

Abstract
The pharmacogenomic effects of a corticosteroid (CS) were assessed in rat skeletal muscle using microarrays. Adrenalectomized (ADX) rats were treated with methylprednisolone (MPL) by either 50 mg/kg intravenous injection or 7-day 0.3 mg/kg/h infusion through subcutaneously implanted pumps. RNAs extracted from individual rat muscles were hybridized to Affymetrix Rat Genome Genechips. Data mining yielded 653 and 2316 CS-responsive probe sets following MPL bolus and infusion treatments. Of these, 196 genes were controlled by MPL under both dosing conditions. Cluster analysis revealed that 124 probe sets exhibited three typical expression dynamic profiles following acute dosing. Cluster A consisted of up-regulated probe sets which were grouped into five subclusters each exhibiting unique temporal patterns during the infusion. Cluster B comprised down-regulated probe sets which were divided into two subclusters with distinct dynamics during the infusion. Cluster C probe sets exhibited delayed down-regulation under both bolus and infusion conditions. Among those, 104 probe sets were further grouped into subclusters based on their profiles following chronic MPL dosing. Several mathematical models were proposed and adequately captured the temporal patterns for each subcluster. Multiple types of dosing regimens are needed to resolve common determinants of gene regulation as chronic exposure results in unexpected differences in gene expression compared to acute dosing. Pharmacokinetic/pharmacodynamic (PK/PD) modeling provides a quantitative tool for elucidating the complexities of CS pharmacogenomics in skeletal muscle.
Keywords: Microarray studies, pharmacokinetics, pharmacodynamics, mathematical models, computational biology
Introduction
Corticosteroids (CS) are used extensively in clinical therapy for treatment of various autoimmune diseases, skin disorders, persistent asthma, and inflammatory diseases.1 Chronic treatment with this class of drug may lead to broad and severe side effects including hyperglycemia, dyslipidemia, arteriosclerosis, muscle wasting, and osteoporosis.2 Understanding of the diverse pharmacological effects and the underlying molecular mechanisms for their adverse effects is incomplete at this time.
There are two major adverse effects of CS in skeletal muscle: insulin resistance and muscle atrophy. Corticosteroids affect the transcription and activity of many proteins involved in insulin signaling, such as insulin receptor substrate 1.3,4 A number of enzymes associated with glucose or fatty acid metabolism are also regulated by CS.5–7 The changes of these enzymes represent a pattern of switching off glucose metabolism and triggering energy consumption using fatty acids instead of carbohydrates. Following CS treatment, muscle protein synthesis is decreased while enzymes involved in muscle protein degradation such as the ubiquitin–proteasome proteolytic system are increased, leading to net muscle protein and mass loss.8 In addition, many transcription factors are also regulated by CS.9 It is likely that, besides the direct effects by CS itself, the alterations of these transcription factors elicit further and more extensive regulatory effects on gene transcription and modify the pharmacological actions of the drugs. This evidence suggests that CS elicit diverse functions through various connected pathways.
High-throughput microarrays have been employed in our laboratory to simultaneously examine many biological markers influenced by CS treatment. This approach was used to assess CS pharmacogenomics in rat liver which revealed six major patterns of gene regulation following 50 mg/kg MPL treatment.10 In the present paper, we expand the study and identify the probe sets that are responsive to both acute and infusion MPL treatments in skeletal muscle. Integrated PK/PD/pharmacogenomic (PG) modeling was carried out to explore and capture the pharmacogenomic profiles common to both dosing regimens.
Materials and Methods
Experimental Procedures
Gastrocnemius muscles were obtained from animal studies previously performed in our laboratory.11,12 In brief, male adrenalectomized (ADX) Wistar rats (Harlan Sprague–Dawley Inc., Indianapolis, IN) were used in both acute and chronic studies. Endogenous plasma corticosterone concentrations in ADX Wistar rats are negligible.13 Thus, complexities from regulatory effects of endogenous corticosterone and its circadian rhythm on gene expression were removed. In the acute study, a single intravenous bolus of 50 mg/kg of MPL (Solu-Medrol, Pharmacia-Upjohn Company, MI) was given to 47 animals. In the 7-day infusion study, Alzet osmotic pumps (model 2001, flow rate 1 μL/h; Alza, Palo Alto, CA) were subcutaneously implanted in rats between the shoulder blades. MPL was reconstituted in the supplied diluent and given to 40 rats at a fixed rate of 0.3 mg/kg/h. Rats were sacrificed by aortic exsanguination at various time points between 0 and 72 h in the acute study and throughout the 7-day period in the infusion study. Four vehicle-treated rats were designated as controls for both studies. Gastrocnemius muscles were rapidly excised, quickly frozen in liquid nitrogen, and stored at −80 °C.
Microarrays
Frozen gastrocnemius muscles were ground into powder. Total RNA was obtained by Trizol–chloroform based extraction and used to prepare biotinylated cRNA target according to manufacturer protocols. The targets from the acute study were hybridized to 51 individual Affymetrix GeneChip RG_U34A (Affymetrix, Santa Clara, CA), which was the first-generation rat genechip and contained 8799 probe sets. The target cRNAs from the chronic study were hybridized to 44 individual Affymetrix GeneChip RAE230A (Affymetrix, Santa Clara, CA), a second-generation rat genechip which contained 15967 probe sets. Cross-validation of these two genechips has been performed by Affymetrix.14 The entire data set has been submitted to the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus database (GSE490 and GSE5101) and is also available online at http://pepr.cnmcresearch.org/.15
Data Mining
The microarray data were examined using a filtering method described by Almon et al.16–18 The initial data acquisition software (Affymetrix Microarray Suite 5.0 (MAS 5.0)) generates a “call” of present (P), absent (A), and marginal (M) for each probe set based on comparison of matched and mismatched pairs, The results were then transferred to GeneSpring 7.0 (Silicon Genetics, Redwood City, CA) for further analysis. The data for each probe set at various time points were normalized by the mean of the four control values (normalized intensity). A series of three filters was applied to the data, which excluded those probe sets that are not expressed in the tissue, not regulated by the drug, and did not pass quality-control criteria. The selection criteria for the acute study was described previously.16 Due to fewer time points, the corresponding criteria in the chronic study were modified. A probe set had to have a call of at least 3 “P” out of the total 44 chips to be considered expressed in the tissue. Additionally, probe sets are considered regulated by the drug if at least 3 out of 11 time points have a normalized intensity higher than 1.5 or lower than 0.66. Besides the quality control filter for the control data, the probe set has to have at least 5 additional time points with replicates having a CV less than 50%. Genes regulated jointly in both acute and chronic MPL studies were selected and grouped as a new data set for cluster analysis.
Cluster Analysis
The probe sets selected were subjected to quality threshold (QT) clustering based on their expression profiles following acute MPL treatment. A cluster had to display a minimum of 10 probe sets to be identified as a cluster. The probe sets in each cluster were then translated into the chronic data set and subjected to QT clustering based on their expression changes during the 7-day treatment. These probe sets were then grouped into several subclusters. A subcluster had a minimum number of 5 probe sets. Pearson correlation with a correlation coefficient of at least 0.4 in each cluster was used as the similarity criterion for cluster analysis.
Theoretical Basis
Pharmacokinetics and Receptor Dynamics
The pharmacokinetics of plasma MPL was described previously.11,12,18 The kinetics of MPL was described by a two-compartment model with a zero-order input k0 into the central compartment.
| (1) |
| (2) |
where
where Ap and At are the drug amounts in the plasma and tissue compartments and CL, Vp, k12, and k21 are clearance, central compartment volume of distribution, and distribution rate constants. For the acute study, k0 was fixed to 0. All parameters were fixed according to our previous values.11,12
Receptor dynamics in rat skeletal muscle was previously described by a mechanism-based receptor-gene mediated PD model (Figure 1).11 The equations describing the glucocorticoid receptor dynamics are as follows:
| (3) |
| (4) |
| (5) |
| (6) |
where symbols represent cytosolic free glucocorticoid receptor density (GR), GR mRNA (GRmRNA), cytosolic GR–drug complex (DR), GR–drug complex in nucleus (DR(N)), zero-order GR mRNA synthesis rate constant (ks_Rm), and first-order rate constants for GR mRNA degradation (kd_Rm), GR synthesis (ks_R), and GR degradation (kd_R). Other rate constants include second-order association rate constant of GR and drug (kon), translocation rate constant of DR from cytosol to nucleus (kT), the overall turnover rate of DR(N) (kre), and the fraction of GR that is recycled from nucleus to cytosol (Rf). The IC50_Rm is the concentration of DR(N) at which the synthesis rate of GR mRNA is reduced to 50% of its baseline level. Assuming steady-state before dosing of MPL, the following equations derive from eqs 3 and 4.
Figure 1.
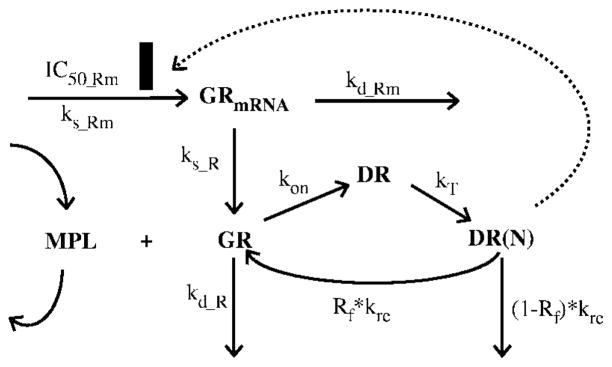
Fifth-generation CS pharmacodynamic model of receptor-gene mediated effects. Symbols and differential equations are defined in eqs 3–8. The solid rectangle represents inhibition of GR mRNA transcription by DR(N).
| (7) |
| (8) |
where GRmRNA0 and GR0 are the baseline levels of GR mRNA and free cytosolic GR at time zero. Receptor mRNA and cytosolic receptor density following acute MPL dosing were obtained from our previous studies and refitted.11 Parameters were used to simulate receptor profiles following chronic MPL dosing and fixed in modeling of genomic data.
Pharmacogenomic Models
Binding of CS with the receptor leads to activation and translocation of the receptor into the nucleus.2 Activated glucocorticoid receptor binds to recognition sites in the promoter region of target genes and activates or inhibits target gene transcription. It has been found that CS may modulate many transcription factors. The altered expression of these transcription factors may further affect expression of other genes. Additionally, other mediators may also affect gene transcription. In the present report, these intermediate transcription factors and other biological factors are referred to as biosignals. Thus, a target gene may be regulated either directly by DR(N), or secondarily through a biosignal, or by both. Various pharmacogenomic models are proposed to describe diverse gene expression profiles following both acute and chronic treatments. In all of these models, DR(N) is assumed to be the driving force influencing the dynamics of gene expression. When drug is absent, the expression of genes (mRNA) is described by a turnover model with a zero-order production rate (ks) and a first-order degradation rate (kd):
| (9) |
Since endogenous CS in ADX rats is negligible, steady-state gene expression was assumed before drug administration.13 The following baseline condition can be derived:
| (10) |
where mRNA0 is the baseline target gene expression level. It was fixed to 1 since all data were normalized as ratios to the baseline. Figure 2 shows the five models proposed to explain our gene expression profiles.
Figure 2.
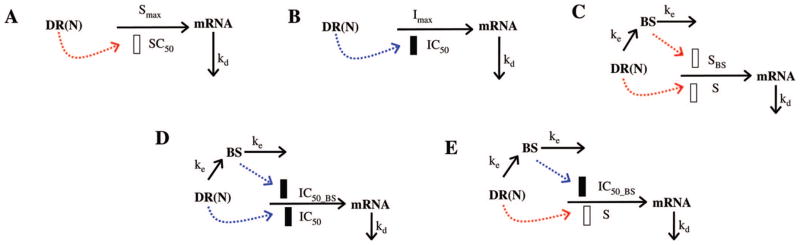
Diverse pharmacodynamic models for CS effects on gene expression. Symbols and differential equations are defined in eqs 9–16. The solid and open rectangles represent inhibition or stimulation of gene transcription by DR(N) or the biosignal (BS).
Model A assumes that DR(N) has a direct stimulatory effect on gene transcription. The differential equation is either:
| (11a) |
| (11b) |
where S defines the stimulatory efficiency of DR(N) on target gene transcription. The effect of DR(N) on some genes is described by a capacity equation where Smax represents the maximum stimulation effect and SC50 is the concentration of DR(N) that leads to half of the maximum stimulation effect. The baselines of mRNA in acute (BmRNA_A) and chronic (BmRNA_C) studies were either fixed to 1 or estimated during modeling.
Model B assumes that DR(N) has a inhibitory effect on gene transcription. The target gene expression is modeled as
| (12) |
where Imax represents the maximum inhibitory effect and IC50 is the concentration of DR(N) that leads to half of the maximum inhibitory effect.
Genes described by model C were characterized by two joint stimulatory effects from DR(N), one directly and the other indirectly through a hypothetical biosignal. In the proposed model, the biosignal (BS) represents DR(N)-induced changes of a transcription factor or other mediator from its original baseline. Equations reflecting these joint effects are:
| (13) |
| (14) |
where ke defines the production of the biosignal, SBS represents the stimulatory efficiency of the biosignal on target gene expression, and γ is a power coefficient amplifying the effect of BS. The initial condition of eq 13 was fixed to 0.
Model D proposes that dual inhibitory pathways, one directly from DR(N) and the other through BS, affect gene expression at the same time. Expression profiles of these genes are described by
| (15) |
where BS is described by eq 13 and IC50_BS is the concentration of DR(N) when the inhibitory effect of DR(N) reaches half of its maximum.
Genes described by model E are controlled by an inductive effect from DR(N) and an inhibitory effect from a biosignal whose own production is affect by DR(N). The equation representing these joint effects is
| (16) |
where the BS is described by eq 13.
Data Analysis
Data taken from individual animals were pooled and averaged at various time points.11 Simultaneously modeling of gene expression versus time profiles of the two treatments was performed for all genes. Since two different rat genome microarray chips RG_U34A and RAE230A were used, a scaling factor (SF) was incorporated in the modeling. The SF accounts for the different sensitivity of two probe sets to the controlling factors.20 The output functions of mRNA in acute (YA(t)) and chronic (YC(t)) studies were
| (17) |
| (18) |
where mRNAA(t) and mRNAC(t) define mRNA expression at various times.
The ADAPT II program21 with the maximum likelihood method was used for all modeling procedures. A linear variance model was used in all fittings
| (19) |
where Vi is the variance of the response at the ith time point, ti is the actual time at the ith time point, and Y(ti) represents the predicted response at time ti from the model. Variance parameters σ1 and σ2 were estimated together with system parameters during fittings. The goodness-of-fit criteria included visual inspection of the fitted curves, sum of squared residuals, Akaike information criterion, Schwartz criterion, and coefficients of variation (CV) of the estimated parameters.
Results
The pharmacokinetics of MPL for both studies were simulated using previously estimated parameters.11,12 The simulated curves are shown in Figure 3 reflecting biexponential disposition (acute dose) and steady-state (infusion study). Receptor dynamics following both studies are shown in Figure 3. The estimated parameters are listed in Table 1.
Figure 3.
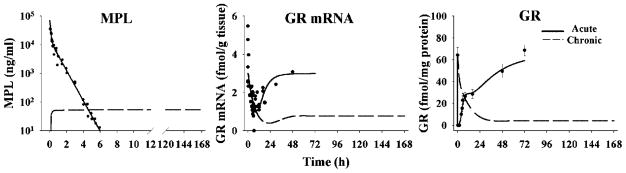
MPL pharmacokinetics and receptor dynamics following 50 mg/kg MPL i.v. bolus administration and 0.3 mg/kg/h 7-day infusion. Kinetic and dynamic profiles were simulated using parameters obtained from previous studies.11,12 Original data (solid dots) were obtained from Sun et al.11 and Ramakrishnan et al.12
Table 1.
Fixed and Estimated Receptor Dynamic Parameters
| parameter (unit) | definition | value | CV (%) |
|---|---|---|---|
| ks_Rm (fmol/g muscle/h) | GR mRNA synthesis rate constant | 0.416 | —a |
| IC50_Rm (fmol/mg protein) | inhibition constant | 0.911 | 126 |
| kd_Rm (1/h) | GR mRNA degradation rate constant | 0.139 | 18 |
| kon (1/nmol/h) | association rate constant | 0.00269 | 73 |
| kre (l/h) | DR(N) overall loss rate constant | 0.618 | 50 |
| Rf | recycling fraction | 0.720 | 12 |
| kd_R (l/h) | GR degradation rate constant | 0.0356 | 27 |
| kT (l/h) | translocation rate constant | 90 | |
| ks_R (fmol/mg/(fmol mRNA/g)/h) | GR synthesis rate constant | 0.777 | a |
| GRmRNA0 (fmol/g muscle) | baseline GR mRNA | 2.99 | 8 |
| GR0 (fmol/mg protein) | baseline GR | 65.3 | 3 |
Secondary parameters.
Data mining yielded 653 probe sets whose transcription is modified by MPL following the intravenous dose and 2316 probe sets during the infusion. Comparison of these probe sets resulted in 196 genes which are regulated by MPL following both conditions. These genes are represented by 241 probe sets in the acute data set and 219 probe sets in the chronic data set since some genes have multiple probe sets.
The purpose of cluster analysis is to group selected genes based on similar expression profiles so that we can presume similarities in regulatory mechanisms and use one PD model to capture all profiles grouped together. QT clustering was used to ensure that probe sets in each cluster passed the minimum correlation threshold. It generated a set of non-overlapping clusters that met both the similarity threshold and the requirement for minimum number of probe sets. Not all probe sets were grouped into clusters. Those probe sets with uncommon dynamic patterns such as biphasic changes and opposite regulatory directions following the two dosing regimens were found but removed in the clustering process. This method identified probe sets with more typical dynamic profiles and made it possible for later PK/PD analysis. Thus, 104 out of 196 genes passed the similarity criteria following both acute and infusion dosing and were grouped into three clusters and eight subclusters.
Clustering based on the acute study yielded three clusters with one up-regulated cluster and two down-regulated clusters. Cluster A contained 75 probe sets, all of which showed increased mRNA expression. The peaks occurred at about 6 h after dosing. These probe sets exhibited similar dynamic profiles as tyrosine aminotransferase (TAT) in liver and glutamine synthetase (GS) in muscle, both of which are prototype CS-stimulated genes.11,12,19,22 Cluster B contained 35 probe sets. All probe sets in this cluster were down-regulated. Their troughs occurred at around 6 h and returned to baseline by 18 h. Probe sets in the last cluster (Cluster C) also exhibited reduced expression after dosing. However, these 14 probe sets had slower down-regulation, later troughs (around 12 h), and did not return to baseline until 30 h. Probe sets that did not pass the similarity criteria were excluded from clustering.
For cluster A, 64 probe sets were further grouped into five subclusters according to their expression following drug infusion. Three prototype genes were selected for each subcluster and their original data and best-fitted curves are shown in Figure 4. The accession numbers in the Affymetrix chips are shown for each gene. The first subcluster A1 had 15 probe sets. These probe sets were stimulated and reached peaks between 13 and 18 h. After peaking, they gradually decreased to steady-states above the baselines. Model A with eq 11a adequately captured the acute and chronic dynamics of cluster A1 probe sets simultaneously. Table 2 shows the estimated parameters and precision for selected genes. The estimated scaling factors are around 1 as expected. Degradation rate constants kd are between 0.05 and 0.08 h−1. The stimulatory coefficient S (0.1 to 0.3 (fmol/mg protein)−1) suggests moderate sensitivity of the target gene to DR(N). Most parameters were estimated with good precision with CVs less than 30%. Baselines were either estimated or fixed during the fitting process.
Figure 4.

Time courses of selected genes in cluster A and their fittings in ADX rat muscle following 50 mg/kg MPL i.v. bolus and 0.3 mg/kg/h infusion treatments. Symbols (solid dots) are the mean gene array data and bars are standard deviations. Solid lines are simultaneously fitted curves for acute and chronic data.
Table 2.
Pharmacodynamic Parameters for Selected Cluster A Genesa
| gene name | accession no.
|
SF
|
kd (h−11)
|
S ((fmol/mg protein)−11) |
Smax
|
SC50 (fmol/mg protein) |
SBS ((fmol/mg protein)−11) |
IC50_BS (fmol/mg protein) |
ke (h−11) |
γ
|
BmRNA_A
|
BmRNA_C
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| acute/chronic | scaling factor |
mRNA degradation rate constant |
stimulatory coefficient |
maximum stimulatory effect |
50% maximum stimulation concentration |
maximum stimulatory effect by biosignal |
50% maximum stimulation concentration |
transit rate constant |
power coefficient |
baseline in acute study |
baseline in chronic study |
|
| Cluster A1 | ||||||||||||
| myogenic factor 6 | M27151_at/1387181_at | 1.634 (23) | 0.0539 (23) | 0.1002 (32) | 0.756 (10) | 1 | ||||||
| phosphatidylserine-specific phospholipase A1 | D88666_at/1370445_at | 0.518 (18) | 0.0838 (19) | 0.3269 (26) | 0.713 (13) | 1 | ||||||
| poly(rC) binding protein 4 | rc_AA891314_at/1371915_at | 1.327 (18) | 0.0643 (20) | 0.09546 (7) | 0.850 (7) | 1 | ||||||
| Cluster A2 | ||||||||||||
| placental growth factor | L40030_at/1368918_at | 0.526 (23) | 0.249 (51) | 2.749 (44) | 2.605 (147) | 0.863 (11) | 1 | |||||
| aryl hydrocarbon receptor nuclear translocator | U08986_s_at/1369244_at | 0.845 (18) | 0.296 (56) | 0.740 (30) | 1.349 (123) | 1 | 1 | |||||
| DnaJ (Hsp40) homologue B5 | rc_AA891542_at/1372144_at | 1.438 (22) | 0.224 (52) | 0.785 (37) | 0.982 (111) | 0.988 (6) | 1 | |||||
| Cluster A3 | ||||||||||||
| multimerin | rc_AI639113_at/1392053_at | 0.809 (8) | 0.065 (93) | 0.0941 (104) | 0.0217 (272) | 0.0155 (19) | 2.85 (66) | 1 | 1 | |||
| phenol sulfotransferase 1A | L19998_at/1370019_at | 1.247 (11) | 0.124 (58) | 0.1898 (57) | 0.0401 (143) | 0.0119 (12) | 3.57 (33) | 0.710 (12) | 1 | |||
| CYP27A1 | M38566mRNA_s_at/1387914_at | 1.576 (13) | 0.113 (64) | 0.0426 (53) | 0.0168 (32) | 0.0144 (10) | 3 | 0.842 (5) | 1 | |||
| Cluster A4 | ||||||||||||
| SH3 domain binding protein CR16 | U25281_at/1370648_a_at | 1.413 (12) | 0.2215 (32) | 0.176 (26) | 3.20 (38) | 0.0088 (47) | 3.89 (166) | 1 | 1 | |||
| peroxisome proliferator activated receptor δ | rc_AI230294_at/1374914_at | 1.01 (17) | 0.0334 (43) | 0.235 (62) | 6.27 (39) | 0.0568 (45) | 2.82 (83) | 1 | 1 | |||
| EST | rc_AA866383_at/1383188_at | 1.117 (37) | 0.0688 (61) | 0.084 (70) | 9.89 (68) | 0.0642 (74) | 3.54 (161) | 0.9173 (7) | 1 | |||
| Cluster A5 | ||||||||||||
| serine/threonine-protein kinase pim-3 | AF086624_s_at/1367725_at | 0.837 (37) | 0.306 (31) | 0.0525 (20) | 0.0273 (76) | 0.00708 (16) | 5 | 0.862 (9) | 0.923 (17) | |||
| endothelin converting enzyme 1 | D29683_at/1367801_at | 0.732 (38) | 0.0704 (69) | 0.0583 (66) | 0.0057 (65) | 0.0106 (14) | 5 | 0.870 (7) | 0.843 (12) | |||
| prostaglandin-endoperoxide synthase 1 | U03388_s_at/1368259_at | 1.499 (37) | 0.0505 (83) | 0.0490 (86) | 0.0034 (77) | 0.0128 (17) | 5 | 1.037 (10) | 0.856 (10) | |||
Values are estimates (CV %).
Cluster A2 contained 8 probe sets which also exhibited enhanced expression during drug infusion. These probe sets reached peaks between 6 and 10 h and then maintained steady-states almost at the peak values. They did not show the late apparent reduction of gene expression observed in Cluster A1 profiles. The dynamics of these probe sets was well captured by model A with eq 11b with both Smax and SC50 estimated. Simulated DR(N) concentrations during drug infusion are above 20, which are much higher than the estimated SC50 (1–3 fmol/mg protein). The fittings suggest that target gene transcription is saturated before 24 h during drug infusion. All parameters except SC50 have reasonable precision. The high CVs associated with estimation of SC50 might be due to insufficient data.
There were 23 probe sets identified for cluster A3, which comprised the largest subcluster in cluster A. This group of probe sets is distinct from clusters A1 and A2 in that they did not show steady-states during drug infusion, although MPL was maintained at a constant level after 6 h. Following peaks at around 30 h, these probe sets exhibited very slow return phases. Compared to cluster A1 probe sets, this group displayed a much longer time delay between acute and chronic peaks. Model C was applied in the simultaneous fitting of both acute and chronic profiles. The model adequately captured the MPL stimulated gene expression changes. Examination of the estimated parameters suggests that most probe sets have a moderate sensitivity to DR(N) stimulation but a relatively weak stimulation by the biosignal. The power coefficient was around 3. Relatively large CVs are seen for several parameters, which might be due to overparameterization. Model fittings of three representative genes are shown in Figure 4. This subcluster also includes glutamine synthetase, interleukin-6 receptor, cystein dioxygenase, and eukaryotic translation initiation factor 4E binding protein 1.
Similar to cluster A1, all 10 probe sets in cluster A4 also exhibited early up-regulation and returning phases in the later period during drug infusion. However, model A could not predict their profiles. Comparison of these two subclusters reveals several unique properties which distinguishes cluster A4 as a separate group. Probe sets in this cluster had relatively sharper stimulation and reached peaks between 6 and 10 h. Following their peaks, most probe set expression profiles fell with an abrupt slope and returned to a low value which was maintained thereafter. A number of probe sets completely returned to or even fell below the baseline, which cannot be explained by model A. A few probe sets presented slight trends of increased expression at late times. Model E was applied to model the probe set dynamics. Addition of a secondary inhibitory effect through the biosignal could explain the sharp returning phases and steady-states reaching baseline or below baseline. The model predicts a slight rising phase at later times for all cluster A4 probe sets. However, this is not seen for some probe sets due to the limited sampling period and very small value of ke. Most parameters (shown in Table 2) were estimated with reasonable precision. Model estimations suggest DR(N) has a moderate efficiency in stimulation of gene expression while these probe sets have relatively high sensitivities to the presumed biosignal.
The last subcluster in cluster A distinguishes itself from other probe sets by exhibiting very slow rising phases and a long time delay between acute and chronic peaks. Containing only six probe sets, this cluster displayed high variability in expression. Most of these probe sets reached peaks between 48 to 96 h after dosing. A few did not return to baseline while the rest only returned at the end of the study. Model C was applied to probe sets in cluster A5. Despite high data variability, the model was able to characterize the major trends of probe set dynamic profiles following the dosing regimens. Our data suggest that similar receptor dynamics can yield dramatically distinct probe set expression profiles in acute and chronic studies.
In cluster B, 30 probe sets were further grouped into two subclusters based on their dynamics following drug infusion. All probe sets in cluster B1 showed reduced expression throughout the 7-day infusion period. They reached troughs at around 10 h and remained at those levels thereafter. Model B was able to characterize their joint dynamics. Four selected probe sets are shown in Figure 5 and their estimated parameters are listed in Table 3. Estimated IC50 values were relatively small, which explained the prolonged transcriptional inhibition even after DR(N) was reduced to low levels during the late period of drug infusion. Parameters were estimated with good precision (CV less than 40%) except IC50, which has a CV higher than 50%. Three of these selected probe sets represent one gene, myristoylated alanine-rich C-kinase substrate. Modeling yielded similar dynamic related parameters except for SF, which may be due to different probe set sensitivities and helps justify the presence of this factor in comparing different genechips. All baselines were fixed to 1 for this analysis.
Figure 5.

Time courses of selected genes in cluster B and their fittings in ADX rat muscle following bolus and infusion MPL treatments. Symbols and lines are as defined in Figure 4.
Table 3.
Pharmacodynamic Parameters for Selected Cluster B and C Genesa
| gene name | accession no.
|
SF
|
kd h−11
|
Imax
|
IC50 fmol/ mg protein |
IC50_BS fmol/ mg protein |
ke h−11
|
γ
|
BmRNA_A
|
BmRNA_C
|
|---|---|---|---|---|---|---|---|---|---|---|
| acute/chronic | scaling factor |
mRNA degradation rate constant |
maximum inhibitory effect |
50% maximum stimulation concentration |
50% maximum stimulation concentration |
transit rate constant |
power coefficient |
baseline in acute study |
baseline in chronic study |
|
| Cluster B1 | ||||||||||
| acyl-CoA synthetase 3 | D30666_at/1368177_at | 1.117 (14) | 0.479 (36) | 0.500 (11) | 1.205 (53) | 1 | 1 | |||
| myristoylated alanine-rich C-kinase substrate | rc_AA955167_s_at/1370948_a_at | 1.192 (17) | 0.233 (31) | 0.679 (17) | 0.724 (71) | 1 | 1 | |||
| myristoylated alanine-rich C-kinase substrate | rc_AA955167_s_at/1370949_at | 1.925 (13) | 0.311 (27) | 0.603 (12) | 0.486 (60) | 1 | 1 | |||
| myristoylated alanine-rich C-kinase substrate | rc_AA955167_s_at/1373432_at | 1.254 (15) | 0.257 (29) | 0.650 (14) | 0.650 (63) | 1 | 1 | |||
| Cluster B2 | ||||||||||
| platelet factor 4 | rc_AI169104_at/1371250_at | 1.434 (17) | 0.553 (31) | 1 | 40.31 (20) | 1.472 (29) | 1 | 1 | ||
| tropomyosin 1R | M60666_s_at/1379936_at | 2.024 (17) | 0.406 (29) | 1 | 28.11 (27) | 1.112 (25) | 1 | 1 | ||
| tropomyosin 1R | M60666_s_at/1390471_at | 1.249 (16) | 0.401 (27) | 1 | 27.71 (26) | 1.084 (24) | 1 | 1 | ||
| Cluster C | ||||||||||
| mitogen-activated protein kinase 6 | M64301_at/1368273_at | 1.073 (16) | 0.814 (178) | 1 | 174.8 (148) | 5.403 (23) | 0.0206 (21) | 2.46 (37) | 1 | 1 |
| glycine amidinotransferase | U07971_at/1367627_at | 0.503 (54) | 0.118 (48) | 1 | 8.53 (188) | 4.043 (32) | 0.0164 (34) | 5.36 (73) | 1 | 1 |
| procollagen XV | rc_AA800298_at/1388939_at | 1.323 (36) | 0.083 (50) | 1 | 22.02 (235) | 3.762 (14) | 0.0244 (33) | 4.60 (59) | 1.126 (8) | 1.282 (17) |
Values are estimates (CV %).
Probe sets in cluster B2, which also exhibited reduced expression, showed a different dynamic pattern compared with cluster B1. After the first troughs between 6 to 10 h, these probe sets displayed a transient increasing phase until 48 h and then a second decrease thereafter. Model D was applied assuming that the biosignal leads to the second down-regulation after 48 h. The ke value was fixed to 0.001 h−1 due to limited information after 168 h, and γ was fixed to 1. Three selected probe sets are shown in Figure 5, two of which represent a single gene, tropomysin 1α. Estimated parameters suggest relatively low sensitivity to DR(N) inhibition but high sensitivity to the biosignal (Table 3).
One subcluster was found for cluster C containing 12 probe sets. Examination of the profiles reveals prolonged inhibition until 96 h. Compared to cluster B probe sets, the initial decreases of Cluster C probe sets were much slower following both acute and infusion treatments. In the infusion study, the troughs did not occur until 30 h. These probe sets were maintained at a trough for almost 60 h before they gradually returned to baselines. Model D was applied in the data analysis. The model well captured the probe set expression dynamics following both treatments. Figure 6 shows the pharmacodynamic data and the best-fitted curves for three selected genes. Their estimated parameters are listed in Table 3. Over parameterization leads to relatively high CVs for some parameters such as IC50.
Figure 6.

Time courses of selected genes in cluster C and their fittings to model D in ADX rat muscle following bolus and infusion MPL treatments. Symbols and lines are as defined in Figure 4.
Discussion
The main purpose of this report is to utilize pharmacokinetic/pharmacodynamic modeling to assess CS pharmacogenomics. This area has been a scientific interest as enhanced or reduced gene expression has been associated with many CS side effects. Our previous research using microarrays found that almost all gene expression profiles returned to baselines at 72 h after single-dose intravenous injection.10 This is consistent with clinical observations that single-dose or short-term CS treatment is rarely associated with severe side effects. Abnormal glucose, lipid, and protein metabolism and their consequences usually accompany long-term drug therapy.2 In order to study CS pharmacogenomics in a more clinically relevant condition, we performed a 7-day MPL infusion study. The selected dose 0.3 mg/kg/h is relevant to the clinically used doses after adjustment for different pharmacokinetics in humans and rats. Significant muscle mass loss and glucose elevation were associated with this dosing regimen in ADX rats.12,23 The majority of probe sets identified did not return to baselines at the end of infusion. Long-term drug treatment results in sustained gene up- or down-regulation, reaching a new homeostasis which might be the basis for occurrence of adverse effects.
We identified 196 genes which exhibited modified expression during acute and chronic treatments with MPL. These genes comprised about 2% of all probe sets on the Affymetrix rat genome microarrays. Examination of these genes reveals many transcription factors and genes involved in glucose and lipid metabolism, insulin signaling, and muscle degradation. These changes are consistent with the development of two major adverse effects of CS in skeletal muscle, insulin resistance and muscle atrophy. Evaluation of these genes and their dynamics under CS treatment might be beneficial in assessing means for preserving the desired functions while avoiding severe side effects especially in screening for new compounds.
It is believed that most metabolic effects of CS are regulated by its receptor-gene mediated pathway.2 Unbound CS binds with cytosolic free receptors and activates the receptor. Drug-receptor complex is translocated into the nucleus where it dimerizes and binds to glucocorticoid response element (GRE) in the target DNA. This leads to enhancement or inhibition of target gene expression. Besides the direct effects through GREs, many transcription factors are controlled by CS.9 These changes may lead to further regulation of their downstream genes, resulting in secondary effects. Stimulation or suppression of other biosignals may also occur. The CS are known to induce elevated cholesterol in plasma after long-term treatment.24 It has been found that high cholesterol leads to decreased tropomyosin 1α expression at the transcriptional level.25 This effect may be the driving force leading to the secondary inhibition of tropomyosin 1α transcription. Our analysis suggests that more than 50% of clustered probe sets are regulated through processes more complex than direct DR(N) stimulation or inhibition. Pharmacogenomic models including direct regulation (models A and B) and addition of a biosignal (models C–E) are capable of characterizing numerous probe sets regulated by MPL. The BS compartment represents either increased or decreased biosignal concentrations above or below the baselines. In our models, it is described as a positive variable. However, it may also represent decreased expression of a biosignal which has an opposite regulatory effects on gene transcription. A decreased biosignal which stimulates target gene transcription is expressed in the model as an increased biosignal which represses target gene transcription. There is insufficient information to identify the exact biosignal for each gene. Thus, an empirical compartment, BS, was created to represent the potential effects of transcription factors and other intermediate driving forces.
All cluster A probe sets exhibited single stimulation phases following MPL bolus dosing. Their profiles can be described by our fifth-generation model with direct stimulation from DR(N).10 This model would produce a profile with an early rising phase and a later above baseline steady-state following 0.3 mg/kg/h MPL infusion. Besides this expected pattern (clusters A1 and A2), our microarray studies also revealed three unexpected patterns (clusters A3, A4, and A5), which cannot be explained by simple stimulation of DR(N). Similarly, all cluster B probe sets exhibited single inhibitory phases following the acute study. However, the infusion study separated them into two different subclusters with diverse dynamic profiles. These results suggest that similar acute time profiles may not predict comparable expression patterns during continuous infusion. Chronic dosing thus brings additional insights and complexities. Simple extrapolation of acute gene expression dynamics to other dosing regimens is risky without understanding the underlying regulatory mechanisms.
Our studies are limited by the availability of probe sets on the microarray chips. The older chip used in the acute study (RG_U34A) contains fewer probe sets than the chip used in chronic study (RAE230A). Additionally, the sensitivity of the probe sets and our data mining analysis may lead to loss of useful information. There were 653 probe sets identified as regulated following MPL acute treatment. Of these, only 217 passed the filters for regulated probe sets in the infusion study.
Besides regulating gene transcription, CS also exert translation or post-translational regulation affecting protein levels and activities.23,26 The stability and half-life of mRNAs of some pro-inflammatory proteins are regulated by CS.26 A membrane-bound GR has been found to mediate GR-specific nongenomic effects.26 These responses were not evaluated in our studies. Glucocorticoid receptor also regulates gene transcription through interference or crosstalk with other transcription factors, such as NF-κB, AP-1 or members of the STAT family. These activities were not assessed in the current study. However, the indirect transcriptional regulation through biosignals might partially be attributed to GR interaction with these transcription factors. During long-term CS infusion, elevated glucose and lipid also affect gene expression in skeletal muscle and add complexity to CS pharmacogenomics.27–29 However, we could not distinguish responses mediated by this mechanism and thus they were not specifically addressed.
In summary, microarray studies were performed to evaluate genes regulated by CS under both bolus and infusion regimens. There were 196 CS-responsive genes identified as regulated by CS and they revealed diverse dynamic patterns. Generalized pharmacogenomic models have been developed to evaluate drug-induced processes for each typical pattern. This approach provides an essential tool in understanding the global actions of CS and helps to formulate hypotheses for processes underlying changes in expression for individual as well as groups of CS-regulated genes.
Acknowledgments
We acknowledge the technical expertise of Ms. Nancy Pyszczynski for the conduct of animal studies. This work was supported by Grant No. GM24211 from the National Institute of General Medical Sciences, National Institutes of Health, and a research grant from NASA.
References
- 1.Barnes PJ. Corticosteroid effects on cell signalling. Eur Respir J. 2006;27:413–26. doi: 10.1183/09031936.06.00125404. [DOI] [PubMed] [Google Scholar]
- 2.Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96:23–43. doi: 10.1016/s0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 3.Giorgino F, Almahfouz A, Goodyear LJ, Smith RJ. Glucocorticoid regulation of insulin receptor and substrate IRS-1 tyrosine phosphorylation in rat skeletal muscle in vivo. J Clin Invest. 1993;91:2020–30. doi: 10.1172/JCI116424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dupont J, Derouet M, Simon J, Taouis M. Corticosterone alters insulin signaling in chicken muscle and liver at different steps. J Endocrinol. 1999;162:67–76. doi: 10.1677/joe.0.1620067. [DOI] [PubMed] [Google Scholar]
- 5.Lewin TM, Granger DA, Kim JH, Coleman RA. Regulation of mitochondrial sn-glycerol-3-phosphate acyltransferase activity: response to feeding status is unique in various rat tissues and is discordant with protein expression. Arch Biochem Biophys. 2001;396:119–27. doi: 10.1006/abbi.2001.2604. [DOI] [PubMed] [Google Scholar]
- 6.Mooney RA, Senn J, Cameron S, Inamdar N, Boivin LM, Shang Y, Furlanetto RW. Suppressors of cytokine signaling-1 and -6 associate with and inhibit the insulin receptor. A potential mechanism for cytokine-mediated insulin resistance. J Biol Chem. 2001;276:25889–93. doi: 10.1074/jbc.M010579200. [DOI] [PubMed] [Google Scholar]
- 7.Cha BS, Ciaraldi TP, Carter L, Nikoulina SE, Mudaliar S, Mukherjee R, Paterniti JR, Jr, Henry RR. Peroxisome proliferator-activated receptor (PPAR) gamma and retinoid X receptor (RXR) agonists have complementary effects on glucose and lipid metabolism in human skeletal muscle. Diabetologia. 2001;44:444–52. doi: 10.1007/s001250051642. [DOI] [PubMed] [Google Scholar]
- 8.Wing SS, Goldberg AL. Glucocorticoids activate the ATP-ubiquitin-dependent proteolytic system in skeletal muscle during fasting. Am J Physiol. 1993;264:E668–76. doi: 10.1152/ajpendo.1993.264.4.E668. [DOI] [PubMed] [Google Scholar]
- 9.Almon RR, DuBois DC, Jin JY, Jusko WJ. Temporal profiling of the transcriptional basis for the development of corticosteroid-induced insulin resistance in rat muscle. J Endocrinol. 2005;184:219–32. doi: 10.1677/joe.1.05953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin JY, Almon RR, DuBois DC, Jusko WJ. Modeling of corticosteroid pharmacogenomics in rat liver using gene microarrays. J Pharmacol Exp Ther. 2003;307:93–109. doi: 10.1124/jpet.103.053256. [DOI] [PubMed] [Google Scholar]
- 11.Sun YN, McKay LI, DuBois DC, Jusko WJ, Almon RR. Pharmacokinetic/Pharmacodynamic models for corticosteroid receptor down-regulation and glutamine synthetase induction in rat skeletal muscle by a receptor/gene-mediated mechanism. Pharmacol Exp Ther. 1999;288:720–8. [PubMed] [Google Scholar]
- 12.Ramakrishnan R, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ. Pharmacodynamics and pharmacogenomics of methylprednisolone during 7-day infusions in rats. Pharmacol Exp Ther. 2002;300:245–56. doi: 10.1124/jpet.300.1.245. [DOI] [PubMed] [Google Scholar]
- 13.del Mar Grasa M, Serrano M, Fernandez-Lopez JA, Alemany M. Corticosterone inhibits the lipid-mobilizing effects of oleoylestrone in adrenalectomized rats. Endocrinology. 2007;148:4056–63. doi: 10.1210/en.2007-0331. [DOI] [PubMed] [Google Scholar]
- 14.Technical note: array design and performance of the genechip rat expression set 230. http://www.affymetrix.com/support/technical/technotesmain.affx
- 15.Almon RR, Chen J, Snyder G, DuBois DC, Jusko WJ, Hoffman EP. In vivo multi-tissue corticosteroid microarray time series available online at Public Expression Profile Resource (PEPR) Pharmacogenomics. 2003;4:791–9. doi: 10.1517/phgs.4.6.791.22816. [DOI] [PubMed] [Google Scholar]
- 16.Almon RR, DuBois DC, Piel WH, Jusko WJ. The genomic response of skeletal muscle to methylprednisolone using microarrays: tailoring data mining to the structure of the pharmacogenomic time series. Pharmacogenomics. 2004;5:525–52. doi: 10.1517/14622416.5.5.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Almon RR, Lai W, DuBois DC, Jusko WJ. Corticosteroid-regulated genes in rat kidney: mining time series array data. Am J Physiol. 2005;289:E870–82. doi: 10.1152/ajpendo.00196.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Almon RR, DuBois DC, Jin JY, Jusko WJ. Pharmacogenomic responses of rat liver to methylprednisolone: an approach to mining a rich microarray time series. AAPS J. 2005;7:E156–94. doi: 10.1208/aapsj070117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun YN, DuBois DC, Almon RR, Jusko WJ. Fourth-generation model for corticosteroid pharmacodynamics: a model for methylprednisolone effects on receptor/gene-mediated gluco-corticoid receptor down-regulation and tyrosine aminotransferase induction in rat liver. J Pharmacokinet Biopharm. 1998;26:289–317. doi: 10.1023/a:1023233409550. [DOI] [PubMed] [Google Scholar]
- 20.Yao Z, Zhao B, Hoffman EP, Ghimbovschi S, DuBois DC, Almon RR, Jusko WJ. Application of scaling factors in simultaneous modeling of microarray data from diverse chips. Pharm Res. 2007;24:643–9. doi: 10.1007/s11095-006-9215-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’Argenio DZ, Schumitzky A. ADAPT II User’s Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software. Biomedical Simulation Resource; Los Angeles: 1997. [Google Scholar]
- 22.Yao Z, Dubois DC, Almon RR, Jusko WJ. Modeling circadian rhythms of glucocorticoid receptor and glutamine synthetase expression in rat skeletal muscle. Pharm Res. 2006;23:670–679. doi: 10.1007/s11095-005-9608-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin JY, DuBois DC, Almon RR, Jusko WJ. Receptor/gene-mediated pharmacodynamic effects of methylprednisolone on phosphoenolpyruvate carboxykinase regulation in rat liver. J Pharmacol Exp Ther. 2004;309:328–39. doi: 10.1124/jpet.103.061515. [DOI] [PubMed] [Google Scholar]
- 24.Boers M, Nurmohamed MT, Doelman CJ, Lard LR, Verhoeven AC, Voskuyl AE, Huizinga TW, van de Stadt RJ, Dijkmans BA, van der Linden S. Influence of glucocorticoids and disease activity on total and high density lipoprotein cholesterol in patients with rheumatoid arthritis. Ann Rheum Dis. 2003;62:842–5. doi: 10.1136/ard.62.9.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci US A. 2003;100:13531–6. doi: 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gold R, Buttgereit F, Toyka KV. Mechanism of action of glucocorticosteroid hormones: possible implications for therapy of neuroimmunological disorders. J Neuroimmunol. 2001;117:1–8. doi: 10.1016/s0165-5728(01)00330-7. [DOI] [PubMed] [Google Scholar]
- 27.Tsintzas K, Chokkalingam K, Jewell K, Norton L, Macdonald IA, Constantin-Teodosiu D. Elevated free fatty acids attenuate the insulin-induced suppression of PDK4 gene expression in human skeletal muscle: potential role of intramuscular long-chain acylcoenzyme A. J Clin Endocrinol Metab. 2007;92:3967–72. doi: 10.1210/jc.2007-1104. [DOI] [PubMed] [Google Scholar]
- 28.Wang B, Li HL, Yang WY, Xiao JZ, Du RQ, Bai XP, Pan L. Effect of lipid infusion on the function of islet β cells and gene expression of insulin signal transduction system. Beijing Da Xue Xue Bao. 2007;39:462–466. [PubMed] [Google Scholar]
- 29.Wu X, Wang J, Cui X, Maianu L, Rhees B, Rosinski J, So WV, Willi SM, Osier MV, Hill HS, Page GP, Allison DB, Martin M, Garvey WT. The effect of insulin on expression of genes and biochemical pathways in human skeletal muscle. Endocrine. 2007;31:5–17. doi: 10.1007/s12020-007-0007-x. [DOI] [PubMed] [Google Scholar]