the clade I/II division in 101 isolates (Table 2) (analysis excluded strains JIR1204 and RGB_17). For 5 of these 12 genes, none of the clade I/II-correlating differences resulted in a difference in the encoded protein. The remaining seven genes included DNO_1167, which encodes the AprV2/AprB2 protease. The DNO_1034 gene had the most number of differences (21 SNPS) that correlated with the clade I/II division (Table 2).
TABLE 2 .
Genes containing SNPs that correlate with the clade I/clade II division
| Genea | No. of variable positionsb | No. of bases in coding region | No. of correlating SNPsc | No. of correlating SNPs resulting in an amino acid difference | Product |
|---|---|---|---|---|---|
| DNO_0246 | 76 | 2,504 | 1 | 0 | Leucyl-tRNA synthetase |
| DNO_0309 | 25 | 1,164 | 6 | 1 | N-Acetylglucosamine-6-phosphate deacetylase |
| DNO_0330 | 117 | 1,842 | 2 | 1 | Competence family protein |
| DNO_0620 | 15 | 192 | 4 | 1 | ABC iron transporter permease |
| DNO_0817 | 13 | 816 | 2 | 0 | 2,3,4,5-Tetrahydropyridine-2-carboxylate N-succinyltransferase |
| DNO_0818 | 17 | 474 | 2 | 0 | GTP cyclohydrolase I |
| DNO_0819 | 48 | 1,236 | 1 | 0 | Two-component phosphate sensor PhoR |
| DNO_0820 | 19 | 675 | 7 | 1 | Conserved hypothetical membrane protein |
| DNO_0823 | 31 | 1,197 | 2 | 1 | Phosphopantothenoylcysteine decarboxylase/phosphopantothenate-cysteine ligase |
| DNO_1034 | 80 | 1,998 | 21 | 5 | Carboxyl-terminal protease family protein |
| DNO_1167 | 29 | 1,806 | 8 | 3 | Acidic extracellular subtilisin-like protease AprV2/B2 |
| DNO_1203 | 18 | 867 | 1 | 0 | Hypothetical protein |
Locus tag from the VCS1703A genome annotation.
Positions where at least one of the 101 isolates has an SNP.
Positions where the sequence difference correlates with the clade I/clade II division.
These data demonstrate the existence of genes that have distinctive alleles found in either presumptively virulent (clade I) or benign (clade II) isolates, which suggests that the encoded proteins have subtly different functions that may play an important role in the difference in disease phenotype between virulent and benign isolates. These SNPs (Table 2) account for only ~500 of the 31,627 core SNPs. The use of a less stringent approach, where some deviation from an exact match with the clade I/II division was allowed, identified an extended list of candidate genes that may have a role in virulence (Table S2). This analysis provides a basis for a new, directed approach to the investigation of virulence in D. nodosus and to the development of new diagnostic tools.
Genes containing SNPs that correlate best (top 1%) with the clade I/clade II division. Table S2, PDF file, 0.1 MB. (54.3KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Eight major regions of sequence variability in D. nodosus.
Eight major regions of sequence variability were identified from our analysis, as shown in the BRIG diagram (Fig. 1), where white regions indicate where these isolates had no sequence similar to the reference VCS1703A sequence. These regions generally corresponded to the regions of atypical trinucleotide composition identified previously (13). The coding sequences associated with each of the variable regions in strain VCS1703A are listed in Table S3 and will now be discussed in turn.
Genes associated with each of the variable regions. Table S3, PDF file, 0.1 MB. (180.1KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
The largest area of variability is the region shown in VCS1703A to be an integrated Mu-like bacteriophage (13) that was previously characterized from another isolate and named DinoH1 (21). Twenty-nine of the strains sequenced here have an identical or a very similar integrated bacteriophage at this position; 18 of these isolates are Australian, 8 are from Norway, and 1 each is from Sweden, Nepal, and Bhutan. Other strains, including most of the benign Swedish strains, show some regions of similarity across this region and therefore may have similar although not identical integrated bacteriophage genomes.
The vrl region previously was identified in Australia as being preferentially associated with virulent isolates (22, 23), but sequencing of this region did not identify any potential virulence genes and the region is most likely the remnant of an inserted bacteriophage genome (24). Only 21 of the isolates sequenced here contained most of the vrl region (14 Australian isolates, 6 Swedish isolates, and 1 Indian isolate), with a further 2 Australian, 2 Swedish, and 2 Norwegian isolates containing a portion of the region (Fig. 1). While this region is still preferentially associated with virulent isolates, we note that of the Australian isolates, the vrl region is present only in strains isolated before 1990, indicating a possible loss of this region from the Australian D. nodosus genetic pool. However, the regional localization of our recent Australian isolates may also explain this difference. It is regardless clear that there is no direct functional association of the vrl region with virulence.
The vap regions were originally identified as genomic islands present in virulent strains of D. nodosus but absent from more than two-thirds of benign strains (23). These regions are found in multiple copies in two of the eight variable regions of the genome in many strains and appear to have arisen by insertion of a bacteriophage or plasmid into the tRNA gene(s), leading to their designation as genomic islands (25). Integrase genes are found adjacent to these vap regions, and further investigation of these regions has led to their designation as intA, intB, intC, and intD elements (26–28). Further studies indicated that there was some correlation between the presence of the intA element and virulence (29), although this was not found in a study by other workers (11). There also appears to be a correlation between the intD element and benign strains (30). In this study, we have found that all of the isolates have at least some of the genes contained within the vap regions, but those that contained the largest vap regions were virulent isolates from Australia and Sweden. In contrast, in the Norwegian strains, the vap regions more commonly were associated with benign isolates, with two virulent serogroup H exceptions (15). From the overall distribution of the vap regions (Fig. 1) among the 103 isolates, there was no correlation between the vap regions and the clade I or clade II designations.
Another variable region encoded a type 1 restriction and modification system that typically consists of three closely linked genes, hsdR, hsdM, and hsdS (31). All three genes were present in 95 of the D. nodosus strains sequenced, and major variation between the strains was observed only in the hsdS gene. In other bacteria, HsdS is the specificity subunit and is known to contain two variable regions, with the rest of the protein being highly conserved (31). The D. nodosus hsdS gene showed similar variation, with the regions encoding amino acids 210 to 256 and 420 to 476 being conserved and other regions being more variable. It has recently been reported that genetic rearrangement by domain shuffling in hsdS drives the diversification of the very common system (32). These data suggest that there is strain-to-strain variability in the DNA sequences recognized by the different Hsd restrictions systems present in D. nodosus isolates.
Many Gram-negative bacteria secrete RTX proteins. These proteins contain repeated C-terminal glycine- and aspartate-rich sequences which form calcium binding sites and are secreted via a type I secretion system (33). The DNO_0334 protein was identified as an RTX-like protein by comparative sequence analysis (13) and by domain searches (33). The gene encoding this protein contains multiple tandem repeats, limiting our ability to accurately assemble contiguous sequences across this region; as a result, it appears as a region of sequence variability. The gene encoding the D. nodosus RTX-like protein lies downstream of a gene encoding an OmpA family protein (DNO_0333) and upstream of genes encoding an ABC-2 transporter and an ABC transporter domain protein (DNO_0335, DNO_0336). While not a typical type I secretion system, these transporter proteins may be involved in the secretion of the putative RTX protein in D. nodosus. The three genes neighboring DNO_0334 are all highly conserved across the 103 sequenced strains.
The Omp1 region of the genome (DNO_0382 to DNO_0385) contains four linked genes that encode four different forms of the major outer membrane protein Omp1. It has been suggested that site-specific inversion events in this region give rise to the antigenic variation of Omp1 (34). The genes on either side of these four genes are highly conserved across all of our isolates.
Finally, the pgr gene (DNO_0690) encodes a protein with a variable number of 9-amino-acid repeat units and was originally identified as being absent from benign isolates (13). However, further work showed that the gene was present in benign isolates but contained a different repeat unit structure (35). The function of the Pgr protein is unknown, and the significance of the variation in repeat structure has not been determined. Our study confirmed the presence of this highly variable gene in each isolate, but once again, contiguous sequences could not be assembled.
Fimbrial biogenesis genes are highly conserved.
A key question was whether the known virulence genes demonstrated variability that correlated with the two clades. D. nodosus type IV fimbriae are known virulence factors, and the major fimbrial subunit gene fimA and the twitching motility genes pilT and pilU are essential for virulence (6, 7). These fimbriae are the major surface antigens and define the 10 serogroups of D. nodosus (5, 36, 37). These serogroups are divided into two classes based on differences in the genetic organizations of the fimA gene region. Class I strains have the fimA and fimB genes (38), but the fimB gene is not required for fimbrial biogenesis (7). Class II strains (serogroups D and H only) contain the fimA and fimZ genes, and fimZ is a potentially redundant fimbrial subunit gene (38). We have inferred a phylogenetic tree that provides a comprehensive overview of the relationship between all available D. nodosus fimA and fimZ genes, including the sequences arising from this study (see Fig. S2 in the supplemental material). The uniformity within serogroup A was striking, with 14 of the Norwegian isolates and three of the Danish isolates having identical fimA sequences, which suggested that there was a close genetic relationship between these isolates. The relationships between taxa within other serogroup clades was in many cases a continuum, with no clear subgroups of isolates; this result suggests that any classification subordinate to that of a serogroup is impractical.
Phylogenetic tree showing the relationship between the fimA and fimZ genes. The data include sequences from the 103 isolates sequenced in this study (indicated by a colored circle beside the taxon label) and 78 additional D. nodosus fimA or fimZ genes available in GenBank (indicated by a colored square beside the taxon label). Accession numbers are presented in Table S7. Serogroups are color coded as follows: serogroup A (gray), serogroup B (red), serogroup C (light green), serogroup D (dark green), serogroup E (yellow), serogroup F (blue), serogroup G (pink), serogroup H (brown), serogroup I (light gray), serogroup M (dark blue), and fimZ sequence taxa (light blue). The multiple-sequence alignment used to infer relationships was trimmed to exclude parts of the 5′ and 3′ coding regions. The evolutionary history was inferred using the minimum evolution (ME) method (A. Rzhetsky and M. Nei, J. Mol. Evol. 35:367–375, 1992, doi:10.1007/BF00161174). The optimal tree with the sum of branch lengths equal to 4.09 is shown. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the maximum composite likelihood method (K. Tamura, M. Nei, and S. Kumar S, Proc. Natl. Acad. Sci. U. S. A. 101:11030–11035, 2004, doi:10.1073/pnas.0404206101), and units are numbers of base substitutions per site. The ME tree was searched using the close-neighbor-interchange algorithm (M. Nei and S. Kumar, Molecular Evolution and Phylogenetics, 2000) at a search level of 1. The neighbor-joining algorithm was used to generate the initial tree. The analysis involved 188 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 288 positions in the final data set. Evolutionary analyses were conducted in MEGA6 (K. Tamura, G. Stecher, D. Peterson, A. Filipski, and S. Kumar, Mol. Biol. Evol. 30:2725–2729, 2013, doi:10.1093/molbev/mst197). Download Figure S2, PDF file, 0.1 MB (112.3KB, pdf) .
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
The other fimbrial biogenesis genes are scattered throughout the genome (13) and are listed in Table S4 in the supplemental material. These genes and their deduced gene products generally were conserved across the 103 sequenced isolates. For example, only one amino acid difference was observed in the essential fimbrial retraction protein PilT among the 103 isolates sequenced, a P52Q substitution in strain 07BKT018497 from Sweden.
Fimbrial biogenesis genes. Table S4, PDF file, 0.1 MB. (118.8KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
The more varied fimbrial biogenesis genes included pilQ, for which taxa were loosely divided into three groups, with one group predominantly comprising Norwegian and Danish isolates. The pilE fimU pilX pilW pilV locus showed variation across the whole locus, with a tendency for the serogroup class II isolates to be located in a separate group. None of the trees inferred using the fimbrial biogenesis genes alone were congruent with the whole core genome phylogeny (Fig. 2), indicating that none of the fimbrial biogenesis genes contribute to the phylogenetic signal that divides clade I and clade II. Therefore, although fimbriae are essential for virulence, it appears that none of the fimbrial components are exclusively present in virulent strains and that none of the allelic forms are exclusive to clade I isolates (Table S5). This finding may be a reflection of a more general role of the fimbriae in D. nodosus; fimbriae are essential not only for virulence but also for more-general functions, including protease secretion and DNA uptake (6, 7).
Details of individual isolates. Table S5, PDF file, 0.2 MB. (260.4KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Sequence variation of the extracellular proteases.
D. nodosus strains produce three extracellular serine proteases that are all putative virulence factors (8). The genes for these proteases are located in highly conserved regions of the chromosome, with aprV5 (DNO_0603) and bprV (DNO_0605) in close proximity and aprV2 (DNO_1167) in a different region. All three genes encode proteases with a pre-pro region and a C-terminal extension, which are cleaved to produce the mature protease (39).
Comparison of the mature protease sequences across the 103 sequenced isolates showed the mature AprV2 protease to be highly conserved, with the only major variation being the previously reported Y92R substitution that correlated strongly with the clade I and clade II groupings of isolates, as already discussed (Fig. 2). The mature AprV5 protease showed more variation, with 4 Norwegian and 5 Swedish isolates having a G216K substitution and another 33 isolates having a D310N change, which placed these strains into phylogenetic groups separate from the remainder (Fig. S3). These groups did not correlate with our clade I and clade II classifications. The mature basic protease BprV also showed more variation, with 11 conservative differences in one or more isolates; 8 of these changes generally correlated with the clade I/II division (Fig. S3). These differences were the same as those previously reported to distinguish BprV and BprB (40), while the remaining variations occurred in only a minority of strains.
Phylogenetic trees showing the relationship between each of the three protease genes: aprV2 (DNO_1167) (A), bprV (DNO_0605) (B), and aprV5 (DNO_0603) (C). The relationships shown were inferred using the minimum evolution method as implemented in MEGA6 (K. Tamura, G. Stecher, D. Peterson, A. Filipski, and S. Kumar, Mol. Biol. Evol. 30:2725–2729, 2013, doi:10.1093/molbev/mst197), and support for nodes is indicated by percentages of the 500 bootstrap replicates that contained the particular node. Taxa are labeled with the name of the isolate, the country of isolation, the serogroup, and the AprV2/AprB2 type. Color coding of taxa are as follows: blue for the AprB2-R92 type (correlating with clade II) and red for the AprV2-Y92 type (correlating with clade I). Download Figure S3, PDF file, 0.2 MB (172.3KB, pdf) .
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Geographic overview. (i) The Norwegian outbreak.
An outbreak of virulent ovine footrot occurred in Norway in 2008, probably arising from a single point introduction of a virulent D. nodosus strain. Subsequently, a range of healthy and diseased sheep were screened for D. nodosus, leading to a detailed characterization of isolates from several locations (15). Each isolate was designated by the diagnostic laboratory as virulent or benign based on its protease stability profile. The virulent isolates were predominantly serogroup A, and pulsed-field gel electrophoresis (PFGE) indicated that they were clonal (16). The benign isolates were more diverse in serogroup and geographical spread. The isolates sequenced were chosen to allow examination of both the clonality of the virulent serogroup A isolates and the diversity of the overall Norwegian isolates. In addition, since a few importations of sheep from Denmark predated the 2008 outbreak, eight Danish isolates, including two isolates with PFGE types similar to the predominant Norwegian PFGE type, were sequenced. BRIG analysis (Fig. 3A) showed that the virulent Norwegian and Danish isolates were very similar; an overview of the relationship between the clade I Norwegian and Danish isolates is shown in Fig. 3B. The near clonality of the Norwegian clade I isolates was in contrast to the genomic diversity of the Danish isolates, probably reflecting a difference between endemic strains and an outbreak situation.
FIG 3 .
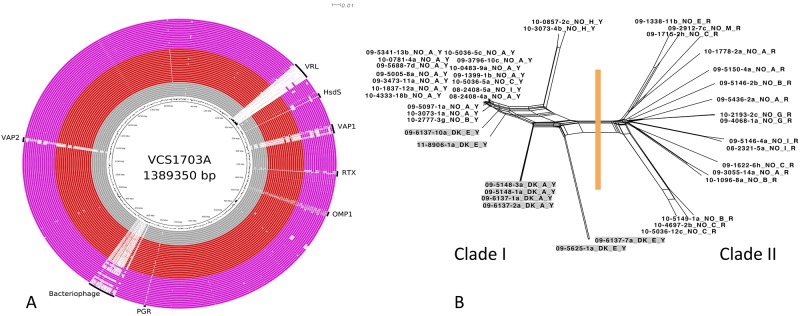
Comparative analysis of sequences from Norway and Denmark. (A) BRIG diagram showing an overview of the genomic relationship between VCS1703A and the 44 sequenced isolates of D. nodosus originating from either Denmark or Norway. Symbols and colors are as described in Fig. 1. (B) Network diagram showing the relationship between the genome sequences of the 44 D. nodosus isolates from Norway and Denmark. The methodology is as described in Fig. 2.
All of the virulent isolates from the Norwegian outbreak (clade I) (Fig. 3B), even isolates from different fimbrial serogroups, were closely related. Our analysis indicated that these differences were likely to have arisen from a small number of genetic recombination events, not from a gradual accumulation of single base mutations. We suggest that these strains were recently derived from a common progenitor. The relationships between the isolates observed in this study were consistent with the hypothesis that the Norwegian outbreak strain originated from Denmark. However, a definitive conclusion to this effect cannot be made due to the lack of any epidemiological link between the isolates that were sequenced.
(ii) The Swedish isolates are diverse.
The Swedish virulent isolates contained the majority of the vrl region and both vap regions, making them more similar to the older clade I Australian isolates than to the other Scandinavian isolates and quite different from the benign Swedish isolates (Fig. 1 and 4A). In contrast, a related, although not identical, bacteriophage genome was present in several benign isolates, while one virulent strain contained an integrated bacteriophage almost identical to that in VCS1703A (Fig. 4A). SNP analysis confirmed these findings, with the clade I isolates from Sweden being most closely related to Australian isolates (Fig. 2). Overall, the Swedish clade I isolates were closely related, whereas the clade II isolates were more diverse (Fig. 4B).
FIG 4 .
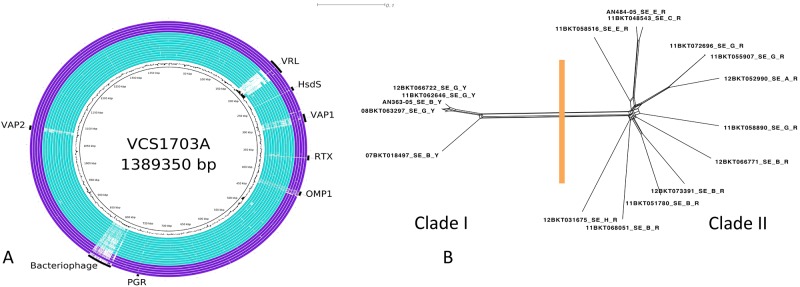
Comparative analysis of sequences from Sweden. (A) BRIG diagram showing an overview of the genomic relationship between VCS1703A and the 17 sequenced isolates of D. nodosus originating from Sweden. Symbols and colors are as described in the legend of Fig. 1. (B). Network diagram showing the relationship between the genome sequences of the 17 D. nodosus isolates from Sweden. The methodology is as described in the legend of Fig. 2.
(iii) Analysis of the Australian isolates demonstrated a historical and geographical distribution.
The vrl region was present in the historical virulent Australian isolates but was absent from the more recent isolates from Tasmania and South Australia (Fig. 5A). SNP analysis (Fig. 5B) revealed that the Australian clade I isolates, but not the clade II isolates, formed clusters, with most of the historical isolates forming one large cluster. The more recent isolates from Tasmania formed two distinct clusters, possibly indicating that these isolates, which remained in the population after a vaccination program, were derived from two different strains. The three South Australian isolates also grouped into a distinct cluster. Finally, two of the isolates from Western Australia grouped into a cluster quite separate from the other historical isolates, which was consistent with previous observation suggesting that isolates from this state were different from other Australian isolates (10). The clustering of distinct sets of related isolates may indicate some sampling bias; the sequencing of more isolates will be required to build a complete picture of the genetic diversity of the virulent D. nodosus isolates present in Australia, where virulent footrot has been endemic over an extended period of time.
FIG 5 .
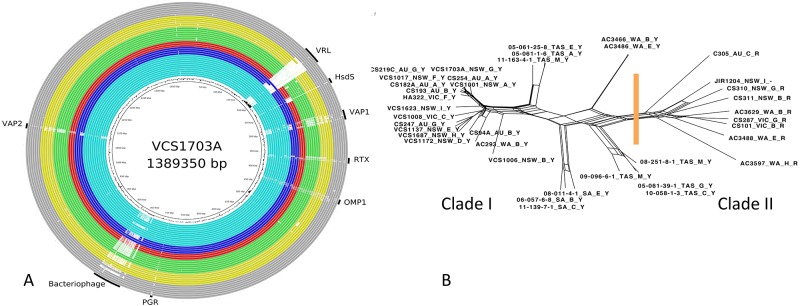
Comparative analysis of sequences from Australia. (A) BRIG diagram showing an overview of the genomic relationship between VCS1703A and the 38 sequenced isolates of D. nodosus originating from Australia. The major color groups are light blue for New South Wales (NSW), dark blue for Victoria (VIC), red for South Australia (SA), green for Tasmania (TAS), yellow for Western Australia (WA), and gray for isolates of unknown origin in Australia (AU). (B) Network diagram showing the relationship between the genome sequences of the 38 D. nodosus isolates from Australia sequenced as part of this study. The methodology is as described in the legend of Fig. 2.
(iv) Analysis of individual isolates from the United Kingdom, Bhutan, India, and Nepal.
The isolates from the United Kingdom, Bhutan, India, and Nepal were included to provide a snapshot of the genetic relationship between isolates from the major regions covered in this study (Scandinavia and Australia) and isolates from two other regions where footrot is prevalent. Analysis of the isolates from Bhutan, India, and Nepal provided an indication that virulent isolates from Asia are similar to those present in other parts of the world, with the India and Nepal isolates being most closely related to isolates from Australia (Fig. 2) and the Bhutan isolate being somewhat unique among the clade I isolates, with this isolate being the only AprB2-type isolate located in clade I. This may be an aberrant isolate or an indicator of an unusual pattern of footrot in Bhutan. The United Kingdom isolate was more closely related to some of the Danish isolates, suggesting that isolates in the United Kingdom may be similar to those found in Europe. Further genomic studies involving a larger number of isolates from these countries are required to confirm these findings.
Conclusions.
The sequencing of 103 genomes of isolates from diverse geographical locations showed that D. nodosus comprises a distinctly bimodal population of strains. Detailed analysis of core SNPs revealed two distinct clades of D. nodosus isolates, with these clades correlating with the presumptive designation of virulent and benign isolates and with a single amino acid difference between the mature AprV2 and AprB2 proteases. Eleven other genes with SNPs (Table 2) that correlated with this phylogenetic differentiation were identified; however, in only six of these genes did these differences result in an amino acid substitution in the putative encoded protein. These genes, along with other genes that have SNPs that generally correlated with the clade I/II division (Table S2), were scattered throughout the genome. This finding indicated that the genetic differences between the clades were deeply embedded.
The relationships between sequences in regions outside those mentioned in Tables 2 and S2 in the supplemental material show no consistent pattern of relationship, except that isolates from the same outbreak are more closely related. This pattern of sequence difference between isolates cannot be explained by the divergence of two independent lineages. This conclusion is supported by the presence of regions of sequence identity between clade I and clade II isolates in the non-clade I/II-conforming regions of the genome. In a naturally transformable organism such as D. nodosus, this type of relationship is consistent with horizontal gene transfer occurring regularly between any two isolates of D. nodosus. Regions of the genome not contributing to the disease phenotype differences are presumably under no selective pressure, in contrast to those regions in Tables 2 and S2, where we propose that these genetic differences in total contribute to niche adaption, leading to the virulent and benign disease phenotypes and resulting in the observed relationship between core SNPs. The relatively low rate at which horizontal gene transfer changes are embedded is indicated by the strong relationship in the core SNP tree between isolates from the Norwegian footrot outbreak. Further analysis of these genome sequences will be used to examine in more detail the genetic legacy of horizontal gene transfer in D. nodosus.
It was previously reported that D. nodosus has one of the smallest genomes of an anaerobic bacterium and lacks any evidence of ongoing genome reduction (13). This observation is in agreement with the results obtained here, where we have shown that the genomes of 103 isolates are 95% conserved. We conclude that this bacterium has refined its genome to what is required for it to survive within its niche, the hoof of the ruminant, and to cause disease. Whether by occupying distinct subenvironments within the hoof or by utilizing distinct life cycles, two genetically definable lineages of D. nodosus coexist in this niche.
MATERIALS AND METHODS
Bacterial isolates and epizootiology.
The D. nodosus isolates used in this study are listed in Table S5. It is important to note differences in the breeds of the animals, the climates, and the husbandry conditions in the regions from which these isolates were obtained.
Sheep in Australia are predominantly Merino, which are very susceptible to footrot. They are kept on pasture throughout the year, and flock sizes of >2,000 sheep are common. The Australian isolates were a collection of virulent and benign isolates from collections held at Monash University, the University of Sydney, and the Commonwealth Scientific and Industrial Research Organisation (CSIRO) and included strains for each of nine serogroups (41). The majority of the isolates were historical and were isolated between 1972 and 1991, except for 10 isolates from South Australia (Kangaroo Island) and Tasmania (King Island), which were isolated between 2005 and 2011 (42).
The Norwegian isolates were from an outbreak of ovine footrot that started in 2008. Virulent isolates were found only in sheep in Rogaland County in southwest Norway, and 96% of these isolates belonged to serogroup A, pointing to a focal disease outbreak (15). The Norwegian sheep industry consists primarily of farms with between 20 and 99 breeding ewes, which is a situation very different from the large sheep farms in Australia. The meat- and wool-producing crossbred Norwegian white sheep is the most common breed. Sheep are usually housed during the winter and kept on mountain pastures with their lambs during the summer.
The Swedish D. nodosus isolates were mainly from a study conducted between 2011 and 2012. Flocks from different geographical locations in Sweden, primarily in the southeast, and with different manifestations of footrot were sampled. As in Norway, but unlike in Australia, sheep are often housed indoors in winter.
Bacteriological methods.
All isolates except those from Sweden were grown at Monash University in a Coy anaerobic chamber (Coy Laboratory Products Inc.) in an atmosphere of 10% (vol/vol) H2, 10% (vol/vol) CO2, and 80% (vol/vol) N2 on Eugon (BBL) yeast extract (EYE) agar with 5% (vol/vol) defibrinated horse blood (Bio-Lab) or in EYE broth, as described previously (43). D. nodosus genomic DNA was prepared by using a Qiagen DNeasy kit according to the manufacturer’s instructions. The Swedish isolates were grown on fastidious anaerobe agar plates (Lab M Ltd.) with 5% defibrinated horse blood (Håtunalab AB). The plates were incubated anaerobically at 37°C for 4 days. DNA extraction was performed with a BioRobot EZ1 system (Qiagen) according to the manufacturer’s instructions using the EZ1 tissue kit and the bacterial protocol from the same manufacturer.
DNA sequencing and sequence compilation.
For each strain, genomic DNA prepared from pure cultures was mechanically sheared to an average size of 300 bp. Libraries were prepared according to Illumina protocols for paired-end sequencing, and libraries were run on either an Illumina GAIIx or a MiSeq instrument. The yield of sequence for each strain, along with details of read length and read coverage, can be found in Table S6 in the supplemental material.
Details of genome sequencing and analysis data. Download Corrected Table S6, DOCX file, 40 KB (39.1KB, docx) .
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Details of genome sequencing and analysis data. Table S6, PDF file, 0.2 MB. (260.7KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
The raw, paired-end sequence read sets have been submitted to the Sequence Read Archive; accession numbers are listed in Table S6. Prior to analysis, read sets were filtered, which involved deleting regions of reads with low-quality base calls or similarity to Illumina adaptors. Subsequently, reads shorter than 20 bases were removed from the analysis. Read sets in which more than 10% of the reads were removed by filtering were manually checked for suitability. Various similarity-searching techniques were used to detect evidence of contaminating reads; the extremely low levels (<10 reads per read set) detected were assessed to have no potential to interfere with the analysis conducted in this study.
The numbers of reads in each of the read sets ranged from 135,899 (strain AC3488, 151 bases, paired ends) to 7,869,851 (strain 10_4697_2b, 101 bases, paired ends), yielding read coverage that ranged from 30- to 1,000-fold. For analysis purposes, read subsets were used to cap read coverage at 100-fold. Each read set was mapped to the genome sequence of D. nodosus strain VCS1703A (GenBank accession number NC_009446.1) using SHRiMP (44) as implemented in our in-house software package Nesoni (Victorian Bioinformatics Consortium). The resultant BAM file was used as the input to call sites of sequence variation in each strain using the “nway” module of Nesoni. For each strain, reads not mapping to the VCS1703A genome sequences were collected into a new data set of unmapped reads.
Reads were assembled de novo using Velvet (17). For each assembly, a coarse optimization of assembly conditions was conducted by varying the k-mer size in the range 35 to 195 in increments of 10. An optimized assembly condition was identified for each data set. PLINK (45) was used to rank the correlation of the pattern of base differences at a particular position with particular groups of isolates.
Comparative analysis of genome sequences.
BLAST Ring Image Generator (BRIG) (18) was used to provide a visual overview of the relationship between the draft genome sequences of various isolates to the genome sequence of D. nodosus strain VCS1703A. SplitsTree (19) and MEGA6 (46) were used to infer relationships between sequences. Muscle (47) was used for alignment of sequences. Specific gene sequences were extracted from the draft genome sequences of each of the strains using BLAST (48). The minimum evolution method, as implemented in MEGA6, was used to infer relationships between individual gene sequences. SplitsTree was used to infer the genome-wide relationship between the strains.
fimA or fimZ sequences obtained from GenBank. Table S7, PDF file, 0.1 MB. (134.3KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
ACKNOWLEDGMENTS
Research at the Norwegian Veterinary Institute and at Monash University was financially supported by a grant from the Research Council of Norway (project 199422). Research at Monash University was supported by the Australian Research Council (ARC), which provided funding to the ARC Centre of Excellence in Structural and Functional Microbial Genomics, and by the Victorian Life Sciences Computation Initiative, an initiative of the Victorian State Government. Research at the University of Sydney was supported by Australian Wool Innovation. The Swedish Farmers’ Foundation for Agricultural Research is acknowledged for financial support.
Synnøve Vatn at the Norwegian Meat and Poultry Research Center and Ulrika König at the Swedish Animal Health Service are thanked for providing samples and clinical information. We thank Øystein Angen, Shakeel Wani, and Elizabeth Wellington for the provision of strains.
Footnotes
This article is a direct contribution from a Fellow of the American Academy of Microbiology.
Citation Kennan RM, Gilhuus M, Frosth S, Seemann T, Dhungyel OP, Whittington RJ, Boyce JD, Powell DR, Aspán A, Jørgensen HJ, Bulach DM, Rood JI. 2014. Genomic evidence for a globally distributed, bimodal population in the ovine footrot pathogen Dichelobacter nodosus. mBio 5(5):e01821-14. doi:10.1128/mBio.01821-14.
REFERENCES
- 1. Stewart DJ, Clark BL, Jarret RG. 1984. Differences between strains of Bacteroides nodosus in their effects on the severity of foot rot, body weight and wool growth on Merino sheep. Aust. Vet. J. 61:349–352. [DOI] [PubMed] [Google Scholar]
- 2. Green LE, George TR. 2008. Assessment of current knowledge of footrot in sheep with particular reference to Dichelobacter nodosus and implications for elimination or control strategies for sheep in Great Britain. Vet. J. 175:173–180. doi: 10.1016/j.tvjl.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 3. König U, Nyman AK, de Verdier K. 2011. Prevalence of footrot in Swedish slaughter lambs. Acta Vet. Scand. 53:27. doi: 10.1186/1751-0147-53-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moore LJ, Wassink GJ, Green LE, Grogono-Thomas R. 2005. The detection and characterisation of Dichelobacter nodosus from cases of ovine footrot in England and Wales. Vet. Microbiol. 108:57–67. doi: 10.1016/j.vetmic.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 5. Kennan RM, Han X, Porter CJ, Rood JI. 2011. The pathogenesis of ovine footrot. Vet. Microbiol. 153:59–66. doi: 10.1016/j.vetmic.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 6. Han X, Kennan RM, Davies JK, Reddacliff LA, Dhungyel OP, Whittington RJ, Turnbull L, Whitchurch CB, Rood JI. 2008. Twitching motility is essential for virulence in Dichelobacter nodosus. J. Bacteriol. 190:3323–3335. doi: 10.1128/JB.01807-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kennan RM, Dhungyel OP, Whittington RJ, Egerton JR, Rood JI. 2001. The type IV fimbrial subunit gene (fimA) of Dichelobacter nodosus is essential for virulence, protease secretion, and natural competence. J. Bacteriol. 183:4451–4458. doi: 10.1128/JB.183.15.4451-4458.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kennan RM, Wong W, Dhungyel OP, Han X, Wong D, Parker D, Rosado CJ, Law RH, McGowan S, Reeve SB, Levina V, Powers GA, Pike RN, Bottomley SP, Smith AI, Marsh I, Whittington RJ, Whisstock JC, Porter CJ, Rood JI. 2010. The subtilisin-like protease AprV2 is required for virulence and uses a novel disulfide-tethered exosite to bind substrates. PLoS Pathog. 6:e1001210. doi: 10.1371/journal.ppat.1001210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Palmer MA. 1993. A gelatin test to detect activity and stability of proteases produced by Dichelobacter (Bacteroides) nodosus. Vet. Microbiol. 36:113–122. doi: 10.1016/0378-1135(93)90133-R. [DOI] [PubMed] [Google Scholar]
- 10. Rood JI, Howarth PA, Haring V, Billington SJ, Yong WK, Liu D, Palmer MA, Pitman DR, Links I, Stewart DJ, Vaughan JA. 1996. Comparison of gene probe and conventional methods for the differentiation of ovine footrot isolates of Dichelobacter nodosus. Vet. Microbiol. 52:127–141. doi: 10.1016/0378-1135(96)00054-5. [DOI] [PubMed] [Google Scholar]
- 11. Dhungyel OP, Hill AE, Dhand NK, Whittington RJ. 2013. Comparative study of the commonly used virulence tests for laboratory diagnosis of ovine footrot caused by Dichelobacter nodosus in Australia. Vet. Microbiol. 162:756–760. doi: 10.1016/j.vetmic.2012.09.028. [DOI] [PubMed] [Google Scholar]
- 12. Stäuble A, Steiner A, Frey J, Kuhnert P. 2014. Simultaneous detection and discrimination of virulent and benign Dichelobacter nodosus in sheep of flocks affected by foot rot and in clinically healthy flocks by competitive real-time PCR. J. Clin. Microbiol. 52:1228–1231. doi: 10.1128/JCM.03485-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Myers GS, Parker D, Al-Hasani K, Kennan RM, Seemann T, Ren Q, Badger JH, Selengut JD, Deboy RT, Tettelin H, Boyce JD, McCarl VP, Han X, Nelson WC, Madupu R, Mohamoud Y, Holley T, Fedorova N, Khouri H, Bottomley SP, Whittington RJ, Adler B, Songer JG, Rood JI, Paulsen IT. 2007. Genome sequence and identification of candidate vaccine antigens from the animal pathogen Dichelobacter nodosus. Nat. Biotechnol. 25:569–575. doi: 10.1038/nbt1302. [DOI] [PubMed] [Google Scholar]
- 14. Riffkin MC, Wang LF, Kortt AA, Stewart DJ. 1995. A single amino-acid change between the antigenically different extracellular serine proteases V2 and B2 from Dichelobacter nodosus. Gene 167:279–283. doi: 10.1016/0378-1119(95)00664-8. [DOI] [PubMed] [Google Scholar]
- 15. Gilhuus M, Vatn S, Dhungyel OP, Tesfamichael B, L’Abée-Lund TM, Jørgensen HJ. 2013. Characterisation of Dichelobacter nodosus isolates from Norway. Vet. Microbiol. 163:142–148. doi: 10.1016/j.vetmic.2012.12.020. [DOI] [PubMed] [Google Scholar]
- 16. Gilhuus M, Kvitle B, L’Abée-Lund TM, Vatn S, Jørgensen HJ. 2014. A recently introduced Dichelobacter nodosus strain caused an outbreak of footrot in Norway. Acta Vet. Scand. 56:29. doi: 10.1186/1751-0147-56-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. 2011. BLAST Ring Image generator (Brig): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 20. Stäuble A, Steiner A, Normand L, Kuhnert P, Frey J. 2014. Molecular genetic analysis of Dichelobacter nodosus proteases AprV2/B2, AprV5/B5 and BprV/B in clinical material from European sheep flocks. Vet. Microbiol. 168:177–184. doi: 10.1016/j.vetmic.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 21. Cheetham BF, Parker D, Bloomfield GA, Shaw BE, Sutherland M, Hyman JA, Druitt J, Kennan RM, Rood JI, Katz ME. 2008. Isolation of the bacteriophage DinoHI from Dichelobacter nodosus and its interactions with other integrated genetic elements. Open Microbiol. J. 2:1–9. doi: 10.2174/1874285800802010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haring V, Billington SJ, Wright CL, Huggins AS, Katz ME, Rood JI. 1995. Delineation of the virulence related locus (vrl) of Dichelobacter nodosus. Microbiology 141 (Part 9):2081–2091. doi: 10.1099/13500872-141-9-2081. [DOI] [PubMed] [Google Scholar]
- 23. Katz ME, Howarth PM, Yong WK, Riffkin GG, Depiazzi LJ, Rood JI. 1991. Identification of three gene regions associated with virulence in Dichelobacter nodosus, the causative agent of ovine footrot. J. Gen. Microbiol. 137:2117–2124. doi: 10.1099/00221287-137-9-2117. [DOI] [PubMed] [Google Scholar]
- 24. Billington SJ, Huggins AS, Johanesen PA, Crellin PK, Cheung JK, Katz ME, Wright CL, Haring V, Rood JI. 1999. Complete nucleotide sequence of the 27-kilobase virulence related locus (vrl) of Dichelobacter nodosus: evidence for an extrachromosomal origin. Infect. Immun. 67:1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rood JI. 2002. Genomic islands of Dichelobacter nodosus. Curr. Top. Microbiol. Immunol. 264:47–60. [PubMed] [Google Scholar]
- 26. Cheetham BF, Tattersall DB, Bloomfield GA, Rood JI, Katz ME. 1995. Identification of a bacteriophage-related integrase gene in a vap region of the Dichelobacter nodosus genome. Gene 162:53–58. doi: 10.1016/0378-1119(95)00315-W. [DOI] [PubMed] [Google Scholar]
- 27. Cheetham BF, Whittle G, Katz ME. 1999. Are the vap regions of Dichelobacter nodosus pathogenicity islands?, p 203–218. In Kaper JB, Hacker J. (ed), Pathogenicity islands and other mobile virulence elements. American Society for Microbiology, Washington, DC. [Google Scholar]
- 28. Bloomfield GA, Whittle G, McDonagh MB, Katz ME, Cheetham BF. 1997. Analysis of sequences flanking the vap regions of Dichelobacter nodosus: evidence for multiple integration events, a killer system, and a new genetic element. Microbiology 143(Part 2):553–562. doi: 10.1099/00221287-143-2-553. [DOI] [PubMed] [Google Scholar]
- 29. Cheetham BF, Tanjung LR, Sutherland M, Druitt J, Green G, McFarlane J, Bailey GD, Seaman JT, Katz ME. 2006. Improved diagnosis of virulent ovine footrot using the intA gene. Vet. Microbiol. 116:166–174. doi: 10.1016/j.vetmic.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 30. Tanjung LR, Whittle G, Shaw BE, Bloomfield GA, Katz ME, Cheetham BF. 2009. The intD mobile genetic element from Dichelobacter nodosus, the causative agent of ovine footrot, is associated with the benign phenotype. Anaerobe 15:219–224. doi: 10.1016/j.anaerobe.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 31. Murray NE. 2000. Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol. Mol. Biol. Rev. 64:412–434. doi: 10.1128/MMBR.64.2.412-434.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Loenen WA, Dryden DT, Raleigh EA, Wilson GG. 2014. Type I restriction enzymes and their relatives. Nucleic Acids Res. 42:20–44. doi: 10.1093/nar/gku456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Linhartová I, Bumba L, Mašín J, Basler M, Osička R, Kamanová J, Procházková K, Adkins I, Hejnová-Holubová J, Sadílková L, Morová J, Sebo P. 2010. RTX proteins: a highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 34:1076–1112. doi: 10.1111/j.1574-6976.2010.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moses EK, Good RT, Sinistaj M, Billington SJ, Langford CJ, Rood JI. 1995. A multiple site-specific DNA-inversion model for the control of Omp1 phase and antigenic variation in Dichelobacter nodosus. Mol. Microbiol. 17:183–196. doi: 10.1111/j.1365-2958.1995.mmi_17010183.x. [DOI] [PubMed] [Google Scholar]
- 35. Calvo-Bado LA, Green LE, Medley GF, Ul-Hassan A, Grogono-Thomas R, Buller N, Kaler J, Russell CL, Kennan RM, Rood JI, Wellington EM. 2011. Detection and diversity of a putative novel heterogeneous polymorphic proline-glycine repeat (Pgr) protein in the footrot pathogen Dichelobacter nodosus. Vet. Microbiol. 147:358–366. doi: 10.1016/j.vetmic.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 36. Claxton PD. 1989. Antigenic classification of Bacteroides nodosus, p 155–166. In Egerton JR, Yong WK, Riffkin GG. (ed), Foot rot and foot abscess of ruminants. CRC Press, Boca Raton, FL. [Google Scholar]
- 37. Ghimire SC, Egerton JR, Dhungyel OP, Joshi HD. 1998. Identification and characterisation of serogroup M among Nepalese isolates of Dichelobacter nodosus, the transmitting agent of footrot in small ruminants. Vet. Microbiol. 62:217–233. doi: 10.1016/S0378-1135(98)00206-5. [DOI] [PubMed] [Google Scholar]
- 38. Hobbs M, Dalrymple BP, Cox PT, Livingstone SP, Delaney SF, Mattick JS. 1991. Organization of the fimbrial gene region of Bacteroides nodosus: class I and class II strains. Mol. Microbiol. 5:543–560. doi: 10.1111/j.1365-2958.1991.tb00726.x. [DOI] [PubMed] [Google Scholar]
- 39. Han X, Kennan RM, Steer DL, Smith AI, Whisstock JC, Rood JI. 2012. The AprV5 subtilase is required for the optimal processing of all three extracellular serine proteases from Dichelobacter nodosus. PLoS One 7:e47932. doi: 10.1371/journal.pone.0047932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong W, Wijeyewickrema LC, Kennan RM, Reeve SB, Steer DL, Reboul C, Smith AI, Pike RN, Rood JI, Whisstock JC, Porter CJ. 2011. S1 pocket of a bacterially derived subtilisin-like protease underpins effective tissue destruction. J. Biol. Chem. 286:42180–42187. doi: 10.1074/jbc.M111.298711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dhungyel OP, Whittington RJ, Egerton JR. 2002. Serogroup specific single and multiplex PCR with pre-enrichment culture and immuno-magnetic bead capture for identifying strains of D. nodosus in sheep with footrot prior to vaccination. Mol. Cell. Probes 16:285–296. doi: 10.1006/mcpr.2002.0427. [DOI] [PubMed] [Google Scholar]
- 42. Dhungyel O, Schiller N, Eppleston J, Lehmann D, Nilon P, Ewers A, Whittington R. 2013. Outbreak-specific monovalent/bivalent vaccination to control and eradicate virulent ovine footrot. Vaccine 31:1701–1706. doi: 10.1016/j.vaccine.2013.01.043. [DOI] [PubMed] [Google Scholar]
- 43. Kennan RM, Billington SJ, Rood JI. 1998. Electroporation-mediated transformation of the ovine footrot pathogen Dichelobacter nodosus. FEMS Microbiol. Lett. 169:383–389. doi: 10.1111/j.1574-6968.1998.tb13344.x. [DOI] [PubMed] [Google Scholar]
- 44. Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. 2009. SHRiMP: accurate mapping of short color-space reads. PLoS Comput. Biol. 5:e1000386. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Network diagram inferred using the same data set as for Fig. 2. Taxon labels show the presumptive disease phenotype of the isolate. Isolates for which no presumptive disease phenotype was available are highlighted in gray. The two Swedish isolates that have a presumptive disease phenotype that is not consistent with an association between clade I (virulent isolates) and clade II (benign isolates) are highlighted in pink. The Bhutan isolate is highlighted in blue and was found to be unique among the isolates because of its aprB2 genotype and clade I grouping. Download Figure S1, PDF file, 0.2 MB (197.7KB, pdf) .
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Summary of SNPs present in the intact AprV2/B2 genes found in the 103 isolates in the study. Table S1, PDF file, 0.3 MB. (303.1KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Genes containing SNPs that correlate best (top 1%) with the clade I/clade II division. Table S2, PDF file, 0.1 MB. (54.3KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Genes associated with each of the variable regions. Table S3, PDF file, 0.1 MB. (180.1KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Phylogenetic tree showing the relationship between the fimA and fimZ genes. The data include sequences from the 103 isolates sequenced in this study (indicated by a colored circle beside the taxon label) and 78 additional D. nodosus fimA or fimZ genes available in GenBank (indicated by a colored square beside the taxon label). Accession numbers are presented in Table S7. Serogroups are color coded as follows: serogroup A (gray), serogroup B (red), serogroup C (light green), serogroup D (dark green), serogroup E (yellow), serogroup F (blue), serogroup G (pink), serogroup H (brown), serogroup I (light gray), serogroup M (dark blue), and fimZ sequence taxa (light blue). The multiple-sequence alignment used to infer relationships was trimmed to exclude parts of the 5′ and 3′ coding regions. The evolutionary history was inferred using the minimum evolution (ME) method (A. Rzhetsky and M. Nei, J. Mol. Evol. 35:367–375, 1992, doi:10.1007/BF00161174). The optimal tree with the sum of branch lengths equal to 4.09 is shown. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the maximum composite likelihood method (K. Tamura, M. Nei, and S. Kumar S, Proc. Natl. Acad. Sci. U. S. A. 101:11030–11035, 2004, doi:10.1073/pnas.0404206101), and units are numbers of base substitutions per site. The ME tree was searched using the close-neighbor-interchange algorithm (M. Nei and S. Kumar, Molecular Evolution and Phylogenetics, 2000) at a search level of 1. The neighbor-joining algorithm was used to generate the initial tree. The analysis involved 188 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 288 positions in the final data set. Evolutionary analyses were conducted in MEGA6 (K. Tamura, G. Stecher, D. Peterson, A. Filipski, and S. Kumar, Mol. Biol. Evol. 30:2725–2729, 2013, doi:10.1093/molbev/mst197). Download Figure S2, PDF file, 0.1 MB (112.3KB, pdf) .
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Fimbrial biogenesis genes. Table S4, PDF file, 0.1 MB. (118.8KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Details of individual isolates. Table S5, PDF file, 0.2 MB. (260.4KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Phylogenetic trees showing the relationship between each of the three protease genes: aprV2 (DNO_1167) (A), bprV (DNO_0605) (B), and aprV5 (DNO_0603) (C). The relationships shown were inferred using the minimum evolution method as implemented in MEGA6 (K. Tamura, G. Stecher, D. Peterson, A. Filipski, and S. Kumar, Mol. Biol. Evol. 30:2725–2729, 2013, doi:10.1093/molbev/mst197), and support for nodes is indicated by percentages of the 500 bootstrap replicates that contained the particular node. Taxa are labeled with the name of the isolate, the country of isolation, the serogroup, and the AprV2/AprB2 type. Color coding of taxa are as follows: blue for the AprB2-R92 type (correlating with clade II) and red for the AprV2-Y92 type (correlating with clade I). Download Figure S3, PDF file, 0.2 MB (172.3KB, pdf) .
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Details of genome sequencing and analysis data. Download Corrected Table S6, DOCX file, 40 KB (39.1KB, docx) .
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Details of genome sequencing and analysis data. Table S6, PDF file, 0.2 MB. (260.7KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
fimA or fimZ sequences obtained from GenBank. Table S7, PDF file, 0.1 MB. (134.3KB, pdf)
Copyright © 2014 Kennan et al.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported license, which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original author and source are credited.